0022-538X/08/$08.00
⫹0 doi:10.1128/JVI.01244-08
Copyright © 2008, American Society for Microbiology. All Rights Reserved.
Transactivation, Dimerization, and DNA-Binding Activity of White
Spot Syndrome Virus Immediate-Early Protein IE1
䌤
Wang-Jing Liu,
1Yun-Shiang Chang,
2Hao-Ching Wang,
3Jiann-Horng Leu,
1Guang-Hsiung Kou,
1* and Chu-Fang Lo
1*
Institute of Zoology, National Taiwan University, Taipei, Taiwan
1; Department of Molecular Biotechnology, Da-Yeh University,
Changhua, Taiwan
2; and Institute of Biological Chemistry, Academia Sinica, Taipei, Taiwan
3Received 16 June 2008/Accepted 20 August 2008
Immediate-early proteins from many viruses function as transcriptional regulators and exhibit
transacti-vation activity, DNA binding activity, and dimerization. In this study, we investigated these characteristics in
white spot syndrome virus (WSSV) immediate-early protein 1 (IE1) and attempted to map the corresponding
functional domains. Transactivation was investigated by transiently expressing a protein consisting of the DNA
binding domain of the yeast transactivator GAL4 fused to full-length IE1. This GAL4-IE1 fusion protein
successfully activated the Autographa californica multicapsid nucleopolyhedrovirus p35 basal promoter when
five copies of the GAL4 DNA binding site were inserted upstream of the TATA box. A deletion series of
GAL4-IE1 fusion proteins suggested that the transactivation domain of WSSV IE1 was carried within its first
80 amino acids. A point mutation assay further showed that all 12 of the acidic residues in this highly acidic
domain were important for IE1’s transactivation activity. DNA binding activity was confirmed by an
electro-phoresis mobility shift assay using a probe with
32P-labeled random oligonucleotides. The DNA binding region
of WSSV IE1 was located in its C-terminal end (amino acids 81 to 224), but mutation of a putative zinc finger
motif in this C-terminal region suggested that this motif was not directly involved in the DNA binding activity.
A homotypic interaction between IE1 molecules was demonstrated by glutathione S-transferase pull-down
assay and a coimmunoprecipitation analysis. A glutaraldehyde cross-linking experiment and gel filtration
analysis showed that this self-interaction led to the formation of stable IE1 dimers.
White spot syndrome virus (WSSV) is the causative agent of
a disease that has led to severe mortalities of cultured shrimps
all over the world (10, 14, 23, 53). WSSV is a large
double-stranded DNA virus which is extremely virulent (23, 38, 39),
has a wide host range (14, 33), and targets various tissues (32,
59). It was recently erected as the type species of genus
Whispo-virus in the family Nimaviridae (56). Although the complete
sequence of the WSSV genome has been known for several
years (7, 55, 60), knowledge of the biological functions of the
viral proteins is still quite poor. The WSSV immediate-early
gene ie1 (31) was recently shown to use a shrimp signal
trans-ducer and activator of transcription (STAT) as a transcription
factor to enhance its expression and contribute to its high
promoter activity in host cells (30). In the present study, we
further investigate the characteristics of WSSV IE1. This is
made more difficult by the fact that no continuous shrimp cell
line is currently available, and while bearing in mind that a
heterologous system might introduce experimental artifacts,
here we follow previous studies (22, 30, 34) and use the Sf9
insect cell system.
Many viral immediate-early genes encode multifunctional
transcriptional regulators that both positively and negatively
modulate gene expression (26, 52, 57). These transcriptional
regulators must possess at least two functional domains,
namely, a DNA binding domain (DBD) that allows attachment
of the transactivator to its target sequence within a gene
pro-moter and a transactivation domain (TAD) that can interact
with the basal transcription machinery and promote the
tran-scription of the target genes. These two domains are often
functionally independent and physically separate. In many
cases, the activity of these transcriptional regulators is
regu-lated by homophilic interactions (35, 42) as well as by the
formation of heterodimers with other transcriptional factors.
We show here that WSSV IE1 exhibits all three of these
tran-scriptional regulator functions, and we also attempt to identify
the domains that are associated with these functions.
While the DBDs are extremely well characterized both
func-tionally and structurally, the activation domains do not share
easily recognizable motifs or structures (54). Therefore, in the
present study, the TAD of WSSV IE1 was investigated by
analyzing the transient expression of GAL4 DBD-IE1 N- or
C-terminal deletion mutants. IE1-DNA binding and the
func-tionality of a previously identified Cys
2/His
2-type zinc finger
DNA binding motif (31) were investigated using
electro-phoretic mobility shift assays (EMSAs). Finally, to investigate
the intermolecular interactions of WSSV IE1, a combination
of in vitro and in vivo assays were performed to test for IE1
homophilic interactions.
MATERIALS AND METHODS
Plasmids. (i) Luciferase effectors.The plasmid pIZ⌬IE/V5-His was used as
a starting point in dual-luciferase reporter assays. It was modified from the commercialized plasmid pIZ/V5-His (Invitrogen) by deleting the OpIE2 (Orgyia pseudotsugata multicapsid nucleopolyhedrovirus ie2) promoter lo-cated in front of the multiple cloning sites. Next, part (⬃2 kbp) of the WSSV IE1 promoter fragment upstream of the ATG was amplified from the WSSV
* Corresponding author. Mailing address: Institute of Zoology,
Na-tional Taiwan University, Taipei 106, Taiwan. Phone: 886-2-33662453.
Fax: 886-2-23638179. E-mail for Chu-Fang Lo: [email protected].
E-mail for Guang-Hsiung Kou: [email protected].
䌤
Published ahead of print on 3 September 2008.
11362
at National Taiwan Univ. on May 8, 2009
jvi.asm.org
genomic DNA (using the primers CGGAATTCGATGATGGTGATGTTTC TAGG and CCGCTCGAGCTTGAGTGGAGAGAGAGAGC [underlined sequences represent the restriction enzyme recognition sites]) and cloned
into pIZ⌬IE/V5-His. The resulting plasmid was designated pWSSV-V5-His
and was used to express the full-length ie1 coding region, the GAL4 DBD (29), and various fusion proteins consisting of the GAL4 DBD plus down-stream, in-frame insertions of different regions of the WSSV ie1 coding sequence (see Table 1 for the ie1 primers). To construct the GAL4 DBD gene plasmid (pWSSV-GAL4-V5-His), the gene sequence encoding GAL4 DBD amino acids (aa) 1 to 147 was amplified by PCR from yeast genomic DNA
(using the primers 5⬘-GCTCTAGAATGAAGCTACTGTCTTCTATC-3⬘ and
5⬘-TCCCCGCGGCGATACAGTCAACTGTCTTTG-3⬘) and then cloned into the XbaI/SacII-digested pWSSV-V5-His plasmid. One of the fusion
protein plasmids, pWSSV-GAL4-IE11–80-V5-His, contained the wild-type
IE1 sequence spanning aa 1 to 80, and this plasmid was used as a template to produce a range of N-terminal mutants. Site-directed mutations of the acidic residues of the amino terminus of IE1 were generated by using rolling-circle PCR (20) to replace the acidic residues with alanine. To confirm that only the acidic amino acids were involved in transactivation, alanine was also used to replace two randomly chosen nonacidic residues (G20A and G41A muta-tions). Mutations were verified for all plasmids by DNA sequencing analysis. The specifically designed mutagenic primers used to generate the IE1 TAD mutants are listed in Table 2.
(ii) Luciferase reporters.The reporter plasmid p35BAS-Luc, which contained
the firefly luciferase reporter gene, was constructed by PCR cloning of the
Autographa californica multicapsid nucleopolyhedrovirus (AcMNPV) p35 basal
promoter into the pGL3-Basic vector (Promega), using the primer pair AcMNPV-p35-F1 and AcMNPV-p35-R1 (Table 1). The other reporter plasmid,
G5p35BAS-Luc, contained five copies of the GAL4 DNA binding site upstream
of the AcMNPV p35 basal promoter, and it was constructed by amplifying five copies of the GAL4 DNA binding site from pG5SEAP (Clontech), using primers
GAL4bs-F and GAL4bs-R (Table 1), and then cloning them into p35BAS-Luc
vector KpnI and XhoI sites.
(iii) Glutathione S-transferase–IE1 (GST-IE1) and GST-VP36B.The plasmids
pGST-IE11–224and pGST-IE181–224were generated by cloning PCR-amplified
WSSV ie1 coding region fragments flanked by EcoRI and XhoI restriction sites into the corresponding sites of predigested pGEX-5X-1 vector (Amersham
Phar-macia Biotech). pGST-IE181–224 C2-H2mutwas constructed by rolling-circle PCR
as described above, using pGST-IE181–224as the template. The plasmid
pGST-VP36B was constructed by cloning the WSSV structural protein pGST-VP36B into the pGEX-5X-1 vector, using primers VP36B-F and VP36B-R. Primer sequences are listed in Table 3.
(iv) IE1 expression plasmids.PCR cloning was used to insert the WSSV ie1 coding region into the vectors pDHsp/V5-His and pDHsp/FLAG-His, which both contain the heat-inducible Drosophila heat shock protein 70 promoter (28). The resulting plasmids, pDHsp/IE1-V5-His and pDHsp/IE1-FLAG-His, ex-pressed the V5 and FLAG tag fusion proteins, respectively. Another IE1 expres-sion plasmid, pcDNA3/IE1, was constructed by PCR cloning the WSSV ie1 coding region into the commercialized vector pcDNA3 (Invitrogen). Primer sequences are listed in Table 3.
Transient transfections and dual-luciferase reporter assay.Transfections of Sf9 insect cells were performed using the Cellfectin reagent (Invitrogen). Briefly,
the Sf9 insect cells were seeded onto a 24-well plate (1⫻ 105
cells/well) and grown in Sf-900 II serum-free medium (Invitrogen) overnight at 27°C. Cells were
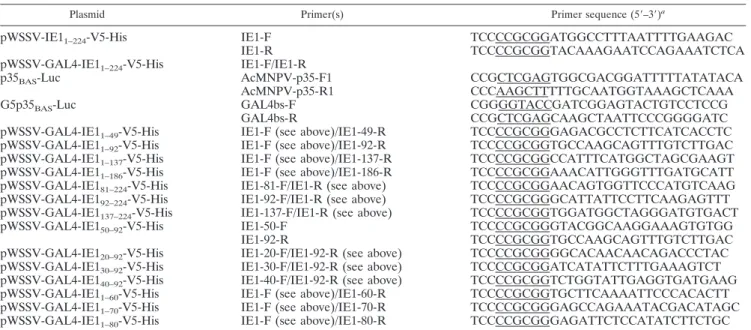
TABLE 1. Primers used for construction of luciferase reporter and effector plasmids
Plasmid Primer(s) Primer sequence (5⬘–3⬘)a
pWSSV-IE1
1–224-V5-His
IE1-F
TCCCCGCGGATGGCCTTTAATTTTGAAGAC
IE1-R
TCCCCGCGGTACAAAGAATCCAGAAATCTCA
pWSSV-GAL4-IE1
1–224-V5-His
IE1-F/IE1-R
p35
BAS-Luc
AcMNPV-p35-F1
CCGCTCGAGTGGCGACGGATTTTTATATACA
AcMNPV-p35-R1
CCCAAGCTTTTTGCAATGGTAAAGCTCAAA
G5p35
BAS-Luc
GAL4bs-F
CGGGGTACCGATCGGAGTACTGTCCTCCG
GAL4bs-R
CCGCTCGAGCAAGCTAATTCCCGGGGATC
pWSSV-GAL4-IE1
1–49-V5-His
IE1-F (see above)/IE1-49-R
TCCCCGCGGGAGACGCCTCTTCATCACCTC
pWSSV-GAL4-IE1
1–92-V5-His
IE1-F (see above)/IE1-92-R
TCCCCGCGGTGCCAAGCAGTTTGTCTTGAC
pWSSV-GAL4-IE1
1–137-V5-His
IE1-F (see above)/IE1-137-R
TCCCCGCGGCCATTTCATGGCTAGCGAAGT
pWSSV-GAL4-IE1
1–186-V5-His
IE1-F (see above)/IE1-186-R
TCCCCGCGGAAACATTGGGTTTGATGCATT
pWSSV-GAL4-IE1
81–224-V5-His
IE1-81-F/IE1-R (see above)
TCCCCGCGGAACAGTGGTTCCCATGTCAAG
pWSSV-GAL4-IE1
92–224-V5-His
IE1-92-F/IE1-R (see above)
TCCCCGCGGGCATTATTCCTTCAAGAGTTT
pWSSV-GAL4-IE1
137–224-V5-His
IE1-137-F/IE1-R (see above)
TCCCCGCGGTGGATGGCTAGGGATGTGACT
pWSSV-GAL4-IE1
50–92-V5-His
IE1-50-F
TCCCCGCGGGTACGGCAAGGAAAGTGTGG
IE1-92-R
TCCCCGCGGTGCCAAGCAGTTTGTCTTGAC
pWSSV-GAL4-IE1
20–92-V5-His
IE1-20-F/IE1-92-R (see above)
TCCCCGCGGGGCACAACAACAGACCCTAC
pWSSV-GAL4-IE1
30–92-V5-His
IE1-30-F/IE1-92-R (see above)
TCCCCGCGGATCATATTCTTTGAAAGTCT
pWSSV-GAL4-IE1
40–92-V5-His
IE1-40-F/IE1-92-R (see above)
TCCCCGCGGTCTGGTATTGAGGTGATGAAG
pWSSV-GAL4-IE1
1–60-V5-His
IE1-F (see above)/IE1-60-R
TCCCCGCGGTGCTTCAAAATTCCCACACTT
pWSSV-GAL4-IE1
1–70-V5-His
IE1-F (see above)/IE1-70-R
TCCCCGCGGGAGCCAGAAATACGACATAGC
pWSSV-GAL4-IE1
1–80-V5-His
IE1-F (see above)/IE1-80-R
TCCCCGCGGGAGATTCTCCATATCTTCTGC
aRestriction enzyme cutting sites are underlined.
TABLE 2. Sequences of mutated oligonucleotides used to generate
point mutations in the WSSV IE1 transactivation domain
Plasmid Primer sequence (5⬘–3⬘)a
pWSSV-GAL4-IE1
1–80...TTTGCCAATATGGACTTGAC
(E6A/D7A)
GAGATTTGTAGAGGCTGCAAA
pWSSV-GAL4-IE1
1–80...TTTGCCAATATGGCCTTGAC
(D16A)
GAGATTTGTAGAGTCTTCAAA
pWSSV-GAL4-IE1
1–80...CGCCCCAATATCATATTCTTT
(D24A)
GGTAGGGGCTGTTGTTGTGCC
pWSSV-GAL4-IE1
1–80...GGGGAGTAGACTTGCAAAGAA
(E34A/E43A)
AACTCTGGTATTGCGGTGATG
pWSSV-GAL4-IE1
1–80...GGGAATTTTGCAGCAAGTGGA
(E59A)
ACACTTTCCTTGCCGTACGAG
pWSSV-GAL4-IE1
1–80...TGGCTCGCAGCTAATGCAGAA
(E71A/D72A)
GAAATACGACATAGCACCTC
pWSSV-GAL4-IE1
1–80...AATGCAGCAGCTATGGAGAATC
(E75A/D76A)
ATCTTCGAGCCAGAAATACGA
pWSSV-GAL4-IE1
1–80...GCAGAAGATATGGCGAATCTC
(E78A)
ATTATCTTCGAGCCAGAAATA
pWSSV-GAL4-IE1
1–80...ACGGCTGCCACAACAACAGAC
(G20A)
CAAGTCCATATTGGCAAAGAG
pWSSV-GAL4-IE1
1–80...AACTCTGCTATTGAGGTGATG
(G41A)
GGGGAGTAGACTTTCAAAGAA
aThe underlined nucleotides show the positions of the mutations. Plasmids
with three or more mutations were constructed by nested PCRs using appropri-ate combinations of the primer sets.
at National Taiwan Univ. on May 8, 2009
jvi.asm.org
cotransfected with 300 ng of the reporter plasmid containing the firefly luciferase gene, 500 ng of one of the different effector plasmids or the empty vector, and 100 ng of the Renilla luciferase gene plasmid, phRL/AcMNPVie1 (30). The phRL/AcMNPVie1 plasmid contains the AcMNPV ie1 promoter to drive the expression of the Renilla luciferase gene and was used to monitor and normalize transfection efficiency. Cells were collected at 48 h posttransfection, and the cell lysates were prepared according to the Promega instruction manual for the dual-luciferase assay system. Luciferase activities were measured with a lumi-nometer (Labsystems). Firefly luciferase activity values were then normalized against the activities of the Renilla luciferase to correct for transfection efficiency, and data were expressed as relative luciferase activities. Luciferase activities were determined for triplicate transfections in two independent experiments, and the means and standard deviations (SD) were calculated. For the point mutation assays, statistically significant differences from the wild-type TAD expression plasmid were identified using paired Student’s t test, with significance set at P
values of⬍0.01.
Cell extracts and Western blot analysis.Total cell lysates were prepared by
directly adding 2⫻ sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) sample buffer (100 mM Tris-HCl [pH 6.8], 200 mM dithiothreitol [DTT], 4% SDS, 0.2% bromophenol blue, 20% glycerol) to cell pellets and then boiling the samples for 10 min. The samples were separated in 15% polyacryl-amide gels, transferred to a polyvinylidene difluoride membrane (MSI),
incu-bated with either anti-V5 antibody (Sigma) or anti--actin antibody (Chemicon),
and then detected with a secondary peroxidase-conjugated antibody. Detected proteins were visualized using an ECL (Perkin-Elmer) detection system.
Expression and purification of GST, GST-VP36B, GST-IE11–224, and GST-IE1 deletion mutants.GST fusion proteins were expressed and purified according to the manufacturer’s manual. After overnight culture of the GST plasmids in transformed Escherichia coli BL21 Codon Plus cells (Stratagene), the cultures
were diluted 1:200 (vol/vol) in Luria-Bertani (LB) medium containing 50g/ml
of ampicillin and then incubated for another 3 h at 37°C. Expression of the fusion proteins was induced by the addition of IPTG (isopropyl--d-thiogalactopyrano-side) to a final concentration of 1 mM, and the cultures were grown for a further 24 h at 15°C. The soluble GST fusion proteins were resuspended in lysis buffer (50 mM Tris-HCl [pH 8.0], 300 mM NaCl, 1 mM DTT, and 1 mM EDTA) and purified by affinity chromatography with an FF 16/10 GST column (Amersham Biosciences). The fusion proteins were eluted from the beads with 50 mM Tris-HCl (pH 8.0), 300 mM NaCl, 1 mM DTT, 1 mM EDTA, and 10 mM reduced glutathione, and then the purified proteins were condensed with an
Amicon Ultra-30 column (Millipore). To obtain the IE11–224protein, the GST
was removed from the GST-IE11–224fusion protein by digestion with factor Xa
(10 units of protease/1 mg GST fusion protein; Amersham Biosciences) in 1⫻ phosphate-buffered saline (PBS) containing 1 mM DTT at 22°C for 16 h. The digested GST was removed with a GST column. Purity of the samples was assessed by SDS-PAGE, and the protein concentration was determined using a Bio-Rad protein assay kit.
DNA binding assay (EMSA).EMSA was performed as described previously (49), with some modifications. Single-stranded oligonucleotides containing a 25-nucleotide random core sequence flanked on each side by 27 nucleotides
[5⬘-GTCGCTCGAGCGGTATGACGAGATCTA(N)25TAGATCTGCGTCAC
TAGTCTAGACTAG-3⬘ (where N can be any of the four deoxyribonucleotides)]
were synthesized (9). A double-stranded [␣-32P]dCTP-labeled oligonucleotide
library was generated by PCR using the forward primer 5⬘-GTCGCTCGAGCG
GTATGACG-3⬘ and the reverse primer
5⬘-CTAGTCTAGACTAGTGACGC-3⬘. Binding reactions were carried out for 30 min at room temperature in 15-l reaction mixtures that contained different concentrations of purified
recombi-nant proteins with 10 mM HEPES (pH 7.9), 1 mM DTT, 5 mM MgCl2, 0.5 mM
ZnCl2, 60 mM KCl, 0.05% NP-40, 200 ng poly(dI-dC), 10% glycerol, and 50
g/ml bovine serum albumin. The DNA-protein complexes were resolved in
7.5% polyacrylamide gels in 0.5⫻ Tris-glycine buffer (12.5 mM Tris and 100 mM
glycine). The gels were dried and visualized by autoradiography. Some EMSA
reactions were run with no ZnCl2in the binding buffer.
In vitro protein synthesis and GST pull-down assay.Coupled in vitro tran-scription-translation reactions were conducted using a TNT kit in accordance with the manufacturer’s protocol (Promega). One microgram of plasmid
pcDNA3/IE1 DNA and 2l of [35S]methionine (1,000 Ci/mmol; 10 mCi/ml)
were added to the TNT mixture (50-l total volume), and reactions were carried out at 30°C for 90 min. To ensure that there was no contamination by nucleic
acids, the purified proteins GST and GST-IE11–224 and the TNT product
[35S]methionine-labeled IE1 were all pretreated with nucleases (1 U DNase I
[Invitrogen] and 0.5g RNase [Sigma]) for 1 h at 25°C in 50 mM Tris-HCl, pH
8, 5 mM MgCl2, 2.5 mM CaCl2, 100 mM NaCl, 5% glycerol, and 1 mM DTT.
Subsequently, equal amounts of the TNT product were incubated with GST-IE11–224(10g) or GST (10 g) bound to glutathione-Sepharose beads in 150
l NETN buffer (20 mM Tris-HCl [pH 8.0], 100 mM NaCl, 1 mM EDTA, 0.5% NP-40, and a cocktail tablet of protease inhibitors [Roche]) in the presence of
ethidium bromide (100g/ml) at 4°C for 3 h. After three 10-min washes with
NETN buffer, the proteins that bound to the beads were resolved by 15% SDS-PAGE, and the gel was dried and exposed to Kodak Biomax MS film.
Coimmunoprecipitation.Sf9 cells were seeded on six-well plates (8⫻ 105
cells/well) and cotransfected with 2g pDHsp/IE1-V5-His and 2 g pDHsp/
IE1-FLAG-His expression plasmid, using Cellfectin reagent. After transfection for 16 to 18 h, the cells were heat shocked in a 42°C water bath for 30 min and then returned to 27°C. Six hours after being heat shocked, the cells were washed
with PBS and lysed in 100l of NP-40 lysis buffer (50 mM Tris-HCl, pH 8.0, 150
mM NaCl, 1% NP-40) supplemented with a protease inhibitor cocktail tablet. The lysis procedure was carried out on ice for 10 min with occasional shaking.
The lysate was centrifuged at 12,000⫻ g for 5 min, and an aliquot of the
supernatant (10l) was reserved for immunoblot analysis to confirm the
expres-sion of the transfected gene. The remaining supernatant (90l) was then
incu-bated with 15l of anti-FLAG M2 affinity gel (Sigma) at 4°C overnight with
rotation. The gel was then washed five times in 150l of NP-40 lysis buffer.
Aliquots of the total cell lysates and immunoprecipitates were separated by 15% SDS-PAGE and transferred to a polyvinylidene difluoride membrane. V5-tagged IE1 fusion proteins were detected with rabbit anti-V5 antibody (Sigma) and goat anti-rabbit immunoglobulin G–horseradish peroxidase conjugate (Sigma). FLAG-tagged IE1 was detected with mouse anti-FLAG monoclonal antibody (Sigma) and goat anti-mouse immunoglobulin G–horseradish peroxidase conju-gate (Sigma).
Gel filtration.To evaluate the native molecular size of IE1, purified IE11–224
was analyzed using a Superdex 200-pg gel filtration column (Amersham Bio-sciences) (using buffer comprised of 500 mM NaCl, 1 mM DTT, 1 mM EDTA,
TABLE 3. Primers used for construction of GST-IE1 fusion proteins and IE1 expression plasmids
Plasmid Primer Primer sequence (5⬘–3⬘)a
pGST-IE1
1–224IE1-1-F
CGGAATTCATGGCCTTTAATTTTGAAGACTC
IE1-224-R
CCGCTCGAGTTATACAAAGAATCCAGAAATC
pGST-VP36B
VP36B-F
GCATGAATTCATGGCGGTAAACTTGGATAATG
VP36B-R
GCAGCTCGAGTTATGTCCAACAATTTAAAAAG
pGST-IE1
81-224IE1-81-F/IE1-224-R (see above)
CGGAATTCAACAGTGGTTCCCATGTCAAG
pGST-IE1
81–224 C2-H2mutIE1-C2-mut-F
TGTAGGGCCAAGTACCCAGGC
IE1-C2-mut-R
CGCATTAGCTACAGAAAACAT
IE1-H2-mut-F
GGTGCTTCTGATTTGACATGT
IE1-H2-mut-R
CACTCCAGCGCCTTCAATAAC
pDHsp/IE1-V5-His
IE1-HindIII-F
CCCAAGCTTCTCAAGATGGCCTTTAATTTTG
IE1-SacII-R
TCCCCGCGGTACAAAGAATCCAGAAATCTCA
pDHsp/IE1-FLAG-His
IE1-HindIII-F (see above)/IE1-SacII-R (see above)
pcDNA3/IE1
IE1-HindIII-F (see above)/IE1-XhoI-R
CGCTCGAGTTATACAAAGAATCCAGAAAT
pET-28b(
⫹)/IE1
IE1-NdeI-F/IE1-XhoI-R (see above)
CCCATATGGCCTTTAATTTTGAAGAC
aRestriction enzyme cutting sites or mutated sites are underlined.
at National Taiwan Univ. on May 8, 2009
jvi.asm.org
and 20 mM sodium acetate, pH 5.5). Gel filtration standard proteins (bovine serum albumin [67 kDa], ovalbumin [43 kDa], chymotrypsinogen A [25 kDa], and RNase A [13.7 kDa]) were used to calibrate the column. For each protein,
the logarithm of molecular mass was plotted against Kav, which was calculated as
follows: Kav⫽ (Ve⫺ Vo)/(Vt⫺ Vo), where Veis the elution volume, Vois the
column void volume using blue dextran 2000, and Vtis the total column bed
volume (120 ml for Superdex 200-pg gel filtration column).
WSSV IE1 antibody preparation.A PCR fragment representing the coding region of ie1 was amplified using the IE1-NdeI-F/IE1-XhoI-R primer set (Table 3), digested with restriction enzymes, and cloned into pET-28b(⫹) (Novagen). The resulting pET clone was transformed into BL21 cells. For protein expression and purification, the cells were grown overnight at 37°C in LB medium
supple-mented with 50g of kanamycin/ml and 34 g of chloramphenicol/ml. The cells
were inoculated into new medium at a ratio of 1:50 and grown at 37°C for 2 to 2.5 h. Expression was induced by the addition of 1 mM IPTG, and incubation was continued for another 1.5 to 3 h. The induced bacteria were spun down at 4°C, suspended in ice-cold PBS containing 10% glycerol and a protease inhibitor cocktail tablet, and then sonicated for 3 min on ice. The insoluble debris was collected by centrifugation, suspended in PBS containing 1.5% sodium lauryl sarcosine, and solubilized by shaking at room temperature for 1 h. The super-natant was clarified by centrifugation and mixed with Ni-nitrilotriacetic acid-agarose beads (Qiagen) on a rotating wheel at 4°C for 16 h or overnight. The beads were then washed several times with ice-cold wash buffer (1 M NaCl, 10 mM Tris-HCl, pH 7.5) to remove unbound material. The fusion proteins were eluted directly from the beads with SDS sample buffer and then subjected to SDS-PAGE analysis. The protein bands containing the fusion proteins were sliced from the gel, minced, mixed with Freund’s adjuvant, and used for antibody production.
Glutaraldehyde cross-linking of proteins.For protein polymerization assays, Sf9 cells were transfected with pDHsp/IE1-V5-His plasmid DNA and heat shocked as described above. The transfected cells were then washed with PBS, lysed in a hypotonic buffer (10 mM Tris-HCl [pH 7.5], 10 mM KCl, and 5 mM
MgCl2), and incubated on ice for 20 min. The swollen cells were passed through
a 25-gauge needle 20 times to disrupt the cells. After centrifugation at 1,000⫻
g, the supernatant was incubated with glutaraldehyde (Sigma) at a final
concen-tration of 0.01% at room temperature for various times. The reactions were
stopped by the addition of an equal volume of 2⫻ SDS sample buffer, and the
samples were subjected to Western blotting using IE1 polyclonal antibody.
RESULTS
WSSV IE1 contains TADs.
As an initial indication of
whether the WSSV IE1 gene product contains a transcriptional
activation domain, the IE1 gene was fused to sequences
en-coding the 147-aa DBD of the yeast transcriptional activator
GAL4 (Fig. 1A). GAL4-IE1
1–224transactivation was
moni-tored in transient expression assays in which Sf9 cells were
transfected with a reporter plasmid that contained the
lucifer-ase gene under the control of the basal promoter of the
AcMNPV p35 gene, with (G5p35
BAS-Luc) or without (p35
BAS-Luc) GAL4 DNA binding sites (Fig. 1B). The results showed
that GAL4-IE1
1–224transactivation of the AcMNPV p35 basal
promoter was about 10 times greater when the reporter
plas-FIG. 1. GAL4-dependent IE1 transactivation. (A) Schematic
rep-resentation of the three effector plasmids. For wild-type (wt) IE1
1–224,
amino acids 1 to 224 from IE1 (dark gray bar) were placed under the
control of the WSSV ie1 promoter. GAL4 DBD had aa 1 to 147 of the
GAL4 DBD (gray bar). For the hybrid GAL4-IE1
1–224, both sequences
were fused as shown. The V5 epitope (arrowhead) was inserted after
DBD residue 147 or IE1 residue 224. (B) The reporter plasmid
p35
BAS-Luc contains the baculovirus p35 basal (BAS) promoter
(TATA box and RNA start site) linked to the firefly luciferase gene
(dark gray arrow). G5p35
BAS-Luc is identical to p35
BAS-Luc except for
the presence of five GAL4 binding sites (solid boxes) upstream of the
TATA element. (C) Transactivation by GAL4-IE1
1–224. Sf9 cells were
cotransfected with 300 ng of the indicated reporter plasmid, 500 ng of
one of the different effector plasmids or empty vector, and 100 ng of
the Renilla luciferase gene plasmid, phRL/AcMNPVie1, to correct for
transactivation efficiency. Relative luciferase activity was normalized to
that of G5p35
BAS-Luc with GAL4-IE1
1–224, which was arbitrarily set to
100%. Data show the means of six repetitions, and error bars show the
SD. (D) Western blot analysis was used to confirm the expression of
chimeric GAL4-IE1
1–224, GAL4 DBD, and IE1
1–224proteins. Protein
extracts corresponding to approximately 1
⫻ 10
5cells/lane were
sep-arated by SDS-PAGE and examined by Western blot analysis with an
anti-V5 antiserum.
at National Taiwan Univ. on May 8, 2009
jvi.asm.org
mid included the five GAL4 binding sites (Fig. 1C). In contrast,
the presence of the GAL4 DNA binding sites had no effect on
the low transactivation activity exhibited by the wild-type IE1
1–224construct, while the GAL4 DBD control also showed only low
levels of transactivation (Fig. 1C). Expression of the various
constructs was confirmed by Western blotting (Fig. 1D). We
concluded that WSSV IE1 contains at least one domain that
functions as a transcriptional activator. This conclusion was
further supported by yeast two-hybrid experiments showing
that WSSV IE1 is a strong autoactivator (data not shown).
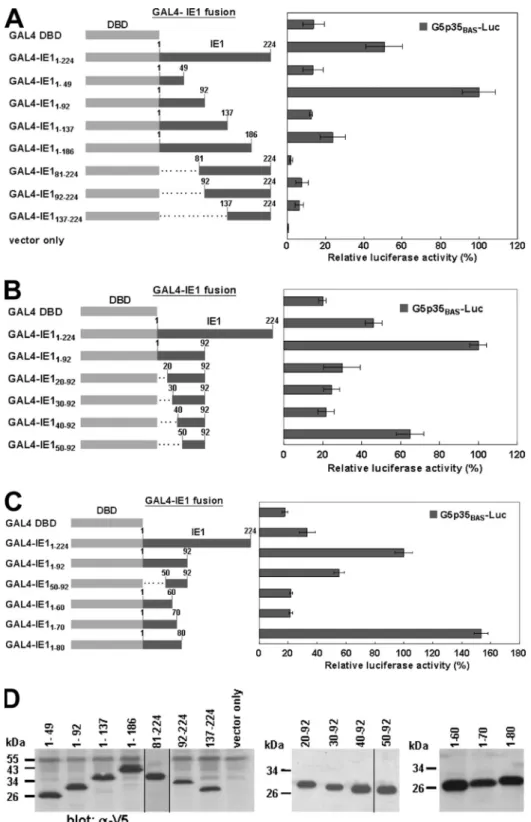
Mapping of the WSSV IE1 TAD.
In order to determine the
essential domains for transactivation, three series of deletion
mutants of WSSV IE1 were generated and constructed as
fusion proteins with the GAL4 DBD at the N terminus (Fig.
2A, B, and C). Additional V5 tags were attached to the C
termini of these fusion proteins for Western blot analysis (Fig.
2D). The various constructs encoding the IE1 deletion mutants
were transfected into Sf9 cells together with the
GAL4-respon-sive reporter plasmid G5p35
BAS-Luc. Surprisingly, coarse
mapping (Fig. 2A) showed that the transactivation activity of
most of the IE1 mutants was almost completely abolished,
except for that of GAL4-IE1
1–92, which exhibited an activity
that was 2 times higher than that of the full-length IE1 fusion
protein and 5.4 times higher than that of the GAL4 DBD
construct. These initial results suggested that WSSV IE1
res-idues 1 to 92 function as a TAD, that resres-idues 50 to 92 may be
critical for this function (compare GAL4-IE1
1–49to
GAL4-IE1
1–92), and that residues 93 to 137 may inhibit this
transac-tivation activity (compare GAL4-IE1
1–92to GAL4-IE1
1–137).
Next, the series of N-terminal and C-terminal truncation
con-structs, shown in Fig. 2B and C, respectively, were designed to
more finely delineate the boundaries of the N-terminal
activa-tion domain. Transfecactiva-tion with these series showed that
resi-dues 1 to 80 exhibited an even stronger transactivation activity
than residues 1 to 92 did (Fig. 2C), which is consistent with the
results for GAL4-IE1
81–224versus GAL4-IE1
92–224(Fig. 2A).
These results suggest that the minimal IE1 TAD may be
lo-cated within aa 1 to 80. Two additional potential inhibitory
domains were also identified. The results of deletions made in
the region of aa 1 to 49 suggested that amino acid residues 41
to 49 might act as an inhibitory domain (Fig. 2B, compare
GAL4-IE1
50–92to
GAL4-IE1
20–92,
GAL4-IE1
30–92,
and
GAL4-IE1
40–92). The other possible inhibitory domain was
identified at residues 81 to 92 (Fig. 2C, compare GAL4-IE1
1–92to GAL4-IE1
1–80), although we note that the different
activa-tion levels might have been due, at least in part, to the larger
quantity of expressed GAL4-IE1
1–80fusion protein. The
West-ern blots in Fig. 2D confirmed that all of these constructs were
successfully expressed. Note that every deletion mutant had a
higher expression level than that of the wild-type IE1 fusion
protein (compare Fig. 2D to Fig. 1D). These higher expression
levels were probably due to the shorter lengths of the
tran-scripts and encoded proteins, which would lead to increased
transcriptional and translational efficiencies.
Negatively charged amino acids are important for IE1
transactivation activity.
Sequence analysis shows that there are
12 acidic amino acids and 6 basic residues in the IE1 minimal
TAD (aa 1 to 80), giving the TAD a net negative charge of
⫺6
and a pI of 4.3. Since most TADs can be classified as either
acidic activators (46), glutamine-rich activators (11), aromatic
and hydrophobic activators (44), or proline-rich activators
(37), the acidity of the WSSV IE1 TAD suggested that it might
fall within the acidic class of activation domains. We
investi-gated this possibility by constructing mutants (Table 2) in
which alanine (A) was used to replace the wild-type aspartate
(D), glutamate (E), or glycine (G) residues. GAL4
transacti-vation assays showed that almost all of these alanine
substitu-tions significantly reduced G5p35
BAS-Luc activation (P
⬍
0.01). Replacement of increasing numbers of acidic residues
led to a further decrease in transactivation activity (Fig. 3A),
while substitutions of nonacidic residues (G20A and G41A)
(Fig. 3A) had no significant effect on transactivation. We
there-fore concluded that the negatively charged residues are critical
for WSSV IE1 transactivation activity. An immunoblot analysis
of the mutated IE1 proteins after a typical transfection showed
that most of the alanine substitution mutants produced
pro-teins at levels close to that of the wild-type construct (Fig. 3B).
Only the E71A/D72A mutant exhibited a very low level of
protein expression. This may have been due to instability and
rapid degradation of the expressed protein, which might in
turn explain why this protein exhibited the lowest
transactiva-tion activity in Fig. 3B. Meanwhile, the results for all other
mutants indicate that in the case of IE1, as with other virus
immediate-early proteins, specific acidic amino acids are
par-ticularly critical to transcriptional function (6, 15).
DNA binding activity of WSSV IE1.
The DNA binding
ac-tivity of WSSV IE1 was investigated by gel mobility shift assays
using a 79-bp double-stranded DNA oligonucleotide
contain-ing a central 25-bp randomized sequence. GST-tagged versions
of IE1 and VP36B (a WSSV structural protein which served as
a negative control) were expressed in E. coli to produce either
GST-IE1
1–224, GST-VP36B, or IE1
1–224alone. These soluble,
well-expressed proteins were then purified with
glutathione-Sepharose beads (Fig. 4A). The entire purification process was
conducted in the presence of EDTA to ensure that the
ex-pressed proteins were free of Zn
2⫹contamination. Purified
IE1
1–224and GST-IE1
1–224proteins both retarded the
migra-tion of the oligonucleotide and formed major discrete bands by
7.5% native PAGE (Fig. 4B, lanes 4 to 6 and 7 to 9,
respec-tively). Furthermore, the intensities of these bands increased
with increasing amounts of purified protein. In contrast, no
complex was formed with the GST or with the purified WSSV
structural protein control GST-VP36B (Fig. 4B, lanes 1 to 3
and 11). The last three lanes in Fig. 4B show that Zn
2⫹is not
required for the DNA binding activity of IE1
1–224. These
pre-liminary data suggested that even though the ie1 coding region
contains a Cys
2/His
2-type zinc finger motif (X
3-Cys-X
3–4-Cys-X
12-His-X
3–4-His-X
4) (31), WSSV IE1 may not in fact be a
zinc finger protein. This was further investigated in the
follow-ing experiments.
The WSSV IE1 C-terminal region is required for DNA
bind-ing.
Since DBDs and TADs usually do not overlap, we
hypoth-esized that the DBD of IE1 was located in the C-terminal
region. To test this hypothesis, we expressed and purified two
GST-IE1 proteins: GST-IE1
81–224was a deletion without the
N-terminal TAD, and GST-IE1
81–224C2-H2mutwas identical
ex-cept for a mutated zinc finger motif (Fig. 5A). An SDS-PAGE
gel of these two fusion proteins is shown in Fig. 5B. The lower
band, at
⬃26 kDa, was confirmed by Western blot analysis to
be nonfused GST (data not shown). The EMSA showed that
at National Taiwan Univ. on May 8, 2009
jvi.asm.org
FIG. 2. Identification of IE1 TAD. (A to C) Three series of deletion assays to identify the location of the IE1 TAD. The GAL4-IE1 fusion
proteins were constructed by joining the GAL4 DBD from amino acids 1 to 147 (gray bar) to the indicated segments of IE1 (dark gray bars). Sf9
cells were cotransfected with reporter plasmid G5p35
BAS-Luc, the indicated GAL4-IE1 fusion plasmids, GAL4 DBD, or empty vector, and 100 ng
of the Renilla luciferase gene plasmid, phRL/AcMNPVie1, and then assayed for luciferase activity. Relative luciferase activities were normalized
with respect to that of GAL4-IE1
1–92, which was arbitrarily set to 100%. Data show the means of six repetitions, and error bars show the SD.
(D) Western blot analysis of chimeric GAL4-IE1 proteins was performed as described in the legend to Fig. 1.
at National Taiwan Univ. on May 8, 2009
jvi.asm.org
when the TAD was deleted, IE1 was still able to bind DNA in
either the presence or absence of Zn
2⫹(Fig. 5C, lanes 2 to 4
and 5 to 7). When the deletion’s zinc finger domain was point
mutated, DNA binding activity was not markedly affected in
either the presence or absence of Zn
2⫹(Fig. 5C, lanes 8 to 10
and 11 to 13). The binding activity of the mutant was not
affected by the presence of Zn
2⫹ions.
IE1 has a strong affinity for self-interaction.
Many virus
immediate-early proteins are in dimeric form when they bind
to DNA (8, 16, 40, 57). We therefore performed an in vitro
biochemical binding assay to determine whether IE1 can also
self-interact directly. For this assay, the GST-IE1
1–224fusion
protein was bound to glutathione-Sepharose beads and
incu-bated with in vitro-translated, [
35S]methionine-labeled IE1.
SDS-PAGE analysis showed that
35S-labeled IE1 bound to the
GST-IE1
1–224fusion protein but not to GST, indicating that
IE1 can interact directly with itself (Fig. 6A). We also studied
the homotypic interaction between IE1 proteins by a
coimmu-noprecipitation assay using V5-tagged and FLAG-tagged
ver-sions of IE1 expressed in Sf9 insect cells. Complexes consisting
of IE1-V5 plus IE1-FLAG were coimmunoprecipitated by
anti-FLAG antiserum and detected by Western blotting using
anti-V5 antibody (Fig. 6B). Both sets of results indicated that
IE1 can undergo specific self-interaction directly.
IE1 protein forms a dimer.
Both gel filtration
chromatogra-phy and chemical cross-linking were used to investigate the
form of IE1 polymerization. Gel filtration chromatography
using a Superdex 200-pg gel filtration column revealed that the
major peak of purified IE1 eluted with an apparent molecular
size of 46 kDa, as calculated from the logarithm of molecular
size against the K
avvalues of protein standards that were
frac-tionated in the same column (Fig. 6C, inset). In addition to the
major elution fraction, some IE1 was also found in fractions
where the 25-kDa chymotrypsinogen A appeared (Fig. 6C).
Since IE1 is composed of 224 aa residues, with a molecular size
of approximately 25 kDa, these results suggest that a portion of
IE1 is in the monomeric state, most likely in dynamic
equilib-rium with the dimeric form. This finding suggested that IE1
exists mainly as a dimer in solution. For the cross-linking study,
Sf9 cells were transfected with an expression plasmid that
contained the full-length IE1 coding region under the control
of the Drosophila heat shock protein 70 promoter. The cellular
lysates were cross-linked with glutaraldehyde, and IE1 was
detected by immunoblotting using anti-IE1 antibody. With
in-FIG. 3. Effects of substituting alanine (A) for the negatively charged acidic amino acids in the IE1 TAD. Labels indicate the original amino
acid(s) (D, aspartate; E, glutamate; and G, glycine [control]) and its location relative to the N terminus of the IE1 coding region. (A) The reporter
plasmid was cotransfected into Sf9 cells with effector plasmids expressing the IE1 wild-type TAD or the indicated point mutants and with the
Renilla luciferase gene plasmid. Relative luciferase activities were normalized with respect to that of GAL4-IE1
1–80(wild-type TAD), which was
defined as 100%. Data show the means of six repetitions, and error bars show the SD. Activities that were significantly different from that of
wild-type TAD are indicated with asterisks (P
⬍ 0.01). (B) Western blot analysis of GAL-IE1 wild-type TAD and its substitution mutants was
performed as described in the legend to Fig. 1. The lower panel shows the internal control: total proteins were probed using anti-
-actin antibody.
at National Taiwan Univ. on May 8, 2009
jvi.asm.org
creasing treatment time, there was a steady accumulation of
dimeric (50 kDa) IE1 (Fig. 6D). Since IE1 was expressed at
extremely high levels under these conditions, the amount of
monomeric polypeptide (25 kDa) did not decrease
signifi-cantly, even when there was extensive dimer formation.
DISCUSSION
Our data suggest that the IE1 residues sufficient for
trans-activation are confined to the N terminus (Fig. 2). The
N-terminal stretch of IE1 from residues 1 to 80 is highly acidic,
with a net charge of
⫺6 and a theoretical pI of 4.3. Acid-rich
transactivation regions are characteristic of acidic
transactiva-tors (26, 46), and to determine which amino acids are involved
in mediating the activity of this domain, point mutations were
introduced into the sequence. Our results (Fig. 3A) showed
that the double mutations generally had a greater effect on
activation than the single mutations did, that the triple and
quadruple mutations had an even greater effect, and that the
glycine mutants were not significantly different from the
wild-type TAD control. This is in broad agreement with previous
mutational analyses of the activation domains of GAL4,
GCN4, and the herpes simplex virus (HSV) transactivator
VP16, all of which revealed a positive correlation between the
number of acidic amino acids and the transcriptional ability of
the acidic activation domains (12, 18, 21). Clearly, however, net
negative charge was not the sole determinant of activity,
be-cause some residues were more critical than others. For
in-stance, the E59A mutation reduced activity further than the
E78A mutation did, and activity was lower with E34A/E43A
mutations than with E75A/D76A mutations. These results
in-dicate that the transcriptional activity is dependent not only on
the net negative charge but also on the position of the acidic
residue in the WSSV IE1 TAD. To explain why some acidic
residues might be more important than others, we note that
acidic activators interact with general transcription factors such
as transcription factor IIB and TATA-box binding protein (3,
25, 45), and we further note that in the case of the TAD of
FIG. 4. DNA binding activity of IE1 and effect of zinc ions on DNA binding. (A) SDS-PAGE analysis of purified proteins used for EMSA.
(B) EMSA was performed with a radiolabeled 79-bp DNA probe and 0.5 to 3
g of the indicated proteins. Lane 10 contained no protein and was
used as a negative control. The bands containing the protein-DNA complexes are indicated.
at National Taiwan Univ. on May 8, 2009
jvi.asm.org
HSV VP16, one specific acidic residue (E476) was shown to be
the most important residue for this transactivator’s interaction
with the human general cofactor PC4 (25). Since IE1 also
presumably interacts with various factors and cofactors, we
therefore hypothesize that each of these interactions is likewise
mediated by specific acidic residues. Alternatively, it is also
possible that mutation of the acidic amino acids in IE1’s TAD
may diminish IE1’s activity by disrupting its secondary
struc-ture, as shown for other transcription factors (13, 48).
In the region of the TAD (aa 1 to 80) of WSSV IE1, three
possible inhibitory domains were identified, at aa 41 to 49, aa
81 to 92, and aa 93 to 137. To date, little is known about
sequences which mediate transcription inhibition and which
are present within transcriptional activators. We note,
how-ever, that the WSSV IE1 potential inhibitory domain from aa
41 to 49 contains a large proportion of positively charged
amino acids (GIEVMKRRL [the three basic residues are
un-derlined]). Slack and Blissard (50) suggested that the
substan-tial concentrations of positively charged amino acids in two
inhibition domains of the baculovirus AcMNPV IE1 may act to
neutralize the adjacent activation region. It is possible that the
basic amino acids in WSSV IE1 aa 41 to 49 are likewise
re-sponsible for negatively regulating WSSV IE1’s transcriptional
activation. The putative WSSV IE1 inhibitory domains at aa 81
to 92 and aa 93 to 137 do not contain large proportions of basic
residues. If they regulate the TAD activity, it is therefore
probable that they do so either by direct interactions with
components of the general transcription factors (4, 51) or by
indirect interactions through secondary “inhibitor” proteins
that mask the activation domain (2, 5).
In addition to the TAD, most transcriptional factors also
require a second region that confers specificity for target
genes. This region may confer target gene specificity either
directly (in the form of a DBD) or indirectly (by serving as an
interface for protein-protein interactions with factors bound to
target genes). Our EMSA results (Fig. 5C) suggested that IE1
has a DBD in the C terminus, and a previous amino acid
sequence analysis of this region identified a putative classic
zinc finger Cys
2/His
2domain between aa 186 and 215 (31).
When this zinc finger motif was mutated, however, the mutant
protein still retained its ability to bind DNA, and the absence
of Zn
2⫹failed to impair this ability (Fig. 5C, lanes 8 to 10 and
11 to 13). We therefore concluded that IE1’s putative zinc
finger motif cannot be directly responsible for its DNA binding
activity. In further support of this conclusion, we also note that
classic zinc finger motifs usually contain a compact
␣
struc-ture (27, 41, 43, 58), but the predicted secondary strucstruc-ture of
the WSSV IE1 zinc finger motif obtained using the NNpredict,
SOPMA, JPRED, and PHD programs (http://ca.expasy.org
/tools/) does not contain this conserved structure (data not
shown).
When there is only a single predicted zinc finger in a
tran-scription factor (for example, human cytomegalovirus
imme-diate-early protein IE2 [1]), it is not always used to bind DNA.
On the contrary, when transcription factors use zinc fingers to
bind DNA, there are usually several (often three or more)
fingers involved (24, 36, 58). Furthermore, the involvement of
zinc finger motifs in other activities has also been documented
for viruses. For instance, the herpesvirus saimiri
immediate-early protein ORF57 is a transcriptional activator with a zinc
finger-like domain in its C terminus, and during a herpesvirus
saimiri infection, this domain is required for transactivation,
repression of viral proteins, and the redistribution of the host
splicing factor SC-35 (19). Other examples include adenovirus
E1A, which has a zinc finger domain that functions in
protein-protein interactions and transactivation activity (17, 47), and
HSV type 1 immediate-early protein ICP27, whose C-terminal
zinc finger domain is required for ICP27 self-interaction (61).
In the case of IE1, several observations are relevant to the
possible function of its zinc finger. Like many other virus
im-mediate-early proteins that bind DNA in a dimeric form (8, 16,
40, 57), our evidence suggests that WSSV IE1 also has this
FIG. 5. The DNA binding domain of IE1 is located in the C terminus,
and the putative zinc finger motif is not essential for DNA binding.
(A) Schematic representation of the GST-IE1 fusion proteins used for the
EMSA reaction. In GST-IE1
81–224, the IE1 N-terminal TAD was deleted;
in GST-IE1
81–224C2-H2mut, the cysteine and histidine residues in the
puta-tive zinc finger motif were also replaced by alanine. (B) SDS-PAGE
analysis of the purified proteins used for EMSA reactions. (C) DNA
binding properties of the IE1 N-terminally truncated fusion proteins.
Both GST-IE1
81–224(0.5 to 3
g) and GST-IE1
81–224 C2-H2mut(0.5 to 3
g)
were detected by EMSA, regardless of whether Zn ions were present in
the DNA binding buffer. The arrows indicate the protein-DNA
com-plexes.
at National Taiwan Univ. on May 8, 2009
jvi.asm.org
characteristic. Starting from the EMSA results (Fig. 4B and
5C), the presence of only a single band of IE1-DNA complex
suggests that only a single form of IE1 was involved in DNA
binding. This single form was most likely the IE1 homodimer,
because when the same purified IE1
1–224construct that was
used for Fig. 4B was subjected to gel filtration
chromatogra-phy, IE1’s apparent molecular size was 46 kDa (i.e.,
approxi-mately double the predicted molecular size of the IE1
mono-mer [
⬃25 kDa]). IE1’s ability to self-interact is also supported
by the GST pull-down and coimmunoprecipitation data (Fig.
6A and B) and by the glutaraldehyde cross-linking analysis
(Fig. 6D). Thus, it is possible that, as in ICP27 (61), the
puta-tive zinc finger motif of WSSV IE1 may be involved in the
formation of the IE1 homodimer. More work will be needed to
investigate this possibility and to elucidate the mechanisms
involved.
In conclusion, we have been able to ascribe several functions
to separate regions of the WSSV IE1 protein. Our data suggest
that WSSV IE1 has at least two distinguishable domains, an
N-terminal region that is essential for transactivation and a
C-terminal region that is required for DNA binding activity.
We also conclude that IE1 probably occurs primarily as a
homodimeric protein.
ACKNOWLEDGMENTS
This investigation was supported financially by a National Science
Council grant (NSC96-2317-B-002-005) and by the Council of
Agri-culture (97AS-14.1.1-AQ-B1).
We are indebted to Paul Barlow for his helpful criticism.
REFERENCES
1. Asmar, J., L. Wiebusch, M. Truss, and C. Hagemeier. 2004. The putative zinc finger of the human cytomegalovirus IE2 86-kilodalton protein is dis-pensable for DNA binding and autorepression, thereby demarcating a con-cise core domain in the C terminus of the protein. J. Virol. 78:11853–11864. 2. Baichwal, V. R., A. Park, and R. Tjian. 1992. The cell-type-specific activator region of c-Jun juxtaposes constitutive and negatively regulated domains. Genes Dev. 6:1493–1502.
3. Blair, W. S., H. P. Bogerd, S. J. Madore, and B. R. Cullen. 1994. Mutational analysis of the transcription activation domain of RelA: identification of a highly synergistic minimal acidic activation module. Mol. Cell. Biol. 14:7226– 7234.
FIG. 6. Homotypic interaction and dimerization of IE1. (A) SDS-PAGE analysis showing the reaction of GST-IE1 fusion protein with
[
35S]methionine-labeled IE1 protein (input). The full-length IE1 protein is indicated by the arrow. (B) Self-interaction of IE1 demonstrated by
coimmunoprecipitation. V5-tagged IE1 was transiently coexpressed in Sf9 cells with FLAG-tagged IE1. Six hours after being heat shocked, the cell
lysates were harvested and immunoprecipitated with anti-FLAG M2 affinity resins. The immunoprecipitated complexes were then subjected to
Western blot analysis with anti-V5 antibody. The detected V5-tagged IE1 is indicated by the arrow. The two panels on the left show the
FLAG-and V5-tagged IE1 inputs. (C) Size exclusion chromatography of IE1 on a Superdex 200-pg gel filtration column monitored at 280 nm. The protein
standards bovine serum albumin (BSA; 67 kDa), ovalbumin (OA; 43 kDa), chymotrypsinogen A (CT; 25 kDa), and RNase A (13.7 kDa) were
fractionated on the same column. The inset shows a plot of the K
avfor each protein against the logarithm of its molecular size (log MWt).
(D) Kinetic study of glutaraldehyde cross-linking of IE1 expressed in Sf9 cells. Transiently expressed V5-tagged IE1 was treated with 0.01%
glutaraldehyde for the indicated times at room temperature and subjected to Western blotting with anti-IE1 antibody. Mock, transfection with
vector plasmid.
at National Taiwan Univ. on May 8, 2009
jvi.asm.org
4. Boyd, J. M., P. M. Loewenstein, Q. Tang, L. Yu, and M. Green. 2002. Adenovirus E1A N-terminal amino acid sequence requirements for repres-sion of transcription in vitro and in vivo correlate with those required for E1A interference with TBP-TATA complex formation. J. Virol. 76:1461– 1474.
5. Brown, H. J., J. A. Sutherland, A. Cook, A. J. Bannister, and T. Kouzarides. 1995. An inhibitor domain in c-Fos regulates activation domains containing the HOB1 motif. EMBO J. 14:124–131.
6. Buczynski, K. A., S. K. Kim, and D. J. O’Callaghan. 1999. Characterization of the transactivation domain of the equine herpesvirus type 1 immediate-early protein. Virus Res. 65:131–140.
7. Chen, L.-L., H.-C. Wang, C.-J. Huang, S.-E. Peng, Y.-G. Chen, S.-J. Lin,
W.-Y. Chen, C.-F. Dai, H.-T. Yu, C.-H. Wang, C.-F. Lo, and G.-H. Kou.2002. Transcriptional analysis of the DNA polymerase gene of shrimp white spot syndrome virus. Virology 301:136–147.
8. Chiou, C.-J., J. Zong, I. Waheed, and G. S. Hayward. 1993. Identification and mapping of dimerization and DNA-binding domains in the C terminus of the IE2 regulatory protein of human cytomegalovirus. J. Virol. 67:6201–6214. 9. Choi, Y.-S., and S. Sinha. 2006. Determination of the consensus
DNA-binding sequence and a transcriptional activation domain for ESE-2. Bio-chem. J. 398:497–507.
10. Chou, H.-Y., C.-Y. Huang, C.-H. Wang, H.-C. Chiang, and C.-F. Lo. 1995. Pathogenicity of a baculovirus infection causing white spot syndrome in cultured penaeid shrimp in Taiwan. Dis. Aquat. Organ. 23:165–173. 11. Courey, A. J., D. A. Holtzman, S. P. Jackson, and R. Tjian. 1989. Synergistic
activation by the glutamine-rich domains of human transcription factor Sp1. Cell 59:827–836.
12. Cress, W. D., and S. J. Triezenberg. 1991. Critical structural elements of the VP16 transcriptional activation domain. Science 251:87–90.
13. Defossez, P. A., J. L. Baert, M. Monnot, and Y. de Launoit. 1997. The ETS family member ERM contains an alpha-helical acidic activation domain that contacts TAFII60. Nucleic Acids Res. 25:4455–4463.
14. Flegel, T. W. 1997. Special topic review: major viral diseases of the black tiger prawn (Penaeus monodon) in Thailand. World J. Microbiol. Biotech. 13:422– 433.
15. Forsythe, I. J., C. E. Shippam, L. G. Willis, S. Stewart, T. Grigliatti, and
D. A. Theilmann.1998. Characterization of the acidic domain of the IE1 regulatory protein from Orgyia pseudotsugata multicapsid nucleopolyhedro-virus. Virology 252:65–81.
16. Gallinari, P., K. Wiebauer, M. C. Nardi, and J. Jiricny. 1994. Localization of a 34-amino-acid segment implicated in dimerization of the herpes simplex virus type 1 ICP4 polypeptide by a dimerization trap. J. Virol. 68:3809–3820. 17. Geisberg, J. V., W. S. Lee, A. J. Berk, and R. P. Ricciardi. 1994. The zinc finger region of the adenovirus E1A transactivating domain complexes with the TATA box binding protein. Proc. Natl. Acad. Sci. USA 91:2488–2492. 18. Gill, G., and M. Ptashne. 1987. Mutants of GAL4 protein altered in an
activation function. Cell 51:121–126.
19. Goodwin, D. J., K. T. Hall, M. S. Giles, M. A. Calderwood, A. F. Markham,
and A. Whitehouse.2000. The carboxy terminus of the herpesvirus saimiri ORF 57 gene contains domains that are required for transactivation and transrepression. J. Gen. Virol. 81:2253–2265.
20. Hemsley, A., N. Arnheim, M. D. Toney, G. Cortopassi, and D. J. Galas. 1989. A simple method for site-directed mutagenesis using the polymerase chain reaction. Nucleic Acids Res. 17:6545–6551.
21. Hope, I. A., S. Mahadevan, and K. Struhl. 1988. Structural and functional characterization of the short acidic transcriptional activation region of yeast GCN4 protein. Nature 333:635–640.
22. Hossain, M. S., S. Khadijah, and J. Kwang. 2004. Characterization of ORF89—a latency-related gene of white spot syndrome virus. Virology 325: 106–115.
23. Inouye, K., S. Miwa, N. Oseko, H. Nakano, and T. Kimura. 1994. Mass mortalities of cultured kuruma shrimp, Penaeus japonicus, in Japan in 1993: electron microscopic evidence of the causative virus. Fish Pathol. 29:149– 158.
24. Iuchi, S. 2001. Three classes of C2H2zinc finger proteins. Cell. Mol. Life Sci.
58:625–635.
25. Jonker, H. R., R. W. Wechselberger, R. Boelens, G. E. Folkers, and R.
Kaptein.2005. Structural properties of the promiscuous VP16 activation domain. Biochemistry 44:827–839.
26. Kovacs, G. R., J. Choi, L. A. Guarino, and M. D. Summers. 1992. Functional dissection of the Autographa californica nuclear polyhedrosis virus immedi-ate-early 1 transcriptional regulatory protein. J. Virol. 66:7429–7437. 27. Lee, M. S., G. P. Gippert, K. V. Soman, D. A. Case, and P. E. Wright. 1989.
Three-dimensional solution structure of a single zinc finger DNA-binding domain. Science 245:635–637.
28. Leu, J.-H., Y.-C. Kuo, G.-H. Kou, and C.-F. Lo. 2008. Molecular cloning and characterization of an inhibitor of apoptosis protein (IAP) from the tiger shrimp, Penaeus monodon. Dev. Comp. Immunol. 32:121–133.
29. Lin, Y.-S., M. Carey, M. Ptashne, and M. R. Green. 1990. How different eukaryotic transcriptional activators can cooperate promiscuously. Nature
345:359–361.
30. Liu, W.-J., Y.-S. Chang, A. H.-J. Wang, G.-H. Kou, and C.-F. Lo. 2007. White
spot syndrome virus annexes a shrimp STAT to enhance expression of the immediate-early gene ie1. J. Virol. 81:1461–1471.
31. Liu, W.-J., Y.-S. Chang, C.-H. Wang, G.-H. Kou, and C.-F. Lo. 2005. Mi-croarray and RT-PCR screening for white spot syndrome virus immediate-early genes in cycloheximide-treated shrimp. Virology 334:327–341. 32. Lo, C.-F., C.-H. Ho, C.-H. Chen, K.-F. Liu, Y.-L. Chiu, P.-Y. Yeh, S.-E. Peng,
H.-C. Hsu, H.-C. Liu, C.-F. Chang, M.-S. Su, C.-H. Wang, and G.-H. Kou.
1997. Detection and tissue tropism of white spot syndrome baculovirus (WSBV) in captured brooders of Penaeus monodon with a special emphasis on reproductive organs. Dis. Aquat. Organ. 30:53–72.
33. Lo, C.-F., C.-H. Ho, S.-E. Peng, C.-H. Chen, H.-C. Hsu, Y.-L. Chiu, C.-F.
Chang, K.-F. Liu, M.-S. Su, C.-H. Wang, and G.-H. Kou.1996. White spot syndrome baculovirus (WSBV) detected in cultured and captured shrimp, crabs and other arthropods. Dis. Aquat. Organ. 27:215–225.
34. Lu, L., H. Wang, I. Manopo, L. Yu, and J. Kwang. 2005. Baculovirus-mediated promoter assay and transcriptional analysis of white spot syndrome virus orf427 gene. Virol. J. 2:71. http://www.virologyj.com/content/2/1/71. 35. Massari, M. E., and C. Murre. 2000. Helix-loop-helix proteins: regulators of
transcription in eucaryotic organisms. Mol. Cell. Biol. 20:429–440. 36. McDowall, J. 2007. Protein of the month: zinc fingers. http://www.ebi.ac.uk
/interpro/potm/2007_3/Page1.htm.
37. Mermod, N., E. A. O’Neill, T. J. Kelly, and R. Tjian. 1989. The proline-rich transcriptional activator of CTF/NF-I is distinct from the replication and DNA binding domain. Cell 58:741–753.
38. Momoyama, K., M. Hiraoka, H. Nakano, H. Koube, K. Inouye, and N.
Oseko.1994. Mass mortalities of cultured kuruma shrimp, Penaeus japonicus, in Japan in 1993: histopathological study. Fish Pathol. 29:141–148. 39. Nakano, H., H. Koube, S. Umezawa, K. Momoyama, M. Hiraoka, K. Inouye,
and N. Oseko.1994. Mass mortalities of cultured kuruma shrimp, Penaeus
japonicus, in Japan in 1993: epizootiological survey and infection trails. Fish
Pathol. 29:135–139.
40. Olson, V. A., J. A. Wetter, and P. D. Friesen. 2003. The highly conserved basic domain I of baculovirus IE1 is required for hr enhancer DNA binding and hr-dependent transactivation. J. Virol. 77:5668–5677.
41. Parraga, G., S. J. Horvath, A. Eisen, W. E. Taylor, L. Hood, E. T. Young, and
R. E. Klevit.1988. Zinc-dependent structure of a single-finger domain of yeast ADR1. Science 241:1489–1492.
42. Patikoglou, G., and S. K. Burley. 1997. Eukaryotic transcription factor-DNA complexes. Annu. Rev. Biophys. Biomol. Struct. 26:289–325.
43. Pavletich, N. P., and C. O. Pabo. 1991. Zinc finger-DNA recognition: crystal structure of a Zif268-DNA complex at 2.1 A. Science 252:809–817. 44. Regier, J. L., F. Shen, and S. J. Triezenberg. 1993. Pattern of aromatic and
hydrophobic amino acids critical for one of two subdomains of the VP16 transcriptional activator. Proc. Natl. Acad. Sci. USA 90:883–887. 45. Roberts, S. G., I. Ha, E. Maldonado, D. Reinberg, and M. R. Green. 1993.
Interaction between an acidic activator and transcription factor TFIIB is required for transcriptional activation. Nature 363:741–744.
46. Sadowski, I., J. Ma, S. Triezenberg, and M. Ptashne. 1988. GAL4-VP16 is an unusually potent transcriptional activator. Nature 335:563–564.
47. Sanchez, T. A., I. Habib, J. L. Booth, S. M. Evetts, and J. P. Metcalf. 2000. Zinc finger and carboxyl regions of adenovirus E1A 13S CR3 are important for transactivation of the cytomegalovirus major immediate early promoter by adenovirus. Am. J. Respir. Cell Mol. Biol. 23:670–677.
48. Schmitz, M. L., M. A. dos Santos Silva, H. Altmann, M. Czisch, T. A. Holak,
and P. A. Baeuerle.1994. Structural and functional analysis of the NF-kappa B p65 C terminus. An acidic and modular transactivation domain with the potential to adopt an alpha-helical conformation. J. Biol. Chem. 269:25613– 25620.
49. Sekimata, M., and Y. Homma. 2004. Sequence-specific transcriptional re-pression by an MBD2-interacting zinc finger protein MIZF. Nucleic Acids Res. 32:590–597.
50. Slack, J. M., and G. W. Blissard. 1997. Identification of two independent transcriptional activation domains in the Autographa californica multicapsid nuclear polyhedrosis virus IE1 protein. J. Virol. 71:9579–9587.
51. Song, C.-Z., P. M. Loewenstein, K. Toth, and M. Green. 1995. Transcription factor TFIID is a direct functional target of the adenovirus E1A transcrip-tion-repression domain. Proc. Natl. Acad. Sci. USA 92:10330–10333. 52. Stenberg, R. M. 1996. The human cytomegalovirus major immediate-early
gene. Intervirology 39:343–349.
53. Takahashi, Y., T. Itami, M. Kondo, M. Maeda, R. Fujii, S. Tomonaga, K.
Supamattaya, and S. Boonyaratpalin.1994. Electron microscopic evidence of bacilliform virus infection in kuruma shrimp (Penaeus japonicus). Fish Pathol. 29:121–125.
54. Triezenberg, S. J. 1995. Structure and function of transcriptional activation domains. Curr. Opin. Gen. Dev. 5:190–196.
55. van Hulten, M. C. W., J. Witteveldt, S. Peters, N. Kloosterboer, R. Tarchini,
M. Fiers, H. Sandbrink, R. K. Lankhorst, and J. M. Vlak.2001. The white spot syndrome virus DNA genome sequence. Virology 286:7–22. 56. Vlak, J. M., J. R. Bonami, T. W. Flegel, G.-H. Kou, D. V. Lightner, C.-F. Lo,
P. C. Loh, and P. J. Walker.2004. Nimaviridae, p. 187–192. In C. M. Fauquet, M. A. Mayo, J. Maniloff, U. Desselberger, and L. A. Ball (ed.), VIIIth report
at National Taiwan Univ. on May 8, 2009
jvi.asm.org
of the International Committee on Taxonomy of Viruses. Elsevier, Amster-dam, The Netherlands.
57. West, J. T., and C. Wood. 2003. The role of Kaposi’s sarcoma-associated herpesvirus/human herpesvirus-8 regulator of transcription activation (RTA) in control of gene expression. Oncogene 22:5150–5163.
58. Wolfe, S. A., L. Nekludova, and C. O. Pabo. 2000. DNA recognition by Cys2His2 zinc finger proteins. Annu. Rev. Biophys. Biomol. Struct. 29:183– 212.
59. Wongteerasupaya, C., J. E. Vickers, S. Sriurairatana, G. L. Nash, A.
Akarajamorn, V. Boosaeng, S. Panyim, A. Tassanakajon, B.
Withyachum-narnkul, and T. W. Flegel.1995. A non-occluded, systemic baculovirus that occurs in the cells of ectodermal and mesodermal origin and causes high mortality in the black tiger prawn Penaeus monodon. Dis. Aquat. Organ. 21:69–77.
60. Yang, F., J. He, X. Lin, Q. Li, D. Pan, X. Zhang, and X. Xu. 2001. Complete genome sequence of the shrimp white spot bacilliform virus. J. Virol. 75: 11811–11820.
61. Zhi, Y., K. S. Sciabica, and R. M. Sandri-Goldin. 1999. Self-interaction of the herpes simplex virus type 1 regulatory protein ICP27. Virology 257:341– 351.

![FIG. 6. Homotypic interaction and dimerization of IE1. (A) SDS-PAGE analysis showing the reaction of GST-IE1 fusion protein with [ 35 S]methionine-labeled IE1 protein (input)](https://thumb-ap.123doks.com/thumbv2/9libinfo/8839437.238420/10.877.131.746.106.584/homotypic-interaction-dimerization-analysis-showing-reaction-methionine-protein.webp)
