Feature Selection and Combination Criteria for
Improving Accuracy in Protein Structure Prediction
Ken-Li Lin, Chun-Yuan Lin, Member, IEEE, Chuen-Der Huang, Hsiu-Ming Chang, Chiao-Yun Yang,
Chin-Teng Lin, Fellow, IEEE, Chuan Yi Tang, and D. Frank Hsu, Senior Member, IEEE
Abstract—The classification of protein structures is essential
for their function determination in bioinformatics. At present, a reasonably high rate of prediction accuracy has been achieved in classifying proteins into four classes in the SCOP database according to their primary amino acid sequences. However, for further classification into fine-grained folding categories, espe-cially when the number of possible folding patterns as those defined in the SCOP database is large, it is still quite a challenge. In our previous work, we have proposed a two-level classification strategy called hierarchical learning architecture (HLA) using neural networks and two indirect coding features to differentiate proteins according to their classes and folding patterns, which achieved an accuracy rate of 65.5%. In this paper, we use a combinatorial fusion technique to facilitate feature selection and combination for improving predictive accuracy in protein structure classification. When applying various criteria in com-binatorial fusion to the protein fold prediction approach using neural networks with HLA and the radial basis function network (RBFN), the resulting classification has an overall prediction accuracy rate of 87% for four classes and 69.6% for 27 folding categories. These rates are significantly higher than the accuracy rate of 56.5% previously obtained by Ding and Dubchak. Our results demonstrate that data fusion is a viable method for feature selection and combination in the prediction and classification of protein structure.
Index Terms—Combinatorial fusion analysis (CFA), data
fu-sion, diversity rank/score graph, hierarchical learning architecture (HLA), neural network (NN), protein structure prediction, radical basis function network (RBFN), rank/score functions.
Manuscript received December 20, 2005; revised September 6, 2006. This work was supported by the National Science Council (NSC) under Contract NSC 93-2321-B-007-001. The work of of C.-Y. Lin was supported by the NSC under Contract NSC93-3112-B-007-008. The work of D.F. Hsu was supported in part by the National Science Foundation under DIMACS Grant STC-91-1999 and in part by the NJ Commission. Asterisk indicates corresponding author.
K.-L. Lin is with the Department of Electrical and Control Engineering, Na-tional Chiao-Tung University, Hsin-chu, Taiwan and Computer Center of Chung Hua University, Hsin-chu,Taiwan (e-mail: [email protected]).
*C.-Y. Lin is with the Department of Computer Science, National Tsinghua University, Hsinchu 300, Taiwan, R.O.C. (e-mail: [email protected]).
C. Y. Tang is with the Department of Computer Science, National Tsinghua University, Hsinchu 300, Taiwan, R.O.C. (e-mail: [email protected]).
C.-D. Huang is with the Department of Electrical Engineering, Hsiuping In-stitute of Technology, Taichung 412, Taiwan, R.O.C. (e-mail: cdhuang@mail. hit.edu.tw).
H.-M. Chang is with the Brain Research Center, National Tsinghua Univer-sity, Hsinchu 300, Taiwan, R.O.C. (e-mail: [email protected]).
C.-Y. Yang is with BenQ Corporation, 18 Jihu Road, Neihu, Taipei 114, Taiwan, R.O.C. (e-mail: [email protected]).
C.-T. Lin is with the Department of Electrical and Control Engineering, National Chiao-Tung University 1001 Ta Hsueh Road, Hsinchu, Taiwan 300, Taiwan, R.O.C. (e-mail: [email protected]).
D. F. Hsu is with the Dept. of Computer and Information Sciences Fordham University, New York, NY 10023 USA (e-mail: [email protected]).
Digital Object Identifier 10.1109/TNB.2007.897482
I. INTRODUCTION
H
IGH-TECHNOLOGY large-scale sequencing projects have produced a massive number of proteins with puta-tive amino acid sequences but much less is known in terms of their 3-D structure. Several popular structure databases, such as the Structural Classification of Proteins (SCOP) [1] and the Class, Architecture, Topology, and Homologous superfamily (CATH) [2], contribute only no more than 32 000 entries in the Protein Data Bank (PDB) (SCOP release version 1.65 [3]: 20619 PDB entries, PDB: 31 217 entries on 7 June 2005). This number constitutes only about 20% of collections in the Swiss-Port (Swiss-Port release version 47.2: 184 304 entries on 7 June 2005). Physically, x-ray diffraction or NMR is used to determine the 3-D structure for a protein. However, each has its limitation [4]. As such, extracting structural informa-tion from the sequence databases becomes an important and complementary alternative, especially for swiftly determining protein functions or discovering new compounds for medical or therapeutic purposes.The classification of protein structures has, more recently, been facilitated with some computer-aided algorithms. Previous research [4], [5] have shown that an accuracy rate of 70%–80% has been achieved to classify most of proteins into four classes according to their amino acid sequence information (i.e., all-alpha , all-beta , alpha/beta and alpha beta
) [6]. In summary, these four classes contain 82.5% folding patterns, 84.7% superfamilies and 88.1% families in the SCOP database (SCOP release version 1.65 [3]). However, less optimal results are obtained if a more complicated category is used such as the one with protein folding patterns in [7].
In [7], Ding and Dubchak proposed a taxonmetric approach for protein folding classification (into 27 folding patterns) beyond four simple classes with a neural network (NN) and support vector machine (SVM) [8]. Their approach attempts to predict the 3-D structure of a protein from its primary amino acid sequence under the assumption that only limited folding patterns are formed in most of these four protein classes and can be used as template. They predicted protein folds according to six single-parameter features C, S, H, P, V, and Z (see Section II-B for details) first, then a combinatorial multiple-parameter features were formed and checked for their prediction accuracy in protein folding classification. They then demonstrated that one multiple-parameter feature CSHP had the highest overall prediction accuracy rate at 56.5% by SVM.
In Huang et al. [9], extra features were defined. We pro-posed two additional indirect coding features B and SB (see Sec-tions II-B and II-C for detail) to correlate neighboring di-peptide
pairs with protein structure classification. In addition to NN and SVM, we also constructed a new computational architec-ture called hierarchical learning architecarchitec-ture (HLA). In HLA, which was the first two-level classification strategy, a protein is classified into one of four classes at first, and then further classified into a folding structure (into one of 27 folding pat-terns). We combined the six single-parameter features proposed by Ding and Dubchak [7] and the outcomes of our two indi-rect coding features to form two new multiple-parameter fea-tures CSHPVZ B and CSHPVZ B SB. With the latter features, Huang et al. [9] improved the prediction accuracy rate by 9%, compared with the result from Ding and Dubchak [7].
In this paper, we apply the technique of data fusion [10]–[14], in particular the combinatorial fusion analysis described in Hsu et al. [13], to perform better protein structure classification and better feature selection and combination. Using data fusion, re-sults from various features are combined to obtain predictions with higher accuracy rate. In addition, the notion of diversity rank/score function is used to select the most suitable features for combination. We start with eight features, six from Ding and Dubchak (C, CS, CSH, CSHP, CSHPV, and CSHPVZ) [7] and two from our previous work (CSHPVZ B and CSHPVZ B SB) [9] to assign protein class and folding pattern. Then, some explicit rules from data fusion in information retrieval (IR) and virtual screening (VS) (see [10]–[14]) are used to-gether with a special diversity rank/score graph to choose the best discriminating features for further combination. It has been demonstrated in IR and VS that using a combination of dis-tinctive features may result in higher prediction accuracy rate than using single features. The proposed rules for proper feature selection are to reduce the complexity at the beginning. Then, we systematically choose the best discriminating features ac-cording to the diversity (see Section II-F for detail) of these fea-tures, which is represented in a diversity rank/score graph. Our experimental results achieve an overall prediction accuracy rate at 87% for predicting protein classes and 69.6% for predicting protein folding patterns which are higher than the previous work at 83.6% and 65.5% by Huang et al. [9], respectively.
Section II of this paper introduces the protein data sets, the features, and the computational architecture used in this paper. Section II also describes the method of data fusion, the rules, and the diversity rank/score graph used to enhance the process of feature selection and combination. Experimental results are included in Section III, while discussions and conclusion are given in Section IV.
II. MATERIALS ANDMETHODS A. Protein Data Sets
We use the data sets from Ding and Dubchak [7] which were originated from the SCOP database for training and testing. Training data set is selected from the database built for the pre-diction of 128 folding patterns in the SCOP database [15]. It is ensured that any pair of two proteins in the training set is less than 35% identical in any aligned subsequence longer than 80 residues. The independent testing set is selected from the PDB-40D set [1], [6], [15], [16]. Moreover, all proteins in the testing set are less than 40% identical to each other. No protein
in the testing set is more than 35% identical to any protein in the training set. The total number of proteins is 698 with 313 and 385 for training and testing, respectively. These proteins will be divided into four classes and 27 folding patterns all together according to their structures. Table I shows the number of pro-teins in different classes and folding patterns used for training and testing in this paper.
B. Features
Features extraction from the data is critical for meaningful re-sults before these features can be subjected to machine learning techniques. Different features may result in different classifi-cations. Two major approaches including direct and indirect coding have been used to extract features from the data. The di-rect one contains a vector for each peptide residue in the chain that characterizes the position, sequence length and so on. In in-direct coding, the vector is assigned for each sequence which is position and length independent [9]. Ding and Dubchak [7] proposed six direct coding features for protein structure classi-fication. These single-parameter features are global descriptions of a peptide chain representing the proteins. These features are based on physical, chemical and structural properties of the con-stituent amino acids.
The proposed six single-parameter features are amino acid composition (C), predicted secondary structure (S), hydropho-bicity (H), normalized van der Waals volume (V), polarity (P), and polarizability (Z). The proposed five multiple-parameter features, CS, CSH, CSHP, CSHPV, and CSHPVZ were con-structed to classify protein folding patterns. Ding and Dubchak [7] finally determined one multiple-parameter feature CSHP with the highest overall accuracy rate for protein structure pre-diction with SVM. The above 11 single and multiple-parameter features all emphasize more on the global properties and struc-tures of amino acid sequences than on the local interactions among neighboring peptides.
In Huang et al. [9], we used the N-gram concept while ex-tracting features from the amino acid sequence of proteins. Two other indirect coding features, generated from the bigram (B) and the spaced-bigram coding (SB) scheme, respectively, were proposed. These features reflect the local interactions among neighboring peptides within the 3-D structure of a protein. We combined the six single-parameter features proposed by Ding and Dubchak [7] and the outcomes of our two indirect coding features to form two new multiple-parameter features CSHPVZ B and CSHPVZ B SB’. We showed that using the feature CSHPVZ B SB together with NN outperformed all single- or multiple-parameter features used by Ding and Dubchak [7] in terms of prediction accuracy rate for protein structure classification.
In this paper, we start with eight features, C, CS, CSH, CSHP, CSHPV, CSHPVZ, CSHPVZ B, and CSHPVZ B SB to assign protein classes or folding patterns. Then, we use the method of data fusion for feature selection and combination in order to improve classification accuracy.
C. The HLA Computational Architecture
The NNs have been commonly used in many machine learning and data mining applications, such as input–output
TABLE I
THEVARIETY INPROTEINSTRUCTURES FORTRAINING ANDTESTING
mapping and bioinformatics [17], [18]. We use NN as a multi-class multi-classifier to build HLA for the purpose of protein structure prediction. The multilayer perceptron (MLP) and the radial basis function network (RBFN) are two popular NN models. The RBFN is a three-layer network with Gaussian function that is suitable to be a classifier [19] since the weights of RBFN are measured and adjusted according to the distance of data. The RBFN is constructed as a kind of hybrid NN network that combines the self-organized-map (SOM) and the back-prop-agation (BP) [9]. It was shown [9] that the overall prediction accuracy rate for protein structure classification using RBFN is better than that using MLP. Therefore, we adopted the RBFN model in this paper where one hidden layer and nodes will be generated automatically. The hidden layer nodes show the coordinate of training sample clusters.
The HLA framework, proposed in Huang et al. [9] consists of a two-level procedure. In the first level, a protein is classified into one of four classes by a multiclass classifier (classifier 1 in Fig. 1). Then, in the second level, it is further classified into one of folding patterns by the corresponding multiclass classifier ( , , and is equal to 6, 9, 9 and 3 in classifier 1, 2, 3, and 4 respectively in Fig. 1).
In Huang et al. [9], it has been shown that the HLA frame-work is an effective learning structure which reduces the number
of classifiers, avoids the voting scheme, and directly indicates the reliability or confidence of the result predicted. Our current study incorporates data fusion in HLA for the testing data set, as shown in Fig. 1. For the training data set, HLA is used without data fusion. To predict which of four classes a protein belongs to with HLA, we use eight individual features to assign class to each protein in the testing data set at first. Then, we use the technique of data fusion to select the best feature and to combine results for the protein class discrimination. Finally, the protein class is predicted with the combined feature. For protein folding patterns associated with each protein class, the eight individual features are used once more to assign protein folding patterns to each protein in the class. Similarly, data fusion is applied again for feature selection and combination in order to improve the prediction of protein folding patterns.
D. Data Fusion and The Diversity Rank/Score Graph
The approach we take to properly select and combine features in protein structure classification is analogous to those used in information retrieval [10], [11], [14], [20], [21], pattern recog-nition [22], molecular similarity searching and structure-based screening [12], [23], and microarray gene expression analysis [24]–[26]. In addition, we adopt some of the notations and ter-minologies from [11]–[13]. Moreover, each feature is viewed as
Fig. 1. The architecture of HLA using data fusion.
a scoring system containing a score function and a rank function on the set of classes.
Our previous work in information retrieval, molecular similarity searching, structure-based virtual screening and microarray gene expression study have demonstrated the following:
Remark 1: For a set of multiple scoring systems, each with a score function and a rank function, we have: (a) the combi-nation of multiple scoring systems would improve the predic-tion accuracy only if: (1) each of the systems has a relatively high performance, and (2) the individual systems are distinctive (or diversified), and (b) rank combination performs better than score combination under certain conditions.
Given a protein sequence and for each feature , let be a function that assign a real number to the class (or folding pattern) in the set of all classes (or folding patterns)
. We view the function as the score func-tion from to (the set of real numbers) with respect to the feature . When treating as an array of real numbers, it would lead to a rank function after sorting the array into descending order and assigning a rank to each of their classes (folding patterns). The resulting rank function is
a function from to .
In order to properly compare and correctly combine score functions from multiple features, the function values have to be normalized. The normalization we used is the
trans-formation from : , to : ,
where , x in D and
in and in .
Suppose we have features (i.e., scoring functions). There are combinatorially combinations for all individual features with rank or score functions. The total number of combinations to be consid-ered for predicting protein class and protein folding pattern
are and respectively in the
HLA architecture. These numbers can become huge when the number of features is large. Moreover, we have to evaluate the predictive power of each combination across all proteins. Because of this complexity, the current paper would start with combining only two features which still retain fairly good prediction power. Combination of more than two features will be considered in our future work.
E. Methods of Combination and Feature Selection
Suppose features , , are given with score function and rank function , there are several different ways of combination. Among others, there are score combina-tion, rank combinacombina-tion, voting, linear average combination and weighted combination [10]–[12], [14], [20]–[27]. Voting is computationally simple and better than simple linear combi-nations when applied to the situation with large number of fea-tures. However, a better alternative is to reduce the number of features to a smaller number and then these features are com-bined. In this paper, we reduce the set of features to those which
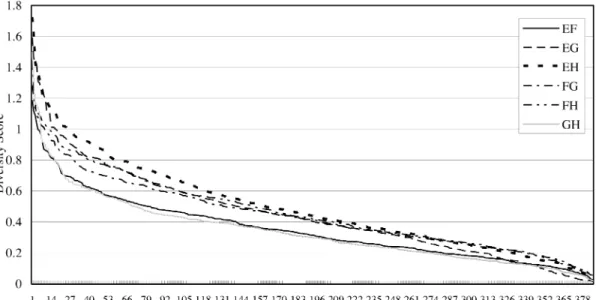
Fig. 2. The diversity rank/score graph for each pair of features from {E,F,G,H} for classifying protein classes.
perform relatively well and then use the diversity rank/score function to decide whether to combine by rank or by score. For the features , rank functions , and score functions , we have the score function and of the rank combination and score combination respectively defined as
and
As we did before, and are then sorted into as-cending and desas-cending order to obtain the rank function of the rank combination and the score combination , respectively.
In this paper, we use the rules (a)(1), (a)(2), and (b) stated in Remark 1 as our guiding principle to select features and to decide on the method of combination. We started with eight features and, in each case, use rule (a)(1) to reduce the number of features to four. A diversity function between features and is then computed using the concept of the rank/score function defined by Hsu et al. [10], [11], [13].
F. The Rank/Score Function and The Diversity Rank/Score Graph
Given a protein sequence and for each feature , we have the score function and rank function . Both and are functions from to [0,1] and respectively, where the set of classes. As in other application domains [10]–[13], we explore the scoring (and ranking) characteristics of feature by calculating the rank/score function, : as follows:
We note that the set is different from the set which is the set of classes (or fold patterns). The set is used as the index set for the rank function value and is indeed the cardinality of . The rank/score function so defined signifies the scoring (or ranking) behavior of the feature and is independent of the classes (or folding patterns) under consideration.
For protein in and the pair of
fea-tures and , the diversity score function is
de-fined as: , where is in
and is the number of classes (or folding pat-terns). When there are features selected (in this paper,
), there are (in this paper, this number is 6) diversity score functions. If we let vary and fix the fea-ture pair , then is the diversity score function
from to . Sorting
into descending order would lead to the diversity rank func-tion . Consequently, the diversity rank/score func-tion is defined as
where is in
We note that the set is different from the set which is the protein set considered. The set is used as the index set for the diversity rank function value and is indeed the cardinality of . The diversity rank/score function
so defined exhibits the diversity trend of the feature pair across the whole spectrum of input set of proteins and is inde-pendent of the specific protein under study.
For two features and , the graph of the diversity rank/score function is called the diversity rank/score graph (or diversity graph in short). Our current study aims to examine all the diversity rank/score graphs to see which pair of features would give the highest diversity measurement. Following rules (a)(2) and (b) in Remark 1, the rank combination of these two features is then calculated to give the final rank function and to choose the class (or folding pattern).
III. RESULTS
The technique of combinatorial fusion (see [13]) is used for protein structure classification on a testing data set with NN using RBFN under the HLA architecture. Initially, we use eight features, C (reworded as A), CS (as B), CSH (as C), CSHP (as
(a)
(b)
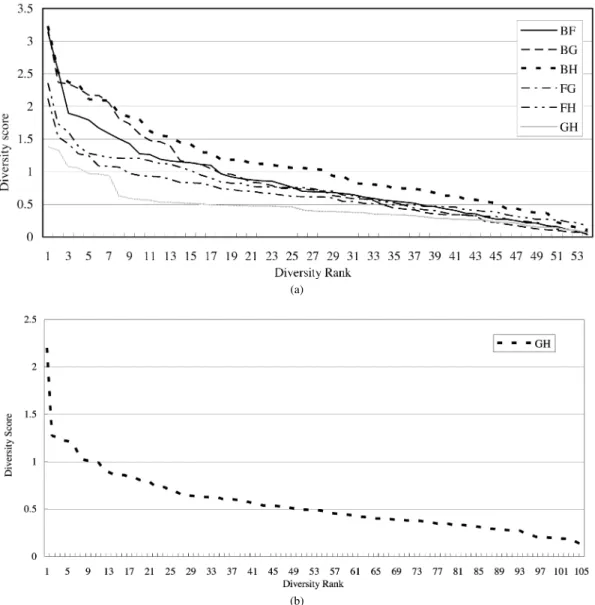
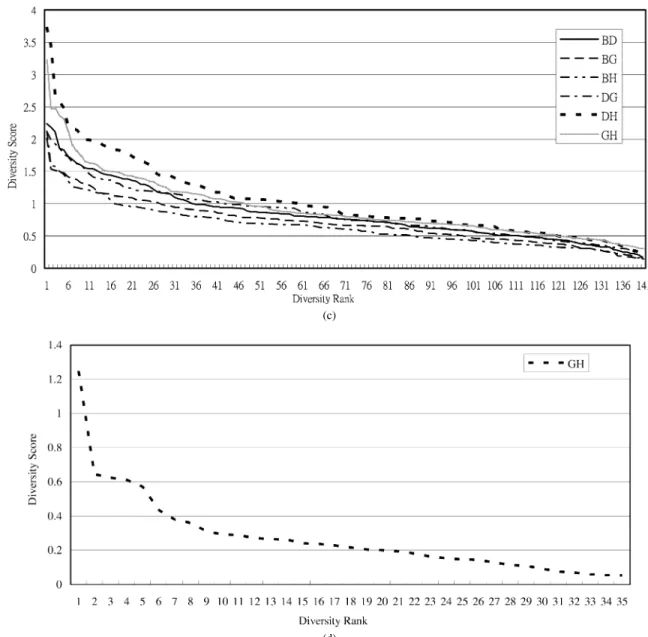
Fig. 3. The diversity rank/score graph for each pair of features in {B,F,G,H} for classifying protein folding patterns in class1; in {G,H} for classifying protein folding patterns in class2; in {B,D,G,H} for classifying protein folding patterns in class3; and in {G,H} for classifying protein folding patterns in class4. (a) Class 1. (b) Class 2.
D), CSHPV (as E), CSHPVZ (as F), CSHPVZ B (as G), and CSHPVZ B SB (as H), to assign protein classes for all pro-teins tested. Following the rule (a)(1) in Section II-D, we select four features E, F, G, and H, for further fusion (or combina-tion) because of their higher accuracy rate than others as demon-strated in [9]. With the help of rule (a)(1), we can reduce combinations to combinations. Following the rules (a)(2) and (b) in Section II-D, we shall use the rank combination of the features to predict the protein class.
As stated in Section II-F, the diversity of any two of features E, F, G, and H can be calculated for all proteins tested and fea-tures E and H are found to have the highest diversity, as shown in Fig. 2, among all six feature combinations. In con-junction with (b) in Remark 1, we use the rank combination of features E and H to predict protein classes for all proteins tested. After the protein classes for all proteins tested have been predicted and categorized, the prediction of protein folding pat-terns follows in the HLA architecture. We use the same rules and a diversity graph to choose the best combined two features in each class for the purpose. Accordingly, we choose a rank combination of features BG, GH, DH, and GH to predict protein
folding patterns in classes 1, 2, 3, and 4, respectively. The diver-sity graph to pick the pair of features (B,G), (G,H), (D,H) and (G,H) for combination and to predict folding patterns in class 1, 2, 3, and 4 are depicted in Fig. 3(a), (b), (c), and (d), respec-tively. In Fig. 3(b) and (d), only the pair of features (G, H) is selected since its accuracy rate is more higher than others. It implies that the features G and H are more suitable than others for classifying proteins, which belong to class 2 or class 4, into folding patterns.
We use the standard percentage accuracy rate [7], [9], [28] to evaluate our work. , where is the number of testing proteins in the th class or folding pattern and is the number of proteins being correctly predicted in the th class or folding pattern. The overall prediction accuracy rate is given
by , where , where is the total
number of proteins tested, and is the number of classes or folding patterns. We compare the overall prediction accuracy rates for protein classes in our previous [9] and current work. These are shown in Table II. The current overall prediction ac-curacy rate is 87%, 3.4% higher than that of our previous work. Table III shows that for prediction of folding pattern, our current
(c)
(d)
Fig. 3. (Continued.) The diversity rank/score graph for each pair of features in {B,F,G,H} for classifying protein folding patterns in class1; in {G,H} for classifying protein folding patterns in class2; in {B,D,G,H} for classifying protein folding patterns in class3; and in {G,H} for classifying protein folding patterns in class4. (c) Class 3. (d) Class 4.
TABLE II
THECOMPARISONS OFOVERALLPREDICTIONACCURACYRATESQFORPROTEINCLASSES
work has an overall prediction accuracy rate of 69.6%, which is 13.1% higher than that of Ding and Dubchak [7], 4.1% higher than that of our previous work.
We summarize the comparisons of prediction accuracy rates of our previous work [9] and our current work in Fig. 4. Our results give prediction accuracy rates ( 80%) in 3 classes, espe-cially in class with accuracy rate reaches 97.2%, all higher than what we achieved previously, shown in Fig. 4(a). For protein folding patterns prediction, the current work gives prediction ac-curacy rates ( 80%) in 9 folding patterns, more than what in our previous work, 7, as shown in Fig. 4(b). Also, the current work
outperforms our previous work in ten folding patterns, especially ( 30% improvement) in folding patterns: (4-helical up-and-down bundle), (viral coat and capsid proteins), (SH3-like barrel), (flavodoxin-like) and (P-loop containing nucleotide). Our previous work has slightly better results only in 5 folding patterns [especially in fold (globin-like)]. Overall, there is an improvement with our current method using the HLA framework and data fusion techniques. In summary, the current method has achieved an accuracy rate of 69.6% for folding pat-tern classification, which is a significant improvement over the result of Ding and Dubchak ([7], 2001) of 56.5%.
TABLE III
THECOMPARISONS OFOVERALLPREDICTIONACCURACYRATESQFORPROTEINFOLDINGPATTERNS
Fig. 4. The comparisons of prediction accuracy ratesQ of our previous work (Huang et al. [9]) (in white) and the current work (in black) (a) for 4 protein classes and (b) for 27 protein folding patterns.
IV. CONCLUSION ANDFUTUREWORK
Methods of combining multiple classification systems or multiple scoring systems have been used in a variety of ap-plications domains including information retrieval, pattern recognition, microarray gene expression analysis, and molec-ular similarity searching [10], [14], [20]–[27]. More recently, criteria to select the classification systems or scoring systems for combination and to decide ways to combine these systems have been discussed and studied [11]–[14], [27]. It has been
demonstrated in Combinatorial Fusion Analysis (see [13] and its references) that (a) the combination of multiple systems (or features) would improve the performance only if (1) each of the individual systems (features or functions) has a relatively high performance, and (2) each individual systems are distinctive (or different), and (b) combination by rank outperform combi-nation by score under certain conditions.
In this paper, we have applied the concept of Combinatorial Fusion (see Remark 1) to improve accuracy in protein struc-ture prediction. In particular, we have successfully improved the
overall predictive accuracy rate of 87% for the second struc-ture (the four classes) and 69.8% for the folding patterns (the 27 folding categories). We improve previous results by Huang et al. [9] (65.5% for folding structure) and Ding and Dubchak [7] (56.5% for folding structure) by incorporating the method of combinatorial fusion in their approach using NN with the RBFN using the HLA.
One of the novelties of our current work is the notion of a di-versity rank/score function between a pair of features and (Figs. 2 and 3). This function characterizes the diver-sity of ranking (or scoring) behavior between features and across the whole spectrum of all protein sequences under con-sideration. This parameter is then used to select appropriate and diverse features for combination. The current work is the first of a series of on-going projects towards the protein structure pre-diction problem using HLA, NN-RBFN, and combination fu-sion analysis. Following the current work, we have observed the following.
The method of combinatorial fusion we used in this paper is computational efficient, able to adapt to different situations and approaches, and scalable to a large number of classes (or folding patterns) and a large number of proteins.
In this paper, we considered only combination of a pair of two features in order to improve the performance. It may be possible to achieve better results with combination of more than two features. However, it is indicated in criteria (a)(1) and (2) that each of these three or more features would have relatively high performance and individual features should be different. As such, the diversity between three or more features should be defined. This will be studied in a latter work.
Although it has been shown (e.g., [31]) that combining mul-tiple predictors or servers improves fold recognition, we note here that combining all the features or multiple scoring sys-tems together may not guarantee optimal performance (see [12] and [13]).
We used rank combination due to criterion (b) which was demonstrated to be better under certain conditions analytically and by simulation in Hsu and Taksa [11]. We observed that score combination does have its merit when the two features combined are similar and homogeneous with respect to their scoring functions, rank function, or rank/score function. We de-cided to use the rank combination because the pair of features to be combined satisfies criteria (a)(1) and (a)(2) and these two items are precisely the conditions stipulated in [11], [12], [14], [25], and [26].
In our feature selection process, we selected top four per-formers out of the original eight features. The ideal case is to select those features which perform much better than the others. That means there is a big difference on the performance between those selected and those not selected.
Our current work represents the first of a series of investiga-tions on the protein structure prediction problem using HLA and combinatorial fusion. It has generated several issues and topics worthy of further study. We summarize some of them here.
Our diversity rank/score function A,B for the feature pair A,B with respect to protein is defined using the variation of the rank/score functions between A and B. As indicated in [11], [13], and [14], variation of the rank functions or the score
function between A and B can be used also to define the diversity score function. We will explore these two other options in a latter work.
The effectiveness of our fusion of multiple features is limited by the set of eight original chosen features. It might be worth-while to study the content of original set of features. For ex-ample, we would like to explore the diversity among the original features such as local versus global, physical versus chemical, and bigram versus trigram scheme.
Related to observation (D) above, one might ask if it is better to expand the scope and the number of features. In this paper, we started with eight features and four are selected using the CFA criteria. In a separate paper [30], eleven features are col-lected and three features are secol-lected according to the criteria (a)(1) and (a)(2) in Remark 1. We have obtained a slightly better overall accuracy rate of 87.8% for four classes and 70.9% for 27 folding categories.
Our results improve previous results by Huang et al. [9] and Ding and Dubchak [7] which used NN with radial basis function in an HLA. Work has been performed to improve those results which used other machine learning technique such as kernel method, SVM and genetic algorithm. For example, Yu et al. [29] has obtained good accuracy rate using SVM with -pep-tide coding schemes and jury voting. Ongoing work has been performed to improve these results using our combinatorial fu-sion approach. These results will be reported in the future.
Due to its importance, protein structure prediction and clas-sification has been studied extensively in the past decade. In particular, protein structure classification using databases of proteins with known structures have been studied (See [32] and [33] and their references). As stated before, our work does not use or rely on any databases of known structures
ACKNOWLEDGMENT
D. F. Hsu would like to thank National Tsinghua University and the Ministry of Education for their support and hospitality during his visit to NTHU, Hsinchu, Taiwan, R.O.C., in Spring 2004.
REFERENCES
[1] L. Lo Conte, B. Ailey, T. J. P. Hubbard, S. E. Brenner, A. G. Murzin, and C. Chothia, “SCOP: A structural classification of proteins data-base,” Nucleic Acids Res., vol. 28, no. 1, pp. 257–259, 2000. [2] F. M. Pearl, D. Lee, J. E. Bray, I. Sillitoe, A. E. Todd, A. P. Harrison,
J. M. Thornton, and C. A. Orengo, “Assigning genomic sequences to CATH,” Nucleic Acids Res., vol. 28, no. 2, pp. 584–599, 2000. [3] A. Antonina, H. Dave, E. B. Steven, J. P. H. Tim, C. Cyrus, and G. M.
Alexey, “SCOP database in 2004: Refinements integrate structure and sequence family data,” Nucleic Acids Res., vol. 32, pp. 226–229, 2004. [4] I. Dubchak, I. Muchnik, S. R. Holbrook, and S. H. Kim, “Prediction of protein folding class using global description of amino acid sequence,” Proc. Nat. Acad. Sci. USA, vol. 92, pp. 8700–8704, 1995.
[5] K. C. Chou and C. T. Zhang, “Prediction of protein structural classes,” Crit. Rev. Biochem. Mol. Biol., vol. 30, no. 4, pp. 275–349, 1995. [6] A. G. Murzin, S. E. Brenner, T. Hubbard, and C. Chothia, “SCOP:
A structural classification of proteins database for the investigation of sequence and structures,” J. Mol. Biol., vol. 247, pp. 536–540, 1995. [7] C. H. Q. Ding and I. Dubchak, “Multi-class protein fold recognition
using support vector machines and neural networks,” Bioinformatics, vol. 17, no. 4, pp. 349–358, 2001.
[8] V. N. Vapnik, The Nature of Statistical Learning Theory. New York: Springer-Verlag, 1995.
[9] C. D. Huang, C. T. Lin, and N. R. Pal, “Hierarchical learning architec-ture with automatic feaarchitec-ture selection for multi-class protein fold clas-sification,” IEEE Trans. NanoBiosci., vol. 2, no. 4, pp. 503–517, Dec. 2003.
[10] D. F. Hsu, J. Shapiro, and I. Taksa, Methods of data fusion in infor-mation retreival: Rank vs. score combination DIMACS Tech. Rep. 58, 2002.
[11] D. F. Hsu and I. Taksa, “Comparing rank and score combination methods for data fusion in information retrieval,” Inf. Retr., vol. 8, pp. 449–480, 2005.
[12] J.-M. Yang, Y.-F. Chen, T.-W. Shen, B. S. Kristal, and D. F. Hsu, “Con-sensus scoring criteria for improving enrichment in virtual screening,” J. Chem. Inf. Model., vol. 45, pp. 1134–1146, 2005.
[13] D. F. Hsu, Y.-S. Chung, and B. S. Kristal, “Combinatorial fusion anal-ysis: Method and practice of combining multiple scoring systems,” in Advanced Data Mining Technologies in Bioinformatics, H.-H. Hsu, Ed. Hershey, PA: Idea Group Inc., 2006, pp. 32–62.
[14] K. B. Ng and P. B. Kantor, “Predicting the effectiveness of naïve data fusion on the basis of system characteristics,” J. Amer. Soc. Inf. Sci., vol. 51, no. 13, pp. 1177–1189, 2000.
[15] I. Dubchak, I. Muchnik, C. Mayor, I. Dralyuk, and S. H. Kim, “Recog-nition of a protein fold in the context of the SCOP classification,” Pro-teins, vol. 35, pp. 401–407, 1999.
[16] L. L. Conte, S. E. Brenner, T. J. Hubbard, S. E. Brenner, A. G. Murzin, and C. Chothia, “SCOP database in 2002: Refinements accommodate structural genomics,” Nucleic Acids Res., vol. 30, no. 1, pp. 264–267, 2002.
[17] P. Baldi and S. Brunak, Bioinformatics: The Machine Learning Ap-proach. Cambridge, MA: MIT Press.
[18] C. H. Wu, Neural Networks and Genome Informatics. Amsterdam, The Netherlands: Elsevier, 2000.
[19] J. Moody and C. J. Darken, “Fast learning in networks of locally tuned processing units,” Neural Comput., vol. 1, no. 2, pp. 281–294, 1989. [20] N. J. Belken, P. B. Kantor, E. A. Fox, and J. A. Shaw, “Combining
evidence of multiple query representation for information retrieval,” Inf. Process. Manage., vol. 31, no. 3, pp. 431–448, 1995.
[21] C. C. Vogt and G. W. Cotrell, “Fusion via a linear combination of scores,” Inf. Retr., vol. 1, pp. 151–172, 1999.
[22] L. Xu, A. Krzyzak, and C. Y. Suen, “Method of combining multiple classifiers and their application to handwriting recognition,” IEEE Trans. Syst., Man, Cybern., vol. 22, no. 3, pp. 418–435, May/Jun. 1992.
[23] C. M. R. Ginn, P. Willett, and J. Bradshaw, “Combination of molecular similarity measures using data fusion,” Perspectives Drug Discov. Des., vol. 20, pp. 1–16, 2000.
[24] M. A. Kuriakose, W. T. Chen, Z. M. He, A. G. Sikora, P. Zhang, Z. Y. Zhang, W. L. Qiu, D. F. Hsu, C. M. Coffran, S. M. Brown, E. M. Elango, M. D. Delacure, and F. A. Chen, “Selection and validation of differentially expressed genes in head and neck cancer,” Cell. Mol. Life Sci., vol. 61, pp. 1372–1383, 2004.
[25] H. Y. Chuang, H. F. Liu, S. Brown, C. M. Coffran, and D. F. Hsu, “Identifying significant genes from microarray data,” in Proc. IEEE Symp. Bioinformatics and Bioengineering (BIBE’04), pp. 358–365. [26] H. Y. Chuang, H. F. Liu, F. A. Chen, C. Y. Kao, and D. F. Hsu,
“Combi-nation methods in microarray analysis,” in Proc. 7th Int. Symp. Parallel Architectures, Algorithms and Networks (I-SPAN ’04), pp. 625–630. [27] D. F. Hsu and A. Palumbo, “A study of data fusion in Cayley graphs
G (Sn, Pn),” in Proc. 7th Intl. Symp. Parallel Architectures, Algorithms and Networks (I-SPAN ’04), pp. 557–562.
[28] B. Rost and C. Sander, “Prediction of protein secondary structure at better than 70% accuracy,” J. Mol. Biol., vol. 232, pp. 584–599, 1993. [29] C. S. Yu, J. Y. Wang, J. M. Yang, P. C. Lyu, C. J. Lin, and J. K. Hwang, “Fine-grained protein fold assignment by support vector machines using generalized n peptide coding schemes and jury voting from multiple-parameter sets,” Proteins, vol. 50, pp. 531–536, 2003.
[30] K.-L. Lin, C. Y. Lin, C.-D. Huang, H.-M. Chang, C. Y. Yang, C.-T. Lin, C. Y. Tang, and D. F. Hsu, “Methods of improving protein struc-ture prediction based on HLA neural network and combinatorial fusion analysis,” WSEAS Trans. Inf. Sci. Appl., vol. 2, no. 12, pp. 2146–2153, 2005.
[31] J. Jundstrom, L. Rychlewski, J. Bujnicki, and A. Elofsson, “Pcons: A neural-network-based consensus predictor that improves fold recogni-tion,” Protein Sci., vol. 10, pp. 2354–2362, 2001.
[32] L. J. McGriffin and D. T. Jones, “Improvement of the GenThreader method for genome fold recognition,” Bioinformatics, vol. 19, no. 7, pp. 874–881, 2003.
[33] P. Daras, D. Zarpalas, A. Axenopoulos, D. Tzovaras, and M. G. Strintzis, “Three-dimensional shape-structure comparison method for protein classification,” IEEE/ACM Trans. Comput. Biol. Bioinform., vol. 3, no. 3, pp. 193–207, 2006.
Ken-Li Lin was born in 1967. He received the B.S. and M.S. degrees from the Department of Control Engineering from National Chiao-Tung University (NCTU), Hsinchu, Taiwan, R.O.C., in 1990 and 1992, respectively. He is currently working toward the Ph.D. degree in the Department of Electrical and Control Engineering, National Chiao-Tung University.
He was a System Engineer with the AT&T Taiwan Telecommunication Corporation from 1994 to 1996, and with the Powerchip Semiconductor Corporation from 1996 to 1998. He is currently with the Computer Center of Chung Hua University, Hsinchu, Taiwan. His research interests are in the areas of informa-tion technology, computainforma-tional intelligence, and bioinformatics.
Chun-Yuan Lin (S’01–M’03) was born in 1977. He received the B.S. degree in information engineering and computer science and the M.S. and Ph.D. degrees from the Department of Information Engineering and Computer Science of Feng Chia University, Taiwan, R.O.C., in 1999, 2000, and 2003, respectively.
He joined the Institute of Molecular and Cellular Biology and the Department of Computer Science at National Tsinghua University, Hsinchu, Taiwan, as a Postdoctoral Fellow in 2003 and 2006, respectively. His research interests are in the areas of parallel and distributed computing, parallel algorithms, algorithm analysis, information re-trieve, proteomics, and bioinformatics.
Dr. Lin is a member of the IEEE Computer Society, the ACM, and Bioinfor-matics Society Taiwan.
Chuen-Der Huang was born in 1958. He received the B.S. degree in electrical engineering and the M.S. degree in control engineering from Feng Chia University, Taiwan, R.O.C., in 1980 and 1983, respectively, and the Ph.D. degree in electrical and control engineering from National Chiao-Tung University, Taiwan.
He is an Associate Professor in the Department of Electrical Engineering at Hsiuping Institute of Technology, Taiwan. His research interests include control, measurement, bioinformatics, information fusion, intelligent systems, and applied engineering.
Hsiu-Ming Chang was born in 1957. He received the B.S. degree in physics from the National Tsinghua University (NTHU), Hsinchu, Taiwan, R.O.C., in 1979 and the Ph.D. degree in physiology from the University of Arizona, Tucson, in 1995.
He had joined research groups in life science and computer science in the NTHU, and has been a re-search fellow in the Brain Rere-search Center, NTHU, since 2004. His research interests are in the areas of neuroscience, bioimaging, and informatics.
Chiao-Yun Yang was born in Taipei, Taiwan, R.O.C. She received the B.S. degree in environmental en-gineering from National Chung-Kung University, Taiwan, in 2003 and the M.S. degree in computer science from National Tsinghua University, Taiwan, in 2005.
She currently works for BenQ Mobile, Taipei, as an mobile software engineer.
Chin-Teng (CT) Lin (S’88–M’91–SM’99–F’05) received the B.S. degree from National Chiao-Tung University (NCTU), Taiwan, R.O.C., in 1986 and the Ph.D. degrees in electrical engineering from Purdue University, West Lafayette, IN, in 1992.
He served as the Director of the Research and De-velopment Office of NCTU from 1998 to 2000, the Chairman of Electrical and Control Engineering De-partment of NCTU from 2000 to 2003, and Associate Dean of the College of Electrical Engineering and Computer Science from 2003 to 2005. He is currently the Chair Professor of Electrical and Computer Engineering, Dean of Computer Science College, and Director of the Brain Research Center at NCTU. He is the coauthor of Neural Fuzzy Systems—A Neuro-Fuzzy Synergism to Intelli-gent Systems (Prentice-Hall), and the author of Neural Fuzzy Control Systems With Structure and Parameter Learning (World Scientific). He has published over 110 journal papers in the areas of neural networks, fuzzy systems, multi-media hardware/software, and soft computing, including about 90 IEEE journal papers. His current research interests are fuzzy neural networks, neural net-works, fuzzy systems, cellular neural netnet-works, neural engineering, algorithms and VLSI design for pattern recognition, intelligent control, and multimedia (in-cluding image/video and speech/audio) signal processing, and intelligent trans-portation system (ITS).
Dr. Lin currently serves as Associate Editor of IEEE TRANSACTIONS ON
CIRCUITS ANDSYSTEMS, PARTI & PARTII, IEEE TRANSACTIONS ONSYSTEMS, MAN, AND CYBERNETICS, IEEE TRANSACTIONS ON FUZZY SYSTEMS, and International Journal of Speech Technology.
Chuan Yi Tang received the B.S. degree from the Department of Electrical Engineering and the M.S. degree from the Department of Computer Science, National Tsinghua University (NTHU), Taiwan, R.O.C., in 1980 and 1982, respectively, and the Ph.D. degree from the Department of Computer Science and Information Engineering, National Chiao-Tung University, Taiwan, in 1985.
In 1985, he joined the Department of Computer Science, NTHU, where he became a Professor in 1992. From 1999 to 2003, he was the chairman of the department. Currently, he is the Deputy Dean of the Academic Affairs, NTHU. Fifty of his papers are published in prestigious journals of computer science. His research interests include the analysis and design of algorithms, computa-tional molecular biology, parallel processing, and computer-aided engineering. In the past years, he developed tools for multiple sequence alignments and evolutionary trees by using the concept of compact set and the technologies of approximation. Recently, he has developed tools that help the enzyme biologist to search active sites, where the functions of these new tools cover the sequence alignment and the consensus finding with structure information. In addition, for the comparative genomics, he designs several different algorithms to predict exons and alternative splicing. He also has plentiful experiences to lead the cooperation of computer scientists and biologists.
D. Frank Hsu (M’88–SM’02) received the M.S. de-gree from University of Texas at El Paso (UTEP) and the Ph.D. degree from the University of Michigan, Ann Arbor, in 1979.
He has held positions as visiting scholar or fac-ulty at MIT, Taiwan University, National Tsinghua University, University of Paris-Sud and CNRS, Keio University as IBM Chair Professor, and JAIST as Ko-matsu Chair Professor. He is currently the Clavius Professor of Science and a Professor of Computer and Information Science at Fordham University, New York. He has served on the editorial board of Networks, IEEE TRANSACTIONS ONCOMPUTERS, International Journal of Foundation of Computer Science, and is currently Editor-in-Chief of the Journal of Interconnection Networks. His re-search interests are combinatorics and algorithms, interconnection networks, and informatics and applications. His interest in applied informatics includes bioinformatics, virtual screening, target tracking and portfolio management.
Dr. Hsu has served as PC or AC member of several conferences and work-shops including I-SPAN, IEEE AINA, and DIMACS workwork-shops. He is a Fellow of New York Academy of Sciences and a Foundation Fellow of the Institute of Combinatorics and Applications.