行政院國家科學委員會專題研究計畫 期中進度報告
評估梨形鞭毛蟲 zinc finger 及 GARP 家族在轉錄及囊體化
過程的角色(1/2)
計畫類別: 個別型計畫 計畫編號: NSC93-2314-B-002-296- 執行期間: 93 年 08 月 01 日至 94 年 07 月 31 日 執行單位: 國立臺灣大學醫學院寄生蟲學科 計畫主持人: 孫錦虹 計畫參與人員: 孫錦虹 李爾博 蘇立欣 報告類型: 精簡報告 處理方式: 本計畫可公開查詢中 華 民 國 94 年 5 月 31 日
評估梨形鞭毛蟲zinc finger 及 GARP 家族在轉錄及囊體化過程的角色
Role of zinc finger and GARP families in transcriptional regulation and encystation of Giardia lamblia
Abstract
In this project, we characterized several giardial proteins, the previously described GARP-like protein 1 (GLP1), its related GLP2, and zinc finger protein 1 (ZFP1) and ZFP2. In the previous study, we showed that glp1 was up-regulated during encystation. However, glp2 and glp3 were down-regulated during encystation. GLP1 could bind to the 32-bp ran promoter element and the cwp1 promoter in the double-stranded configurations in electrophoretic mobility shift assays. Using mutated plasmid and transfection assays we demonstrated that the GLP1 binding sites are positive cis-acting elements of the cwp1 and ran gene promoters in both trophozoites and encysting cells. The results indicate that GLP1 could function as a transcriptional activator. In this project, we found that GLP2 could also bind to the same sequence in the cwp1 and ran gene promoters, suggesting that GLP2 could also function as a transcriptional activator. We also identified another gene (glp4) by in silico screening of the Giardia lamblia genome data base. The glp4 gene was down-regulated during encystation. The zfp1 and zfp2 genes were found previously by in silico screening of the G. lamblia genome data base. In the previous study, we showed that ZFP1 localized to the two nuclei in both vegetative and encysting trophozoites. In this project, we found that both zfp1 and zfp2 mRNA levels increased during encystation. We also found that the C-terminal region of ZFP1 containing the glutamine rich domain functioned as a transactivation domain in Giardia when combined with the DNA-binding domain of yeast GAL4. ZFP1 promoted expression of the reporter gene driven by the cwp1 promoter in Giardia in cotransfection assays. The result suggests that ZFP1 could function as a transcriptional activator. On the other hand, we
observed that the luciferase activity was increased ~25 or ~269-fold when cotransfected with pRANneo or 5’∆5N-Pac plasmid, respectively. The increased expression of luciferase gene could occur at translation level but not transcription level. We are currently investigating the potential function of the GLPs and ZFPs in transcriptional regulation in G. lamblia.
中文摘要
本研究計畫中,我們鑑定了幾個梨形鞭毛蟲的蛋白質,包含已知的
GARP-like protein 1 (GLP1) 和其相關的 GLP2 以及 zinc finger protein 1 (ZFP1)和 ZFP2。在先前的研究中,我們發現 glp1 基因在囊體化時有增強調控的現象,然 而 glp2 和 glp3 基因在囊體化時有減弱調控的現象。我們以電泳凝膠位移分析發
現GLP1 可以結合至雙股結構之 32-bp ran 啟動子及 cwp1 啟動子,我們更進一 步利用變異的質體做轉染分析,發現 ran 啟動子及 cwp1 啟動子之 GLP1 結合序 列在vegetative 及 encysting trophozoite 皆為正調節區域。這些結果顯示 GLP1 的
功能可能為轉錄活化子。本研究計畫中,我們發現GLP2 也可以結合至 cwp1 啟
動子及 ran 啟動子的相同序列,顯示 GLP2 的功能也可能為轉錄活化子。我們由 梨形鞭毛蟲基因資料庫也找到另一個基因 glp4,並發現 glp4 基因在囊體化時有 減弱調控的現象。在先前的研究中,我們由梨形鞭毛蟲基因資料庫找到 zfp1 和 zfp2。我們發現 ZFP1 在 vegetative 及 encysting trophozoites 的兩個細胞核,本研 究計畫中,我們發現 zfp1 及 zfp2 兩者的 mRNA 量在囊體化時有增加的現象。
我們也發現ZFP1 C 端具有許多 Glutamine 的區域與酵母菌 GAL4 DNA 結合區域
結合時具有轉錄活化功用,由共轉染實驗發現ZFP1 可促進 cwp1 啟動子所引導
的報導基因表現,這些結果顯示ZFP1 可能是轉錄活化子。另一方面,我們觀察
到當共轉染pRANneo 或 5’ ∆5N-Pac 質體時 luciferase 活性會增加~25 或~269
倍,此現象可能發生在轉譯時期而非轉錄時期。目前我們正在研究這些GLP 及
ZFP 蛋白質在梨形鞭毛蟲中的轉錄調控之潛在的功能。 關鍵詞(keywords)
Transcription 轉錄, encystation 囊體化, transcriptional activator 轉錄活化子
Introduction
G. lamblia is an important human intestinal pathogen that causes outbreaks of waterborne diarrhea (Adam, 2001; Wolfe, 1992). Giardia belongs to one of the earliest diverging eukaryotic lineages (Cavalier-Smith, 1991; Hashimoto et al., 1994; 1995; Sogin et al., 1989). There is little understanding of the biology of the origins of eukaryotes, particularly of transcriptional regulation during Giardia encystation. The rapid progress in analyses of the Giardia genome greatly helps us to find certain candidates in studies (McArthur et al., 2000; www.mbl.edu/Giardia).
During encystation, G. lamblia cyst wall is formed and both nuclei divide simultaneously, resulting in a cyst with four nuclei (Reiner et al., 1989; Lujan et al., 1997). Synthesis and secretion of certain components, such as cyst wall protein, are required for the formation of a protective cyst wall (Lujan et al., 1995; Mowatt et al., 1995). The life cycle of G. lamblia may provide perspectives on the cell
differentiation in response to different environments. We have learned to complete the life cycle of G. lamblia in vitro, making possible study of the cellular and molecular biology of differentiation (Eichinger, 2001; Gillin et al., 1987; Gillin and Reiner, 1996; Lujan et al., 1997).
suggests that Giardia may have diverged early and represents a transition during the evolution of eukaryatic transcription systems (Best et al, 2004). Many giardial transcription factors diverge at a higher rate than those of crown group eukaryotes (Best et al, 2004). G. lamblia has many unusual features with regard to transcription. No consensus TATA boxes or other cis-acting elements identified in higher eukaryotic promoters have been observed in the promoters of many protein-coding genes
(Elmendorf et al., 2001; Holberton and Marshall, 1995; Knodler et al., 1999; Lujan et al., 1995; Mowatt et al., 1995; Sun and Tai, 1999; Yee et al., 2000). Giardia has a highly divergent TBP and lacks most of the canonical RNA Polymerase II
tranascription factors (Best et al, 2004). AT-rich sequences have been found around the transcription start sites of many genes (Elmendorf et al., 2001; Holberton and Marshall, 1995; Knodler et al., 1999; Lujan et al., 1995; Mowatt et al., 1995; Sun and Tai, 1999; Yee et al., 2000). The AT-rich sequences are essential for promoter activity and play a predominant role in the determining the positions of the transcription start sites, functionally similar to the initiator element in higher eukaryotes (Elmendorf et al., 2001; Sun and Tai, 1999; Yee et al., 2000). The promoter regulatory mechanism could be unusual because very short 5’-flanking regions (<65 bp) are sufficient for the expression of some genes (Elmendorf et al., 2001; Sun and Tai, 1999; Yee et al., 2000).
Despite the importance of cyst wall biogenesis during Giardia encystation, the molecular mechanisms governing transcriptional regulation remain poorly understood. Expression of genes encode three cyst wall structural proteins (CWP1, 2, and 3; Lujan et al., 1995; Mowatt et al., 1995; Sun et al., 2003) and an enzyme in the cyst wall polysaccharide biosynthetic pathway (glucosamine-6-phosphate isomerase-B, G6PI-B) (Knodler et al., 1999; Van Keulen et al., 1998) increases with similar kinetics
(Knodler et al., 1999; Lujan et al., 1995; Mowatt et al., 1995; Van Keulen et al., 1998), suggesting the importance of regulation at transcriptional level. We also identified a gmyb2 gene whose expression increases during encystation (Sun et al., 2002). gMyb2 binding sites are present in the promoter regions of all of the key encystation-induced genes, cwp1, cwp2, cwp3, g6pi-b, and gmyb2 itself and are known to mediate induction of gene expression during encystation.
We hope to study the differential regulation of gene expression as well as transcriptional mechanisms in G. lamblia. When our previous grant was funded, we started to clone and identify transcription factors that might be involved in
encystation-induced genes or that might interact with gMyb2 by querying the Giardia Genome Project Database (NSC92-2314-B-002-354). We found several proteins containing zinc-finger motifs and GARP domains which function as parts of DNA-binding in plants and higher eukaryotes. Since many of the zinc-finger and GARP proteins have been implicated in important biological processes in plants and higher eukaryotes (Makino et al., 2000; Imamura, 1999; Sakai et al., 2001; Kerstetter
et al., 2001; Takatsuji, 1998; Krempler and Brenig, 1999), these proteins could be involved in the regulation of important biological processes that are unique to G. lamblia, such as encystation, excystation, and host-parasite interaction. It is also possible that G. lamblia might have some novel classes of zinc fingers and GARP domains to adapt them to specific developmental processes.
In this study, we characterized several possible giardial transcription factors, the previously described GARP-like protein 1 (GLP1), its related GLP2, and zinc finger protein 1 (ZFP1) and ZFP2. We also observed that the luciferase activity was highly increased when cotransfected with pRANneo or 5’∆5n.pac plasmid. We further investigated the mechanism responsible for this phenomemon.
Experimental procedures
Giardia culture. Trophozoites of G. lamblia isolate WB (ATCC 30957), clone C6, were cultured in modified TYI-S33 medium (Keister, 1983). Encystation was induced according to our published procedure (Sun et al., 2003).
Plasmid construction. All glps, zfps, and cwp1 fragments were amplified from
genomic DNA by PCR. Oligonucleotide sequences are available upon request. All constructs were verified by DNA sequencing (Applied Biosystems). Plasmid 5’∆5N-Pac was a gift from Dr Steven Singer and Dr Theodore Nash (Singer et al., 1998). Plasmid pRANneo, pNG, pNGI, -409/-1, GAL4ran32, and pNGAL4 have been described before (Sun et al., 1998; report for NSC grant). For constructing pNG5, the 5’-flanking region of the glp1 gene was amplified with oligonucleotides G5NF and G5BR, digested with NheI/BspHI, and ligated in place of the NheI/NcoI-excised 32-bp ran promoter and two copies of a 19-bp tet operator sequence in pNLop2-1 (Sun et al., 2002). The resulting plasmid, pNG5, contained the luciferase gene under the control of the glp1 promoter. For constructing pNG, the NheI/BspHI digested 5’-flanking region of the glp1 gene described above was ligated in place of the NheI/NcoI-excised 32-bp ran promoter and two copies of a 19-bp tet operator sequence in pNGI, resulting in pG5T. Then the 3’-flanking region of the glp1 gene was amplified with oligonucleotides G3EF and G3SR, digested with EcoRI/SalI, and ligated in place of the EcoRI/SalI-excised 3’-flanking region of the ran gene in pNG5T, resulting in pNG. For constructing pNTG, the glp1 gene coding region was amplified with oligonucleotides GNF and GAUER, digested with NcoI/EcoRI, and ligated in place of the NcoI/EcoRI-excised luciferase gene in pNT5 (Sun et al., 2002). The resulting plasmid, pNTG contained the glp1 gene controlled by the α2-tubulin promoter with an AU1 tag fused at its C terminus. For constructing pNGm1 (mutation of region 1, residues 7-8), a PCR product generated with oligonucleotides Gm1F and
GAUER was digested with BamHI and EcoRI. Another PCR product generated with oligonucleotides GNF and Gm1R was digested with NcoI and BamHI and cloned into NcoI/EcoRI digested pNGI with the BamHI/EcoRI fragment. Similar strategy was used to clone pNGm2 (mutation of region 2, residues 153-155) and pNGm3 (mutation of region 3, residues 33, 36, and 38). For constructing pNMybGLPC, a PCR product for the 5’-flanking region of the gmyb2 gene and gMyb2 N-terminal region was generated with oligonucleotides Myb2NF and MD3BR and then digested with NheI and BglII. Another PCR product for C-terminal region of GLP1 was generated with oligonucleotides GD1BF and GAUER, digested with BglII and EcoRI, and cloned into NheI/EcoRI digested pNT5 (Sun et al., 2002) with the NheI/BglII fragment. The resulting plasmid, pNMybGLPC, contains a fusion of N-terminal region of gMyb2 and C-terminal region of GLP1 under the control of the gmyb2 promoter. For constructing pNZFP1, the zfp1 gene and its 5’-flanking region was amplified from genomic DNA with oligonucleotides Z5NF and ZAUER, digested with NheI/EcoRI, and ligated in place of the NheI/EcoRI-excised luciferase gene, 32-bp ran promoter and two copies of a 19-bp tet operator sequence in pNLop2-1 (Sun et al., 2002). The resulting plasmid, pNZFP1 contained the zfp1 gene controlled by its own promoter with an AU1 tag fused at its C-terminus. For constructing pNGAL4-ZFP1C, N, or FL, a PCR product for C-terminal, N-terminal, or full-length portions of the ZFP1 coding region generated with specific oligonucleotides was digested with EcoRI and cloned into EcoRI digested and phosphatase treated pNGAL4. During the cloning process of pPW156/51m (not shown), the PCR resulted in two nucleotide changes of the
luciferase gene, in which Leu 411 was mutated to a termination codon and Asp 500 was mutated to Asn. We also constructed pPW1m by replacing the luciferase gene of the pPW1 to the mutant with NcoI digestion. For constructing pPW1s, the partial luciferase gene was amplified with oligonucleotides cwp15BF and lucXR on pPW1m, digested with SphI/XbaI, and ligated in place of the SphI/XbaI-excised partial
luciferase gene in pPW1m. The resulting plasmid, pPW1s, contained the truncated luciferase gene with a termination codon at aa 410. For constructing pNW1m, the luciferase gene with flanking region in pPW1m was excised with KpnI/ClaI and ligated in place of the KpnI/ClaI-digested pRANneo.
Transfection and luciferase assay. Cells transfected with pN series plasmids were
selected with G418 as described (Sun et al., 1998). Stable transfectants were maintained at 150 µg/ml G418. Cells transfected with other plasmids containg pac gene were selected and maintained with 54 µg/ml puromycin. The luciferase activity was determined as described (Knodler et al., 1999). Luciferase activity was measured with an Optocomp I luminometer (MGM Instruments). Western blots were probed with anti-AU1 monoclonal antibody (BAbCO,1/5000 in blocking buffer) and detected with Peroxidase conjugated goat anti-mouse IgG (Pierce, 1/5000) and enhanced
chemiluminescence (Amersham).
Immunofluorescence assay. The stable transfectants were cultured in growth specific
medium under G418 selection. Cells were harvested after 0 hr or 24 hr encystation, washed in PBS, and attached to glass coverslips (2×106 cells/coverslip) then fixed and stained (Abel et al., 2001). Cells were reacted with anti-AU1 monoclonal antibody (BAbCO, 1/300 in blocking buffer), and anti-mouse ALEXA 568 (Molecular Probes, 1/500 in blocking buffer) as the detector. GLP1 was localized on a Zeiss LSM
510 laser scanningconfocal microscope.
Expression and purification of recombinant GLP2 protein. The genomic glp2
gene was amplified using oligonucleotides G2F and G2R and the product was cloned into the expression vector pCRT7/CT-TOPO (Invitrogen) to generate plasmid pG2. The pG2 plasmid was freshly transformed into Escherichia coli BL21(DE3)pLysS (QIAexpressionist, Qiagen). An overnight pre-culture was used to start a 250-ml culture. E. coli cells were grown to an A600 of 0.6, and then induced with 1 mM
isopropyl-D-thiogalactopyranoside (Promega) for 6 hr. Bacteria were harvested by centrifugation and sonicated in 10 ml of buffer A (50 mM sodium phosphate, pH8.0, 300 mM NaCl) containing 10 mM imidazole and complete protease inhibitor cocktail (Roche). The samples were centrifuged and the supernatant was mixed with 1 ml of a 50% slurry of Ni-NTA superflow (Qiagen). The resin was washed with buffer A containing 20 mM imidazole and eluted with buffer A containing 250 mM imidazole. Fractions containing GLP2 were pooled, dialyzed in 25 mM HEPES pH 7.9, 40 mM KCl, 0.1 mM EDTA, and 15 % glycerol, and stored at -70°C. Protein purity and concentration were estimated by Coomassie Blue and silver staining compared with bovine serum albumin. GLP2 was purified to apparent homogeneity (>95%).
Electrophoretic mobility shift assay. Double-stranded oligonucleotides specified
in the text were 5’-end-labeled as described (Sun and Tai, 1999). Binding reaction mixtures contained the components as described (Sun and Tai, 1999), except that Tris buffer was replaced by 10 mM HEPES buffer (pH 7.9). Labeled probe (0.02 pmol) was incubated for 15 min at 25°C with 5 ng of purified GLP2 protein in a 20 µl volume supplemented with 0.5 µg of poly (dI-dC) (Sigma). Competition reactions contained 200-fold molar excess of cold oligonucleotides. In an antibody supershift assay, 0.8 µg of an anti-V5-HRP (Invitrogen) antibody was added to the binding reaction mixtures.
Results
Identification of the glp genes.
domain, we named GLP1, 2, and 3 (GenBank accession number AY822599,
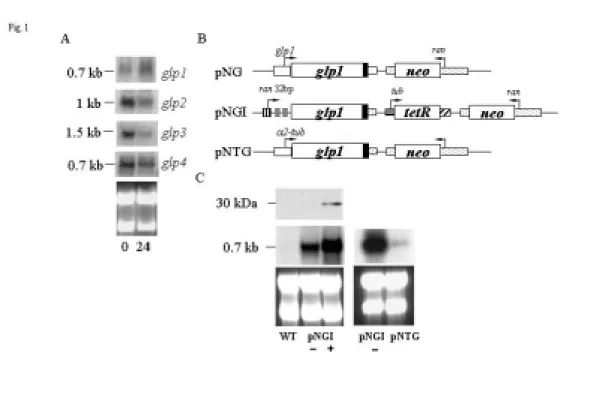
AY822600, and AY822601, respectively). To identify more glp genes from Giardia, we searched the G. lamblia genome database (http://www.mbl.edu/Giardia, McArthur et al., 2000) with the GARP domain from Arabidopsis ARR1 as a query sequence (GenBank accession no. AY056099). Amino acid sequences with similarity to the GARP domain were found in one protein we named GLP4 (GenBank accession number AY822602). We found that the glp1 gene was slightly up-regulated, but the glp2, glp3, and glp4 genes were down-regulated during encystation (Fig. 1A).
To determine the expression of GLP1 protein, we prepared construct pNG in which the glp1 gene is controlled by its own promoter and contains an AU1 epitope tag at its C terminus (Fig. 1B) and stably transfected it into Giardia . The GLP1 protein was not detected in either vegetative or encysting cells in Western blots (data not shown), suggesting that the expression of the glp1 gene under its own promoter may be very low. We therefore prepared construct pNGI in which the glp1 gene is controlled by a tetracycline regulatable 32-bp ran promoter and contains an AU1 epitope tag at its C terminus (Fig. 2B). A ~30-kDa protein, slightly larger than the predicted ~26.3 kDa molecular mass of GLP1 with the AU1 tag (~0.8 kDa), was detected with anti-AU1 antibody after tetracycline induction (Fig. 1C). The pNGI transfectants showed no phenotypic difference in the presence or absence of tetracycline, suggesting that expressed GLP1 is not toxic. The glp1 mRNA was detected at low and high levels in the absence and presence of tetracycline (Fig. 1C). We also prepared a pNTG
construct in which the glp1 gene is controlled by the well-known strong α2-tubulin promoter and contains an AU1 epitope tag at its C-terminus. However, the GLP1 protein was not detected in both vegetative and encysting cells in Western blot or immunofluorescence assays (data not shown). The glp1 mRNA expressed from the pNTG construct was much lower than that from pNGI in the absence of tetracycline (Fig. 1C).
Localization of the GLP1 protein.
The cellular locations of GLP1 were examined using tagged fusion proteins in the
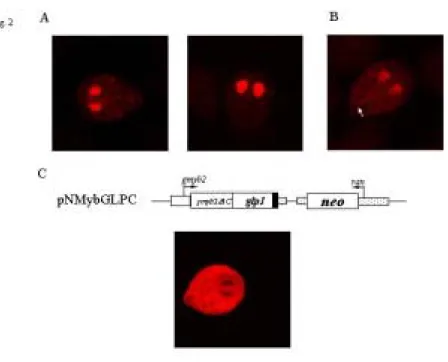
pNGI transfectants. As shown in Fig 2A, the AU1-tagged GLP1 could be detected exclusively in the nucleus in tetracycline induced cells (Fig. 2A, right panel),
indicating that GLP1 is a nuclear protein in Giardia. Expression was ~1% and ~50% positive in tetracycline noninduced and induced cells, respectively. There was also a weak cytosolic staining in the 1% of stained noninduced cells (Fig. 2A, left panel) but not in the induced cells. We also found that the GLP1 expressed by the pNG construct in which the glp1 gene is controlled by its own promoter localized to the nuclei and weakly in cytosol and the proportion of stained cells increased (5% vs 50%) during encystation (data not shown). The staining intensity of the non-regulated pNG
used the pNGI transfectants to identify the portion of GLP1 that is sufficient to direct the protein to the nuclei.
No typical nuclear localization signal was predicted in GLP1 using the PSORT software (http://psort.nibb.ac.jp/, Nakai and Kanehisa, 1992). Mutation of the N-terminal basic amino acids (region 1, residues 7-8, pNGm1). or a stretch of basic amino acids in the GARP-like domain (region 2, residues 153-155, pNGm2) did not affect the nuclear localization (data not shown). Mutation of the three lysine residues near the N-terminus (region 3, residues 33, 36, and 38, pNGm3) resulted in protein targeting to the nuclei and more targeting to the cytosol, including faint staining of an axonemal structure in the presence of tetracycline (Fig. 2B), suggesting that these three lysine residues may play a role in the exclusively nuclear localization. Deletion of the C-terminal region containing the GARP-like domain (residues 143-232, pNG∆C) or deletion of the N-terminal region (residues 2-141, pNG∆N) led to an absence of immunofluorescence (data not shown), indicating that these GLP1 deletion mutants were expressed at very low levels or were unstable proteins. We wanted to know if the C-terminal GARP-like domain of GLP1 could also help the nuclear localization of another protein. Previously, we showed that the gMyb2 transcription factor with a deletion of its C-terminal Myb repeats could not target to the nuclei but was unevenly distributed in the cytosol (pNMyb∆C, Sun et al., 2002). Therefore, we constructed a pNMybGLPC plasmid which contains a fusion of the N-terminal region of gMyb2 and the C-terminal region of GLP1 under the control of the gmyb2
promoter, which is upregulated in encystation. The chimeric protein was localized exclusively in the cytosol during encystation (Fig. 3C), indicating that the C-terminal region of GLP1 which contains the GARP-like domain does not contain a functional NLS.
Identification of the GLP2 binding sites.
We have shown that GLP1 binds to the 32-bp ran promoter element and the cwp1 promoter in the double-stranded configurations in electrophoretic mobility shift assays. The 32-bp ran promoter element was previously shown to play a predominant role in determining promoter activity and positions of the transcription start sites (Sun and Tai, 1999), indicating that GLP1 may be involved in transcriptional regulation. Mutation analysis revealed that (A/G)ATCN sequences were required for the binding of GLP1.
To understand the potential function of GLP2 in G. lamblia, we further investigated the ability of the GLP2 to bind to DNA. We expressed GLP2 with a C-terminal V5 tag in E. coli and purified it to >95% homogeneity as assessed in silver stained gels (data not shown). To determine whether purified GLP2 binds DNA, we performed
electrophoretic mobility shift assays with double-stranded DNA sequences from the 5’-flanking region of an encystation-induced gene, cwp1, and a constitutive gene, ran.
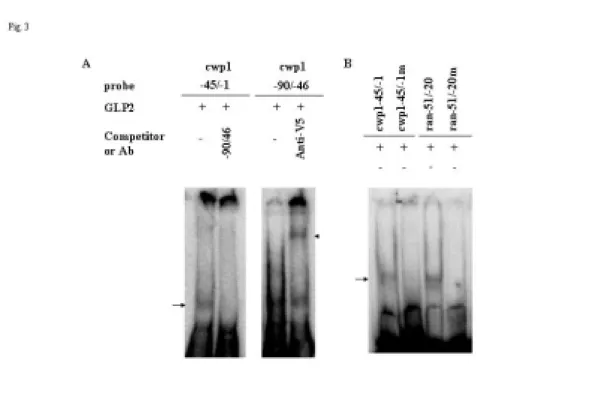
Incubation of a labeled double-stranded DNA probe cwp1 -45/-1 and -90/-46 (Table 1) with GLP2 resulted in the formation of retarded bands (Fig. 3A). GLP2 did not bind to either single strand of the cwp1 -90/-46 probe (data not shown). Within this region, GLP2 bound to the 3’ region, cwp -68/-46, but not to cwp1 -90/-69 (Table 1). GLP2 also bound to cwp1 -45/-1 and to a well-characterized constitutive core promoter, ran -51/-20, but not to ran -30/-1 (Table 1).
The binding specificity was confirmed by competition and supershift assays (Fig. 3A, additional data not shown). The formation of the retarded cwp1 -45/-1 bands was almost totally competed by a 200-fold molar excess of unlabeled cwp1 -90/-46, but not by the same excess of a nonspecific competitor (NS), ran -30/-1 (data not shown). The bound form on cwp1 -90/-46 could also be supershifted by an anti-V5-HRP antibody (Fig. 3A).
The cwp1 -68/-46 probe contains the sequence AATCT, which is the consensus binding sites of GLP1. Mutation analysis of the cwp1 -68/-46 probe revealed that the short AATCT sequence was required for binding (Table 1, cwp1 -68/-46m). Mutation analysis of the cwp1 -45/-1 probe (Table 1, cwp1 -45/-1m) and ran -51/-20 (Table 1, ran -51/-20m), revealed that the GATCA and AATCG sequences were required for binding, respectively (Fig. 3B). These results indicate that the (A/G)ATC(A/G/T) sequence could be the GLP2 binding sites. The other probes tested, cwp1 -90/-69, g6pi-b -30/-1, g6pi-b -60/-31, ran -30/-1, and ran -81/-52, neither contained the (A/G)ATC(A/G/T) sequence nor bound to GLP2 (Table 1, data not shown). Scanning mutagenesis of the 5-bp AATCT sequence in the cwp1 -68/-46 probe showed that any single substitution within the AATC sequence abolished the
DNA-protein interaction (data not shown), but mutations of the fifth T to A or G did not completely eliminate binding (data not shown). Mutations of the fifth T to C abolished binding (data not shown). In summary, GLP2 can bind to
(A/G)ATC(A/G/T). This is similar to the sequence of GLP1 binding sites, (A/G)ATCN.
Identification and characterization of zfp1 and zfp2.
The zfp1 and zfp2 genes were found previously by in silico screening of the G. lamblia genome data base. In the previous study, we showed that ZFP1 localized to the two nuclei in both vegetative and encysting trophozoites. In this project, we found that both zfp1 and zfp2 mRNA levels increased during encystation (Fig. 4). ZFP1 promoted expression of the reporter gene driven by the cwp1 promoter in Giardia in cotransfection assays, indicating that the gMyb2 can transactivate the cwp1 promoter (Fig 5). We also used the GAL4-based system to determine the transactivation domain of ZFP1. We found that transactivation by ~4.2-fold occurred when the effector contained the C-terminal region of ZFP1 containing glutamine rich domain (Fig. 6). The full-length and N-terminal portions of GLP1 showed no transactivation ability.
Effect of G418 and puromycin on G. lamblia.
Despite extensive usage of G418 and puromycin for selection of stable transfectants in the lab, very little is known about what causes toxicity of these
antibiotics in Giardia stable transfectants. To understand the effect of these drugs on G. lamblia transcription or translation, we made two kinds of luciferase mutant, a
truncated luciferase comprised of only 1-410 amino acids (luc+s) and a mutated luciferase gene (luc+m). The luc+m differed from the normal one at two positions, in which Leu 411 was mutated to a termination codon and Asp 500 was mutated to Asn. Trophozoites transfected with a pPW1s construct containing the luc+s gene under the control of the cwp1 promoter displayed only a background level of activity, indicating that the truncated luciferase lacks enzymic activity (Fig. 7). However, trophozoites transfected with a pPW1m construct containing the luc+m gene under the control of the cwp1 promoter exhibited ~200-fold of background activity (Fig. 7). The levels of luc+m were about five orders of magnitude reduction than wild type luc+ in luciferase activity. The results indicate that the nonsense mutation in the luc+m was supressed and the lucifease activity was restored in G. lambila trophozoites.
To understand whether pRANneo influences luciferase expression, pPW1m was stably cotransfected with pRANneo. The luciferase activity in the cotransfectants increased by ~25-fold than pPW1 transfectants, indicating that pRANneo could suppress nonsense mutation. However, cotransfection of pRANneo transiently to pPW1 transfectants without of addition of G418 selection did not increase luciferase activity (data not shown). Since the aminoglycoside gentamicin G418 was used to select and maintain stable transfectants, this effect could be from G418 treatment. We further tested the effect of G418 by adding of G418 to pPW1m transfectants. As shown in Fig. 7, G418 treatment significantly increased luciferase activity by
~63-fold. However, treatment of tetracycline to pPW1 transfectants did not influence luciferase activity (data not shown).
To understand whether 5’∆5N-Pac also influences luciferase expression, we made a pNW1m constuct in which the pac gene was used instead of neo gene. The NW1 stable transfectants displayed ~2-fold of luciferase activity than pPW1 (Fig. 7). Cotransfection of pNW1 with 5’∆5N-Pac increased luciferase activity by ~269-fold than pNW1m transfectants, indicating that 5’∆5N-Pac could also suppress nonsense mutation (Fig. 7). We did not success in testing the effect of puromycin because the treatment of puromycin resulted cell death.
To understand whether the increase of luciferase activity was due to the increase of mRNA level, we further detected the luciferase mRNA of different transfectants by Northern blot analysis. As shown in Fig. 8A, similar high levels of luciferase mRNA were found in the pPW1m and pPW1s transfectants. This indicates that the lack of activity of luc+s was not due to the low mRNA levels. Similar high levels of
luciferase mRNA were also found in the pPW1m transfectants and its pRANneo cotransfectants (Fig. 8B). In addition, similar high levels of luciferase mRNA were also found in the pNW1m transfectants and its 5’∆5N-Pac cotransfectants (Fig. 8B). The results indicate that the higher activity in the pRANneo or 5’∆5N-Pac
cotransfectants was not due to higher mRNA levels.
Discussion
Identification and characterization of the glp genes.
The constitutive and encystation-induced giardial promoter sequences defined to date contain A/T-rich Inr-like elements and are short (Holberton and Marshall, 1995; Lujan et al., 1995; Mowatt et al., 1995; Elmendorf et al., 2001; Sun and Tai, 1999; Knodler et al., 1999; Yee et al., 2000; Davis-Hayman et al., 2003). In earlier studies, unidentified nuclear extract proteins bound only to single-stranded oligonucleotide targets, suggesting a fundamental divergence from transcription mechanisms in other eukaryotes (Sun and Tai, 1999; Yee et al., 2000). Our previously identified gmyb2 gene whose expression increased during encystation could be important for
transcriptional activation of encystation genes, and may help co-ordinate synthesis of cyst wall proteins and polysaccharide (Sun et al., 2002; Sun et al., unpublished data). In this study, we have identified a transcription factor GLP1/2 which binds specific sequences in double-stranded configuration. We found that the core promoter region of a constitutive ran gene and the proximal upstream region of an encystation-induced cwp1 gene contained GLP1/2 binding sites.
The GARP homologues constitute a large family of plant DNA-binding proteins that perform a variety of cellular functions. These GARP domains may have similar sequence specificities for DNA binding, because DNA bound to ARR1 and CCA1 contains GGATT and AGATT sequences, respectively (Sakai et al., 2000; Wang et al., 1997). Although divergent from plant GARP proteins, GLP1 still maintains the ability to bind DNA in a sequence-specific manner. The GLP1 binding sites (A/G)ATCN and the GLP2 binding sites (A/G)ATC(A/G/T) are similar to that of Arabidopsis ARRs binding sites (A/G)ATC(T/C), although ARR is on the noncoding strand (Sakai et al., 2000; Hosoda et al., 2002). This indicates that the binding sites of the members of GARP family have been conserved in evolution. ARR1 has a strong preference for a C or T at position 5; GLP1 tolerates either A, T, C, or G at position 5; GLP2 tolerates either A, T, or G at position 5. The key residues contacting position 5 are E and A of ENVA in Arabidopsis ARRs (Hosoda et al., 2002). However, they are predicted to be Q and E of QNVE in GLP1; K and Q of KHVQ in GLP2 (data not shown). The non-conservative substitutions might explain why GLP1/2 tolerates more nucleotides at position 5. The best GLP1/2 target sequennces were identified as AATCT, GATCA, and AATCG. The ability of binding activity of GLP1/2 to bind GATCA could be
attributed to the creation of another ATC sequence in the complementary strand as a result of the replacement of A at the first position with G. As the specificity
determined by a 4 or 5 bp sequence does not seem high enough for choosing target genes on the giardial genome, it is likely that additional transcription factors interacting with GLP1/2 help increase the specificity for transcriptional regulation. Unlike Arabidopsis ARR1 and other GARP proteins, which contain central GARP domains, the GARP-like domains of giardial GLPs are located near C terminus. The purified C-terminal region containing the GARP-like domain of GLP1 contained the DNA-binding activity of GLP1 (report for grant). It is expected that other members in Giardia containing the GARP-like domain, such as GLP3 and GLP4, may also able to bind DNA with similar sequences. Since the resemblance of GLP1-4 does not extend beyond this domain region, the GARP-like domain may be a functional unit for DNA binding in the giardial GARP family of proteins.
GARP domains have been found to confer the ability to direct nuclear localization in Arabidopsis (Hosoda et al., 2002). However, the GARP-like domain and its C-terminal sequence of GLP1 did not direct the truncated gMyb2 protein to nuclei. No probable nuclear localization signals except some basic amino acids are present in GLP1. The basic sequences may be functionally redundant in terms of the nuclear targeting. This is consistent with our results which showed that mutation of two stretches of basic amino acids (regions 1 and 2, residues 7-8 and residues 153-155) did not affect the nuclear localization. It is also possible that regions 1 and 2 are not responsible for nuclear localization. Mutation of three lysine residues near N-terminal (region 3) resulted in slight protein targeting to the cytosol and axonemes, as well as the nuclei. Although the specific signal sequence by which GLP1 localize to nuclei was not determined completely, our identified region 3 (residues 33, 36, and 38), presumably contributes at least in part to nuclear localization.
Although many plant GARP proteins have been classified as type B two-component response regulators (Sakai et al., 1998; Sakai et al., 2000; Imamura, 1999) and
pseudo-response regulators (Makino et al., 2000; Makino et al., 2001). Giardial GLPs do not contain the amino-terminal receiver domain conserved in this class of proteins. A number of GARP proteins in plants can function as transcriptional activators and are involved in the adaptive response and differentiation processes (Makino et al., 2000; Imamura, 1999; Sakai et al., 2001; Kerstetter et al., 2001). Since Giardia colonizes the intestinal tract, it must constantly react to changing signals from the host environment (Adam, 2001). GLP1/2 binding sites are present in the proximal
5’-flanking regions of some encystation-induced genes such as cwp1 and other genes which are not induced during encystation. This suggests that GLP1/2 may be involved in transcriptional regulation of many different genes. The ran and cwp1 gene
promoters were activated by GLP1/2, suggesting that GLP1/2 is a transactivator in vivo. Other genes whose promoter regions contain the GLP1/2 recognition sequences
are possibly transactivated by GLP1/2. An as yet unidentified signal may modulate the capability for transactivation and/or DNA binding.
The garp genes have been found only in plant genomes to date. Although it is divergent, GLP1/2 is clearly recognizable as a member of the GARP family. This suggests that members of the GARP family evolved before divergence of Giardia from the main eukaryotic line of descent. GARP domains may be important in regulating gene expression in the earliest eukaryotes. The similarity in the binding targets of GLP1/2 and Arabidopsis ARR1 may be co-incidental or may reflect similar structure or function. Further understanding awaits the exploration of other GARP proteins in other deep-branching protist, such as Trichomonas vaginalis and Entamoeba histolytica.
Identification and characterization of zfp1 and zfp2.
Our results showed that cotransfected ZFP1 could increase the expression of cwp1 gene promoter by ~8-fold in vegetative trophozoites. The results indicate that ZFP1 could function as a transcriptional activator. We are trying to understand if other gene promoters can also be upregulated by the overexpressed ZFP1. We also found that the domain responsible for in vivo transactivation of ZFP1 was localized within the C-terminal region of ZFP1. This indicates that the a stretch of glutamine-rich amino acids may have transactivation ability. Glutamine-rich characteristics are frequently associated with the transactivation domain of eukaryotic transcription factors. We are analyzing the biological function of zfp1/2 gene which is up-regulated during
encystation.
Effect of G418 and puromycin on G. lamblia.
The firefly luciferase molecule is folded into two compact domains (Conti et al., 1996). The large N-terminal domain, comprising residues 4-436, consists of a distorted antiparallel beta-barrel and two beta-sheets. The C terminus of the protein (440-544) forms a distinct domain, which is separated from the N-terminal domain by a wide cleft. In our study, the luciferase deletion mutant (luc+s) that lacks the
C-terminal residues 411-544 did not display any activity, suggesting that the C terminus of luciferase is important for enzymic activity. However, transfection of a luciferase mutant (luc+m) in which Leu 411 was mutated to a termination codon and Asp 500 was mutated to Asn exhibited ~200-fold of background levels. The luciferase activity in this mutant was much lower than the wild type activity (~10-5 of the wild type activity). The results indicate that the nonsense mutation in the luc+m was
supressed in G. lambila trophozoites. We also found that stably cotransfection of luc+m containg construct with pRANneo highly increased luciferase activity. However, addition of the aminoglycoside G418 but not transient transfection of pRANneo significantly increased in luciferase activity, suggesting that the G418 treatment induced suppression of premature stop mutation of the luc+m. In addition, mRNA levels in the luc+m stable transfectants and their pRANneo cotranfectants were similar in Northern blot analysis. These results indicate that G418 can suppress the premature stop mutation. We also found that puromycin, a protein synthesis inhibitor, can also suppress the premature stop mutation to a significant extent. Aminoglycosides bind to ribosomal RNA and cause a conformational change in ribosomal RNA (Eustice and Wilhelm, 1984; Schroeder et al., 2000). They disturb codon-anticodon recognition at the aminoacyl-tRNA acceptor site, dissrupting high-fidelity reading of the genetic code. Aminoglycosides cause the ribosome to introduce missense mutations and translates through termination codons in both bacterial and eukaryotic cells. Growth of mammalian cells in the presence of an aminoglycoside antibiotic can promote translation read-through of the nonsense codon (Burke, J.F., and Mogg, A.E. 1985.). Our results also correlated the potentiation of misreading of the Gentamicin G418 in Giardia.
Puromycin is an analog of the aminoacyl terminus of an aminoacyl-tRNA. In higher eukayotes, its amino group linked non-specifically to the carbonyl group of growing polypeptide chains to form an adduct that dissociates from the ribosome, thereby causing premature termination of translation (Nathans et al., 1964; Kirillov et al., 1997). Our results showed that puromycin can suppress the premature stop
mutation in G. lamblia. This is quite different from the effect of puromycin in higher eukaryotes. We are currently working on analyzing the potential mechanism of puromycin in G. lamblia.
References
Abel, E.S., Davids, B.J., Robles, L.D., Loflin, C.E., Gillin, F.D., and Chakrabarti, R. (2001) Possible roles of protein kinase A in cell motility and excystation of the early diverging eukaryote Giardia lamblia. J Biol Chem 276: 10320-10329.
Adam R.D. (2001) Biology of Giardia lamblia. Clin Microbiol Rev 14: 447-475. Best, A.A., Morrison, H.G., McArthur, A.G., Sogin, M.L., and Olsen, G.J. (2004) Evolution of eukaryotic transcription: insights from the genome of Giardia lamblia. Genome Res 14:1537-1547.
Burke JF, Mogg AE. Suppression of a nonsense mutation in mammalian cells in vivo by the aminoglycoside antibiotics G-418 and paromomycin. Nucleic Acids Res. 1985 Sep 11;13(17):6265-72.
Molecules, and Culture. Eds. S. Osawa, T. Honjo, p. 271-304, Tokyo: Springer-Verlag.
Conti, E., Franks, N.P., and Brick, P. Structure. (1996) Crystal structure of firefly luciferase throws light on a superfamily of adenylate-forming enzymes. Structure 4: 287-298.
Eichinger, D. (2001) Encystation in parasitic protozoa. Curr Opin Microbiol 4: 421-426.
Davis-Hayman SR, Hayman JR, Nash TE. Encystation-specific regulation of the cyst wall protein 2 gene in Giardia lamblia by multiple cis-acting elements. Int J Parasitol. 2003 Sep 15;33(10):1005-12.
Elmendorf, H.G., Singer, S.M., Pierce, J., Cowan, J., and Nash, T.E. (2001) Initiator and upstream elements in the α2-tubulin promoter of Giardia lamblia. Mol Biochem Parasitol 113: 157-169.
Eustice DC, Wilhelm JM. Fidelity of the eukaryotic codon-anticodon interaction: interference by aminoglycoside antibiotics. Biochemistry. 1984 Mar 27;23(7):1462-7. Gillin, F.D. and Reiner, D.S. (1996) Cell biology of the primitive eukaryote Giardia lamblia. Annu Rev Microbiol 50: 679-705.
Gillin, F.D., Reiner, D.S., Gault, M.J., Douglas, H., Das, S., Wunderlich, A., and Sauch, J.F. (1987) Encystation and expression of cyst antigens by Giardia lamblia in vitro. Science 235: 1040-1043.
Hashimoto, T., Nakamura, Y., Nakamura, F., Shirakura, T., Adachi, J., Goto, N., Okamoto, K., and Hasegawa, M. (1994) Protein phylogeny gives a robust estimation for early divergences of eukaryotes: phylogenetic place of a mitochondria-lacking protozoan, Giardia lamblia. Mol. Biol. Evol. 11: 65-71.
Hashimoto, T., Nakamura, Y., Kamaishi, T., Nakamura, F., Adachi, J., Okamoto, K., and Hasegawa, M. (1995) Phylogenetic place of mitochondrion-lacking protozoan, Giardia lamblia, inferred from amino acid sequences of elongation factor 2. Mol. Biol. Evol. 12: 782-93.
Holberton, D.V., and Marshall, J. (1995) Analysis of consensus sequence patterns in Giardia cytoskeleton gene promoters. Nucleic Acids Res 23: 2945-2953.
Hosoda, K., Imamura, A., Katoh, E., Hatta, T., Tachiki, M., Yamada, H., et al. (2002) Molecular structure of the GARP family of plant Myb-related DNA binding motifs of the Arabidopsis response regulators. Plant Cell14:2015-2029.
Imamura, A., Hanaki, N., Nakamura, A., Suzuki, T., Taniguchi, M., Kiba, T., et al. (1999) Compilation and characterization of Arabidopsis thaliana response regulators implicated in His-Asp phosphorelay signal transduction. Plant Cell Physiol 40: 733-742.
Keister, D.B. (1983) Axenic culture of Giardia lamblia in TYI-S-33 medium supplemented with bile. Trans R. Soc Trop Med Hyg 77: 487-488.
KANADI regulates organ polarity in Arabidopsis. Nature 411: 706-9.
Knodler, L.A., Svard, S.G., Silberman, J.D., Davids, B.J., and Gillin, F.D. (1999) Developmental gene regulation in Giardia lamblia: first evidence for an
encystation-specific promoter and differential 5' mRNA processing. Mol Microbiol 34: 327-340.
Kirillov S, Porse BT, Vester B, Woolley P, Garrett RA. Movement of the 3'-end of tRNA through the peptidyl transferase centre and its inhibition by antibiotics. FEBS Lett. 1997 Apr 14;406(3):223-33.
Krempler, A., and Brenig, B. (1999) Zinc finger proteins: watchdogs in muscle development. Mol. Gen. Genet.. 261: 209-215.
Lujan, H.D., Mowatt, M.R., Conrad, J.T., Bowers, B., and Nash, T.E. (1995)
Identification of a novel Giardia lamblia cyst wall protein with leucine-rich repeats. Implications for secretory granule formation and protein assembly into the cyst wall. J Biol Chem 270: 29307-29313.
Lujan, H.D., Mowatt, M.R., Nash, T.E. (1997) Mechanisms of Giardia lamblia differentiation into cysts. Microbiol Mol Biol Rev 61: 294-304.
Makino, S., Kiba, T., Imamura, A., Hanaki, N., Nakamura, A., Suzuki, T., et al. (2000) Genes encoding pseudo-response regulators: insight into His-to-Asp phosphorelay and circadian rhythm in Arabidopsis thaliana. Plant Cell Physiol41: 791-803. Makino, S., Matsushika, A., Kojima, M., Oda, Y., and Mizuno, T. (2001) Light response of the circadian waves of the APRR1/TOC1 quintet: when does the quintet start singing rhythmically in Arabidopsis? Plant Cell Physiol 42: 334-339.
McArthur, A.G., Morrison, H.G., Nixon, J.E., Passamaneck, N.Q., Kim, U., Hinkle, G., et al. (2000) The Giardia genome project database. FEMS Microbiol Lett 189:
271-273.
Mowatt, M.R., Lujan, H.D., Cotton, D.B., Bower, B., Yee, J., Nash, T.E., and Stibbs, H.H. (1995) Developmentally regulated expression of a Giardia lamblia cyst wall protein gene. Mol Microb 15: 955-963.
Nakai, K. and Kanehisa, M. (1992) A knowledge base for predicting protein localization sites in eukaryotic cells. Genomics 14: 897-911.
NATHANS D. PUROMYCIN INHIBITION OF PROTEIN SYNTHESIS:
INCORPORATION OF PUROMYCIN INTO PEPTIDE CHAINS. Proc Natl Acad Sci U S A. 1964 Apr;51:585-92.
Reiner, D.S., Douglas, D., and Gillin, F.D. (1989) Identification and localization of cyst-specific antigens of Giardia lamblia. Infect Immun 57: 963-968.
Sakai, H., Aoyama, T., and Oka, A. (2000) Arabidopsis ARR1 and ARR2 response regulators operate as transcriptional activators. Plant J. 24: 703-711.
Sakai, H., Aoyama, T., Bono, H., and Oka, A. (1998) Two-component response regulators from Arabidopsis thaliana contain a putative DNA-binding motif. Plant Cell Physiol 39: 1232-1239.
Sakai, H., Honma, T., Aoyama, T., Sato, S., Kato, T., Tabata, S., and Oka, A. (2001) ARR1, a transcription factor for genes immediately responsive to cytokinins. Science
294: 1519-1521.
Singer SM, Yee J, Nash TE. Episomal and integrated maintenance of foreign DNA in Giardia lamblia. Mol Biochem Parasitol. 1998 Apr 1;92(1):59-69.
Schroeder R, Waldsich C, Wank H. Modulation of RNA function by aminoglycoside antibiotics. EMBO J. 2000 Jan 4;19(1):1-9.
Sogin, M.L., Gunderson, J.H., Elwood, H.J., Alonso, R.A., and Peattie, D.A. (1989) Phylogenetic meaning of the kingdom concept: an unusual ribosomal RNA from Giardia lamblia. Science 243: 75-77.
Sun, C.H., and Tai, J.H. (1999) Identification and characterization of a ran gene promoter in the primitive protozoan pathogen Giardia lamblia. J Biol Chem 274: 19699-19706.
Sun, C.H., Chou, C.F., and Tai, J.H. (1998) Stable DNA transfection of the primitive protozoan pathogen Giardia lamblia. Mol Biochem Parasitol 92: 123-132.
Sun, C.H., McCaffery, J.M., Reiner, D.S., and Gillin, F.D. (2003) Mining the Giardia lamblia genome for new cyst wall proteins. J Biol Chem 278: 21701-21708.
Sun, C.H., Palm, D., McArthur, A.G., Svard, S.G., and Gillin, F.D. (2002) A novel Myb-related protein involved in transcriptional activation of encystation genes in Giardia lamblia. Mol Microbiol 46: 971-984.
Takatsuji, H. Zinc-finger transcription factors in plants. (1998) Cell Mol. Life Sci. 54: 582-596.
Van Keulen, H., Steimle, P. A., Bulik, D. A., Borowiak, R. K., and Jarroll, E. L. (1998) Cloning of two putative Giardia lamblia glucosamine 6-phosphate isomerase genes only one of which is transcriptionally activated during encystment. J. Eukaryot. Microbiol. 45: 637-642.
Wang, Z.Y., Kenigsbuch, D., Sun, L., Harel, E., Ong, M.S., and Tobin, E.M. (1997) A Myb-related transcription factor is involved in the phytochrome regulation of an Arabidopsis Lhcb gene. Plant Cell 9: 491-507.
Wolfe, M.S. (1992) Giardiasis. Clin. Microbiol. Rev. 5: 93-100.
Yee, J., Mowatt, M.R., Dennis, P.P., and Nash, T.E. (2000) Transcriptional analysis of the glutamate dehydrogenase gene in the primitive eukaryote, Giardia lamblia. Identification of a primordial gene promoter. J Biol Chem 275: 11432-11439.
Fig. 1. Analysis of glp1 gene expression. (A) Northern blot analysis of glps
transcription using specific probes. G. lamblia cells were cultured in growth (0 hr) or 24hr in encystation medium (upper panel). A ribosomal RNA loading control is in the lower panel. (B) Diagrams of the pNG, pNGI, and pNTG plasmids. A neo gene (open box) is under the control of the 5’- and 3’-flanking regions of the ran gene (dotted box). The glp1 gene (open boxes) is under the control of its own 5’- and 3’-flanking regions (open boxes) in pNG or it is under the control of the 32-bp ran promoter (striped box) and the 3’-flanking of the ran gene (dotted box) in pNGI. Two tet operators (gray box) were inserted into the promoter context in the pNGI plasmid. The filled box indicates the coding sequence of the AU1 epitope tag. In pNTG, the glp1 gene (open boxes) is under the control of the 5’-flanking regions (open boxes) of the α2-tubulin gene the 3’-flanking of the ran gene (dotted box) in pNGI. The tet repressor (tetR) is flanked by α2-tubulin promoter (open box) and the 3’-flanking region of the ran gene (dotted box). The arrows show the directions of gene transcription. (C) GLP1 protein expression in pNGI stable transfectants. The pNGI transfectants were cultured in growth medium with (+) or without tetracycline (-) and harvested at 24 hr. AU1 tagged GLP1 protein was detected by anti-AU1 antibody. WT means nontransfected wild type cells. Northern blot analysis of glp1 transcription in pNGI and pNTG stable transfectants are shown in the middle panel. Ribosomal RNA loading controls are in the lower panel.
Fig. 2. (A) Nuclear localization of GLP1. The pNGI transfectants were cultured in growth medium with (right panel) or without (left panel) tetracycline for 24 hr and then subjected to immunofluorescence analysis, using anti-AU1 antibody for detection. (B) Nuclear localization of GLP1 mutant protein in the pNGm3
transfectants. It shows that the product of pNGm3 localizes to nuclei, cytosol, and axonemes (arrow) in tetracycline induced cells. (C) Nuclear localization of GLP1 mutant proteins in the pNMybGLPC transfectants. To construct pNMybGLPC, we fused the C-terminal region of GLP1 containing the GARP-like domain (residues 143-232) to the C terminus of the truncated version of the gMyb2 which lacked the Myb repeats and was directed by the gmyb2 promoter. The AU1 tag was also fused to the C terminus of the protein. It shows that the product of pNMybGLPC localizes in cytosol during encystation.
Fig. 3. DNA-binding ability of GLP2 revealed by electrophoretic mobility shift assays. (A) Detection of GLP2 binding sites. Electrophoretic mobility shift assays were
performed using purified GLP2 and various 32P-end-labeled oligonucleotide probes (see Table 1). Components in the binding reaction mixtures are indicated above the lanes.
Fig. 4. Northern blot analysis of zfp1 and zfp2 transcription using specific probes. G. lamblia cells were cultured in growth (0 hr) or 24hr in encystation medium (upper panel). A ribosomal RNA loading control is in the lower panel.
Fig. 5. Transactivation by ZFP1 in the cotranfection system. After stable transfection with these constructs, luciferase activity was measured in vegetative cells. The luciferase activity was expressed as relative light units (RLU) per µg protein. At least three independent experiments were performed for each construct. The activity of cotransfectants (+) relative to single transfection (-) was presented.
Fig. 6. Analysis of transcription acivity by a GAL4 system. The luciferase reporter gene (luc +) is under the control of the 32-bp ran promoter as shown. Five GAL4 binding sequences (open boxes) were inserted upstream of the promoter in
GAL4ran32 reporter plasmid. The pac gene is under the control of 5’- and 3’-flanking region of the gdh gene as shown. The C-terminal, N-terminal, or full-length portions of ZFP1 were fused to the DNA-binding domain of yeast GAL4 which is driven by the α2-tubulin promoter. Single reporer plasmid was transfected to test the promoter activity (-). Transactivation was examined by stable transfection of a reporter plasmid GAL4ran32 first and then stable transfection of an effector plasmid (+). Luciferase activity was measured in vegetative cells. Results are expressed as the mean +/- SD of at least three separate experiments. The basal level of luciferase activity of
GAL4ran32 and pNGAL4 cotransfectants was arbitrarily set to 1.
Fig. 7. Induction of luciferase activity by pRANneo and 5’ ∆5N-Pac in the cotranfection system. After stable transfection with these constructs, luciferase activity was measured in vegetative cells. The luciferase activity was expressed as relative light units (RLU) per µg protein. At least three independent experiments were performed for each construct. The activity of cotransfectants (+) relative to pPW1m transfection (-) was presented.
Fig. 8. Northern blot analysis of luciferase transcription. Specific G. lamblia transfectants were cultured in growth medium and detected using luciferase probe (upper panel). Ribosomal RNA loading controls are in the lower panel.
Table 1. Oligonucleotides and electrophoretic mobility shifts.
Name Sequence (5' to 3') Binding
Activitya cwp1 -45/-1 GTTTACAACTCTCAGATCAAGAAACACAGAAATAAAATATCAGGG + cwp1 -45/-1m GTTTACAACTCTCACTAGTAGAAACACAGAAATAAAATATCAGG - cwp1 -90/-46 CTGGCTAACAGTCTACAGTCTACAATTTACAGTATACAATCTACA + cwp1 -90/-69 CTGGCTAACAGTCTACAGTCTA - cwp1 -68/-46 CAATTTACAGTATACAATCTACA + cwp1 -68/-46m CAATTTACAGTATACTTAGAACA - g6pi-b -30/-1 TATATATGTAACTGCCTCGCCAAAATAAAA - g6pi-b -60/-31 TCCTGTAAGGGTTTACTACAGCAGTAGCAG - ran -30/-1 TAAAACTTTAACCCAGGAACCGAGAGTAGC - ran -51/-20 TAAAATAAATTAAATCGAAATTAAAACTTTAA +
ran -51/-20m TAAAATAAATTATTAGCAAATTAAAACTTTAA -
ran -81/-52 CAGAAAAAGTTGTCAAGGAGGTGATAGCCG -
a The ability of oligonucleotides to bind to the GLP2 protein. “+” and “-“ represent
binding and no binding, respectively. The putative GLP2 binding sequences are underlined. Base changes in the mutants tested are indicated in boldface. 計畫成果自評
In this study, we cloned and identified other putuative transcription factors that may be important for Giardia gene expression. We also characterized previously identified transfection factors GLP1 and ZFP1 and found that they functioned as transactivators. The studies in this proposal may reveal basic insights into formation and function of the G. lamblia encystation. Since cystic stages are key to transmission of many parasites, this differentiation may be a target for interruption of the life cycle. The difference of amino acid sequence and function of transcription factors in G. lamblia and higher eukaryotes will provide an indication of the degree of evolutionary variation. We are wring two papers for the data from this study. This study also provided the master students to learn how to do research work on parasites and learn cellular and molecular biology techniques.