Sesquiterpenoids and Artificial 19-Oxygenated Steroids from the Formosan Soft Coral
Nephthea erecta
Shi-Yie Cheng,
†Chang-Feng Dai,
‡and Chang-Yih Duh*
,†,§Department of Marine Biotechnology and Resources, National Sun Yat-sen UniVersity, Kaohsiung 804, Taiwan, Republic of China, Institute of Oceanography, National Taiwan UniVersity, Taipei, Taiwan, Republic of China, and Center of Asia-Pacific Marine Researches, National Sun Yat-sen UniVersity, Kaohsiung, Taiwan, Republic of China
ReceiVed April 25, 2007
Chemical investigations on the acetone and MeOH solubles of the soft coral Nephthea erecta have afforded five new sesquiterpenoids (1–5), one known sesquiterpene, kelsoene (6), and two known 19-oxygenated steroids (10 and 11). In addition, three unexpected artificial 19-oxygenated steroids (7–9) were obtained by letting 10 and 11 stand in CDCl3for prolonged periods of time. The structures of 1–9 were elucidated by extensive spectroscopic analyses, and their cytotoxicity against selected cancer cells was measured in Vitro.
The family Nephtheidae has been proved to be a rich source of bioactive compounds.1–9The ongoing search for bioactive
con-stituents prompted us to reinvestigate the secondary metabolites of the soft coral Nephthea erecta Kükenthal (Nephtheidae).9
Com-pounds 1–6,10,11,14–1710,12and 1113were isolated from the soft
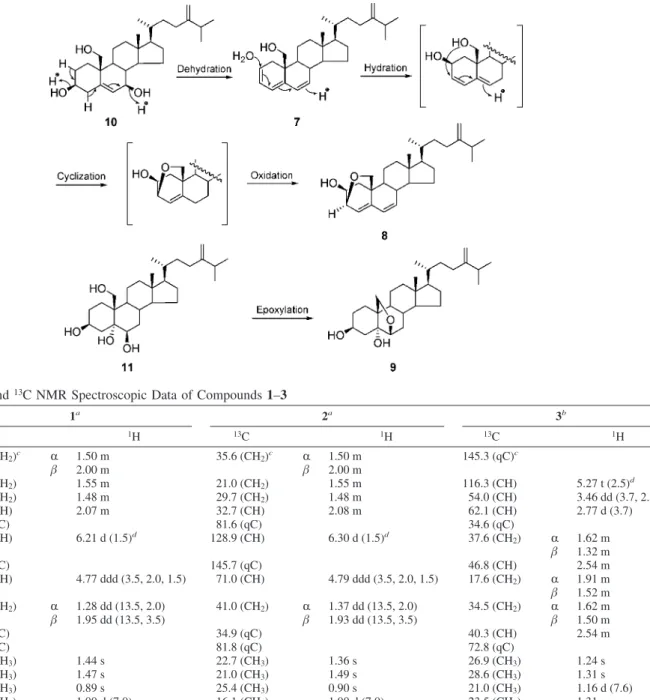
coral N. erecta while 7–9 are artifacts obtained by allowing 10 and 11 to stand in CDCl3for prolonged periods of time. Compound
7 was obtained from 10 by letting the latter stand in CDCl3 overnight. Compound 7 was subsequently converted to 8 after 7 days. In the same conditions, 11 was transformed into 9 in CDCl3 after a week through epoxylation (Scheme 1). However, no reactions occurred when 10 and 11 were treated in pyridine-d5for 2 months, implying 10 and 11 were stable under slightly basic solvent. In this article, we report the structure elucidation and the cytotoxicity of these metabolites.
Results and Discussion
Compound 1 was isolated as a colorless, viscous oil. HRESIMS of 1 exhibited a [M + Na]+peak at m/z 275.1625 and established a molecular formula of C15H24O3, implying four degrees of unsaturation. The1H NMR spectrum of 1 (Table 1) showed signals corresponding to an oxygenated methine proton [δH4.77 (1H, ddd, J ) 3.5, 2.0, 1.5 Hz)], an olefinic proton [δH6.21 (1H, d, J ) 1.5 Hz)], a secondary methyl [δH1.00 (3H, d, J ) 7.0 Hz)], and three tertiary methyls [δH 1.44 (3H, s);δH1.47 (3H, s);δH0.89 (3H, s)], respectively. The13C NMR displayed 15 carbon resonances, and the DEPT spectrum (Table 1) was consistent with the presence of a methine [δC71.4 (CH)], a quaternary carbon [δC81.5 (qC)] bearing a peroxide ring, a quaternary carbon [δC70.7 (qC)] bearing a hydroxyl, and trisubstituted olefinic carbons [δC124.2 (CH) and 149.5 (qC)], as well as four methyls, four methylenes, three methines, and four quaternary carbons. The above data of 1 were similar to those of 5R,8R-epidioxy-6-eudesmene,14except for the
presence of a tertiary hydroxyl at C-11. This was supported by the HMBC spectrum, which shows correlations from H3-12 and H3-13 to C-11 (Figure 1). On the basis of the above evidence, the planar structure of 1 was unambiguously established. The computer-modeled structure of 1 was generated by CS Chem 3D version 9.0 using MM2 force field calculations for energy minimization (Figure 2). The results were consistent with the stereochemistry of 1 as established by the NOESY experiments. The NOESY correlations
between H3-14 and all protons of H-1R, H-4, H-8, and H-9R positioned all these protons on the same side of the molecule and revealed the β-orientation of H3-15. Therefore, the structure of 5β,8β-epidioxy-11-hydroxy-6-eudesmene was characterized as 1. 5β,8β-Epidioxy-11-hydroperoxy-6-eudesmene (2) was isolated as a colorless, viscous oil, and its molecular formula was determined to be C15H24O4, as deduced from HRESIMS spectroscopic data. The1H NMR spectrum of 2 showed a signal atδ
H7.72 (1H, br s) that suggested the presence of a hydroperoxy group, while the hydroperoxyl could be located at C-11, as a result of the HMBC correlations (Figure 1). The carbon signals of Me-12 and Me-13 of 1 were at a lower field when compared to 2, and the signal of C-11 was shifted downfield (∆δC 11.1 ppm). The 13C NMR
* To whom correspondence should be addressed. Tel: 886-7-5252000, ext. 5036. Fax: 886-7-5255020. E-mail: [email protected].
†National Sun Yat-sen University. ‡National Taiwan University.
§Center of Asia-Pacific Marine Researches.
10.1021/np070189t CCC: $37.00 2007 American Chemical Society and American Society of Pharmacognosy Published on Web 09/11/2007
spectroscopic data of 1 and 2 were in good accordance with those of compounds with a similar side chain with a hydroxyl or a hydroperoxyl.16,17Consequently, the structure of 2 was deduced
unambiguously.
Compound 3 was obtained as a colorless, viscous oil, which analyzed for the molecular formula C15H24O2by HRESIMS coupled
with the DEPT and13C NMR spectroscopic data (Table 1). A broad IR spectrum absorption at 3321 cm-1indicated the presence of a hydroxy group. The 1H and 13C NMR spectra of 3 contained resonances for a trisubstituted double bond at C-1 and C-2 [δH5.27 (ddd, J ) 3.5, 2.0, 1.5 Hz, 1H);δC145.3 (qC) and 116.3 (CH)]
Scheme 1. Suggested Pathway for Conversion of 7 to 8 and 11 to 9
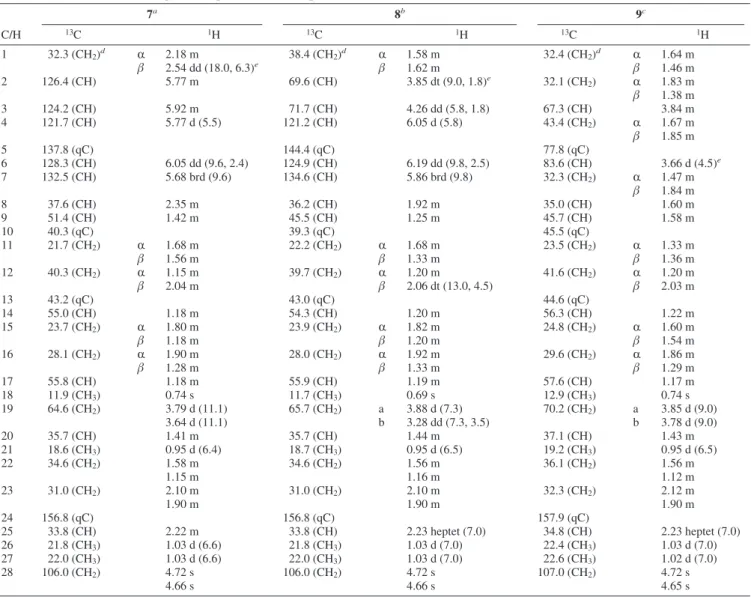
Table 1. 1H and13C NMR Spectroscopic Data of Compounds 1–3
1a 2a 3b C/H 13C 1H 13C 1H 13C 1H 1 35.6 (CH2)c R 1.50 m 35.6 (CH2)c R 1.50 m 145.3 (qC)c β 2.00 m β 2.00 m 2 21.0 (CH2) 1.55 m 21.0 (CH2) 1.55 m 116.3 (CH) 5.27 t (2.5)d 3 29.3 (CH2) 1.48 m 29.7 (CH2) 1.48 m 54.0 (CH) 3.46 dd (3.7, 2.5) 4 32.7 (CH) 2.07 m 32.7 (CH) 2.08 m 62.1 (CH) 2.77 d (3.7) 5 81.5 (qC) 81.6 (qC) 34.6 (qC) 6 124.2 (CH) 6.21 d (1.5)d 128.9 (CH) 6.30 d (1.5)d 37.6 (CH 2) R 1.62 m β 1.32 m 7 149.5 (qC) 145.7 (qC) 46.8 (CH) 2.54 m 8 71.4 (CH) 4.77 ddd (3.5, 2.0, 1.5) 71.0 (CH) 4.79 ddd (3.5, 2.0, 1.5) 17.6 (CH2) R 1.91 m β 1.52 m 9 41.6 (CH2) R 1.28 dd (13.5, 2.0) 41.0 (CH2) R 1.37 dd (13.5, 2.0) 34.5 (CH2) R 1.62 m β 1.95 dd (13.5, 3.5) β 1.93 dd (13.5, 3.5) β 1.50 m 10 35.0 (qC) 34.9 (qC) 40.3 (CH) 2.54 m 11 70.7 (qC) 81.8 (qC) 72.8 (qC) 12 28.0 (CH3) 1.44 s 22.7 (CH3) 1.36 s 26.9 (CH3) 1.24 s 13 28.1 (CH3) 1.47 s 21.0 (CH3) 1.49 s 28.6 (CH3) 1.31 s 14 25.5 (CH3) 0.89 s 25.4 (CH3) 0.90 s 21.0 (CH3) 1.16 d (7.6) 15 16.1 (CH3) 1.00 d (7.0) 16.1 (CH3) 1.00 d (7.0) 23.5 (CH3) 1.31 s OOH 7.72 br s
aSpectra were measured in CDCl
3 (1H, 300 MHz and 13C, 75 MHz). bSpectra were measured in CDCl3 (1H, 500 MHz and 13C, 125 MHz).
cMultiplicities are deduced by HSQC and DEPT experiments.dJ values (in Hz) are in parentheses.
Figure 1.1H–1H COSY and key HMBC correlation of 1–5.
and a disubstituted epoxide [δH3.46 (dd, J ) 3.7, 2.5 Hz, 1H) and 2.77 (d, J ) 3.7 Hz, 1H);δC50.4 (CH) and 62.1 (CH)] at C-3 and C-4. From the above evidences, 3 was suggested to be a tricyclic sesquiterpenoid. From the COSY spectrum of 3 (Figure 1), it was possible to establish that the proton sequence connects from H-2 to H-4 and from H2-6 to H3-14. The1H–1H COSY correlations further observed between H-2 and H-10 showed further the allylic coupling of the above protons. The connectivities between C-1/C-5, C-4/C-C-1/C-5, and C-5/C-6 were confirmed by the HMBC correlations of H3-15 with C-1, C-4, C-5, and C-6. In addition, the HMBC correlations (Figure 1) from H3-12 and H3-13 to C-7 and C-11 proved the attachment of the isopropyl at C-7. On the basis of this evidence, the planar structure of 3, possessing a pseudoguaiane15
skeleton, was unambiguously established. The relative configuration of 3 was determined through inspection of the NOESY spectrum as well as a computer-generated lower energy conformation using MM2 force field calculations (Figure 2). From the NOESY spectrum of 3, H-9β was found to show NOE correlations with both H3-12 and 13, and H-3 exhibited NOE correlations with H-4 and H3-12, indicating theβ-orientations of H-3 and H-4 and the β-orienta-tion of the isopropanol group attached at C-11. Furthermore, NOE correlations could be observed between H-9R and H3-14, H3-14, and H3-15. Therefore, H3-14 and H3-15 should be placed on the R-face. From the aforementioned observations, 3 was formulated
as (3R*,4S*,5R*,7R*,10R*)-3,4-epoxy-11-hydroxy-1-pseudoguaiene.
The molecular formula of 4 was assigned as C15H24O, as derived from its HRESIMS and in agreement with the NMR data. By comparison of the13C NMR spectroscopic data of 4 with those of the known sesquiterpene prespatane,10,11it was found that C-8 (δ
C 50.3 d) in prespatane was converted to a tertiary hydroxyl (δC 87.8 s) in 4, as also confirmed by the HMBC correlations (H3-13/ C-8, C-11, and C-12). Thus, the structure of 4 was established and named 8β-hydroxyprespatane. From the NOESY spectrum of 4, cross-peaks for the signals with H-2, H-6, and H-10β (δ 1.60) fixed the three rings in a stair or chair conformation, while the NOE interactions between 15 and all protons of H-7, 13, and H3-14, which in turn showed correlation with H-10R, positioned the above protons on the same side of the molecule and revealed the β-orientation of the 8-OH.
HRESIMS of 8β-hydroperoxyprespatane (5), a colorless, viscous oil, established a molecular formula of C15H24O2. By comparison of the NMR spectroscopic data of 5 with those of 4, it was found that the1H and13C spectroscopic data of both compounds were nearly the same, except that the carbon shift of the tertiary hydroperoxyl at C-8 (δC100.2, s) of 5 was shifted downfield relative to the signal of C-8 (δC87.8, s) of 4.16,17Thus, the structure of 5 was established unambiguously.
Compound 7 was obtained from 10 by letting the latter stand in CDCl3overnight. The 1H and 13C NMR spectroscopic data of 7 revealed the presence of a conjugated triene (δC126.4 CH and 124.2 CH;δH5.77 m and 5.92 m;δC121.7 CH and 137.8 qC;δH5.77 d, J ) 5.5 Hz;δC128.3 CH and 132.5 CH;δH6.05 dd, J ) 9.6, 2.4 Hz and 5.68 d, J ) 9.6 Hz) in rings A and B. This was confirmed by 2D NMR spectroscopic analyses. Interpretation of the1H–1H COSY spectrum led to partial structures I, II, and III (Figure 3). Partial structures I–III were connected by HMBC correlations.
24-Methylenecholesta-4,6-dien-3β,19-epoxy-2β-ol (8) was ob-tained from 7 by letting the latter stand in CDCl3for 1 week. The 13C NMR and DEPT spectroscopic data of 8 showed signals for six olefin carbons, four methyl carbons, eight methylene carbons, eight methine carbons, and two quaternary carbons. The above functionalities account for three of the eight degrees of unsaturation, suggesting that 8 is a tetracyclic compound with a 3β,19-epoxy moiety. The1H–1H COSY spectrum correlations of 8 were similar
Figure 3.1H–1H COSY and key HMBC correlations of 7–9.
Table 2. 1H and13C NMR Spectroscopic Data of Compounds 4 and 5
4a 5a C/H 13C 1H 13C 1H 1 42.3 (qC)b 42.1 (qC)b 2 44.6 (CH) 2.13 t (7.0)c 45.1 (CH) 2.18 m 3 27.9 (CH2) R 1.70 m 27.9 (CH2) R 1.70 m β 1.43 m β 1.45 m 4 34.6 (CH2) R 1.71 m 34.6 (CH2) R 1.72 m β 1.30 m β 1.29 m 5 37.1 (CH) 1.82 m 37.0 (CH) 1.82 m 6 41.0 (CH) 1.84 m 41.4 (CH) 1.95 m 7 50.4 (CH) 1.80 m 47.1 (CH) 1.72 m 8 87.8 (qC) 100.2 (qC) 9 35.4 (CH2) R 1.65 m 29.2 (CH2) 2.26 m β 2.40 td (12.5, 7.4) 10 39.9 (CH2) R 1.85 td (12.5, 7.4) 39.3 (CH2) R 1.75 m β 1.60 td (12.5, 7.4) β 1.58 m 11 146.4 (qC) 143.3 (qC) 12 111.3 (CH2) 4.87 br s 114.7 (CH2) 5.04 br s 4.94 br s 5.09 br s 13 19.3 (CH3) 1.77 br s 19.5 (CH3) 1.75 br s 14 21.0 (CH3) 1.04 s 20.4 (CH3) 0.97 s 15 14.5 (CH3) 0.92 d (6.0) 14.5 (CH3) 0.90 d (6.3)c OOH 7.71 br s
aSpectra were measured in CDCl
3(1H, 300 MHz and 13C, 75 MHz).bMultiplicities are deduced by HSQC and DEPT experiments.cJ values (in
to those of 7. These data, together with the HMBC correlations (Figure 3), established the structure of 8.
HRESIMS of 24-methylenecholesta-6β,19-epoxy-3β,5R-diol (9) was obtained from 11 by letting the latter stand in CDCl3for 1 week. By comparison of the NMR spectroscopic data (Table 3) of
9 with those of 11,13it was found that hydroxy groups attached to
C-6 and C-19 in 11 were converted to a 6β,19-oxide ring in 9. The position of the oxide group at C-6/C-19 was confirmed by the HMBC correlation (Figure 3) from H-6 to C-19.
Preliminary cytotoxicity screening revealed that 2, 5, and 6 exhibited significant cytotoxicty against P-388 (mouse lymphocytic leukemia) and HT-29 (human colon adenocarcinoma) cells (Table 4). The other tested compounds were not cytotoxic to P-388 and HT-29 cells. The results of further biological activity screening will be reported elsewhere in the future.
Experimental Section
General Experimental Procedures. Optical rotations were
deter-mined on a JASCO P1020 polarimeter. UV spectra were obtained on a Hitachi U-3210 spectrophotometer, and IR spectra were recorded on a JASCO FT/IR-4100 spectrophotometer. The NMR spectra were recorded on a Bruker Avance 300 NMR spectrometer at 300 MHz for
1H and 75 MHz for13C or on a Varian Unity INOVA 500 FT-NMR at
500 MHz for1H and 125 MHz for13C, respectively, using TMS as
internal standard. Chemical shifts are given inδ (ppm) and coupling
constants in Hz. ESIMS were recorded by ESI FT-MS on a Bruker
APEX II mass spectrometer. Si gel 60 (Merck, 230–400 mesh) was used for column chromatography; precoated Si gel plates (Merck, Kieselgel 60 F254, 0.25 mm) were used for TLC analysis.
Animal Material. The soft coral N. erecta was collected by hand
using scuba at Green Island located on the southeast coast of Taiwan, in July 2005, at a depth of 10 m, and was stored in a freezer for 5 weeks until extraction. A voucher specimen (GN-80) was deposited in the Department of Marine Biotechnology and Resources, National Sun Yat-sen University.
Extraction and Isolation. A specimen of N. erecta was extracted
sequentially with acetone and MeOH. After removal of solvent in Vacuo, the acetone-soluble residue was partitioned between H2O and EtOAc.
Table 3. 1H and13C NMR Spectroscopic Data of Compounds 7–9
7a 8b 9c C/H 13C 1H 13C 1H 13C 1H 1 32.3 (CH2)d R 2.18 m 38.4 (CH2)d R 1.58 m 32.4 (CH2)d R 1.64 m β 2.54 dd (18.0, 6.3)e β 1.62 m β 1.46 m 2 126.4 (CH) 5.77 m 69.6 (CH) 3.85 dt (9.0, 1.8)e 32.1 (CH 2) R 1.83 m β 1.38 m 3 124.2 (CH) 5.92 m 71.7 (CH) 4.26 dd (5.8, 1.8) 67.3 (CH) 3.84 m 4 121.7 (CH) 5.77 d (5.5) 121.2 (CH) 6.05 d (5.8) 43.4 (CH2) R 1.67 m β 1.85 m 5 137.8 (qC) 144.4 (qC) 77.8 (qC) 6 128.3 (CH) 6.05 dd (9.6, 2.4) 124.9 (CH) 6.19 dd (9.8, 2.5) 83.6 (CH) 3.66 d (4.5)e 7 132.5 (CH) 5.68 brd (9.6) 134.6 (CH) 5.86 brd (9.8) 32.3 (CH2) R 1.47 m β 1.84 m 8 37.6 (CH) 2.35 m 36.2 (CH) 1.92 m 35.0 (CH) 1.60 m 9 51.4 (CH) 1.42 m 45.5 (CH) 1.25 m 45.7 (CH) 1.58 m 10 40.3 (qC) 39.3 (qC) 45.5 (qC) 11 21.7 (CH2) R 1.68 m 22.2 (CH2) R 1.68 m 23.5 (CH2) R 1.33 m β 1.56 m β 1.33 m β 1.36 m 12 40.3 (CH2) R 1.15 m 39.7 (CH2) R 1.20 m 41.6 (CH2) R 1.20 m β 2.04 m β 2.06 dt (13.0, 4.5) β 2.03 m 13 43.2 (qC) 43.0 (qC) 44.6 (qC) 14 55.0 (CH) 1.18 m 54.3 (CH) 1.20 m 56.3 (CH) 1.22 m 15 23.7 (CH2) R 1.80 m 23.9 (CH2) R 1.82 m 24.8 (CH2) R 1.60 m β 1.18 m β 1.20 m β 1.54 m 16 28.1 (CH2) R 1.90 m 28.0 (CH2) R 1.92 m 29.6 (CH2) R 1.86 m β 1.28 m β 1.33 m β 1.29 m 17 55.8 (CH) 1.18 m 55.9 (CH) 1.19 m 57.6 (CH) 1.17 m 18 11.9 (CH3) 0.74 s 11.7 (CH3) 0.69 s 12.9 (CH3) 0.74 s 19 64.6 (CH2) 3.79 d (11.1) 65.7 (CH2) a 3.88 d (7.3) 70.2 (CH2) a 3.85 d (9.0) 3.64 d (11.1) b 3.28 dd (7.3, 3.5) b 3.78 d (9.0) 20 35.7 (CH) 1.41 m 35.7 (CH) 1.44 m 37.1 (CH) 1.43 m 21 18.6 (CH3) 0.95 d (6.4) 18.7 (CH3) 0.95 d (6.5) 19.2 (CH3) 0.95 d (6.5) 22 34.6 (CH2) 1.58 m 34.6 (CH2) 1.56 m 36.1 (CH2) 1.56 m 1.15 m 1.16 m 1.12 m 23 31.0 (CH2) 2.10 m 31.0 (CH2) 2.10 m 32.3 (CH2) 2.12 m 1.90 m 1.90 m 1.90 m 24 156.8 (qC) 156.8 (qC) 157.9 (qC) 25 33.8 (CH) 2.22 m 33.8 (CH) 2.23 heptet (7.0) 34.8 (CH) 2.23 heptet (7.0) 26 21.8 (CH3) 1.03 d (6.6) 21.8 (CH3) 1.03 d (7.0) 22.4 (CH3) 1.03 d (7.0) 27 22.0 (CH3) 1.03 d (6.6) 22.0 (CH3) 1.03 d (7.0) 22.6 (CH3) 1.02 d (7.0) 28 106.0 (CH2) 4.72 s 106.0 (CH2) 4.72 s 107.0 (CH2) 4.72 s 4.66 s 4.66 s 4.65 s
aSpectra were measured in CDCl
3(1H, 300 MHz and13C, 75 MHz).bSpectra were measured in CDCl3(1H, 500 MHz and13C, 125 MHz).cSpectra
were measured in CD3OD (1H, 500 MHz and13C, 125 MHz).dMultiplicities are deduced by HSQC and DEPT experiments.eJ values (in Hz) are in
parentheses. Table 4. Cytotoxicitya of Compounds 1–9 cell lines ED50(µg/mL) compound HT-29 P-388 1 >10 >10 2 0.4 0.2 3 >10 >10 4 >10 >10 5 0.5 0.3 6 3.2 2.8 7 >10 >10 8 >10 >10 9 >10 >10
aFor significant activity of pure compounds, an ED
50of e4.0µg/mL
The dried EtOAc extract (35.0 g) was chromatographed over a Si column using n-hexane, n-hexane/EtOAc, and EtOAc/MeOH mixtures of increasing polarity. Elution with n-hexane gave fractions containing compound 6, that with n-hexane/EtOAc (90:10) gave fractions contain-ing compounds 4 and 5, that with n-hexane/EtOAc (85:15) gave fractions containing compound 3, that with EtOAc/MeOH (20:1) gave fractions containing compound 10, and that with EtOAc/MeOH (10:1) gave fractions containing compound 11. Compound 6 (3 mg) was further purified by HPLC (Si) by eluting with n-hexane. Compounds 4 (2 mg) and 5 (5 mg) were further purified by HPLC (Si) by eluting with n-hexane/EtOAc (90:10) and n-hexane/CH2Cl2(50:50),
respec-tively. Compound 3 (2 mg) was purified by repeated HPLC (Si) by eluting with n-hexane/EtOAc (85:15). Compound 10 (7.0 mg) was further purified by RP-HPLC by eluting with MeOH/H2O (85:15).
Compound 11 (5.0 mg) was further purified by RP-HPLC by eluting with MeOH/H2O (85:15).
Compound 10 was fully transformed into compound 7 during NMR experiments in CDCl3. Compound 7 was then converted to a
mixture containing compounds 7 and 8. Compound 8 (2 mg) was further purified by RP-18 HPLC by eluting with MeOH/H2O
(90:10). Under the same conditions, compound 11 was transformed into a mixture containing compounds 11 and 9 in CDCl3in 7 days.
Compound 9 (2 mg) was further purified by RP-18 HPLC by eluting with MeOH/H2O (85:15).
The MeOH-soluble residue (320 mg) was partitioned between H2O
and EtOAc. The dried EtOAc layer was then subjected to column chromatography on silica gel using CH2Cl2and CH2Cl2/MeOH mixtures
of increasing polarity. Elution with CH2Cl2/MeOH (80:1) gave fractions
containing compounds 1 and 2. Compounds 1 (7 mg) and 2 (5 mg) were purified by RP-HPLC by eluting with MeOH/H2O (60:40).
5β,8β-Epidioxy-11-hydroxy-6-eudesmene (1): colorless, viscous oil; [R]23
D+10 (c 0.7, CH2Cl2); IR (KBr)νmax3322, 2947, 1640, 1457,
1384, 1239, 1040, 927, 739 cm-1;1H NMR and13C NMR data, see
Table 1; ESIMS m/z 275 [M + Na]+; HRESIMS m/z 275.1625 [M + Na]+(calcd for C15H24O3Na, 275.1623).
5β,8β-Epidioxy-11-hydroperoxy-6-eudesmene (2): colorless, vis-cous oil; [R]23
D+4 (c 0.5, CH2Cl2); IR (KBr)νmax3318, 2952, 1640,
1536, 1458, 1385, 1239, 1036, 927, 734 cm-1;1H NMR and13C NMR
data, see Table 1; ESIMS m/z 291 [M + Na]+; HRESIMS m/z 291.1573 [M + Na]+(calcd for C15H24O4Na, 291.1572).
(3R*,4S*,5R*,7R*,10R*)-3,4-Epoxy-11-hydroxy-1-pseudo-guaiene (3): colorless, viscous oil; [R]23
D+13 (c 0.3, CH2Cl2); IR
(KBr)νmax3321, 2948, 1645, 1536, 1463, 1384, 1239, 1041, 931, 734
cm-1;1H NMR and13C NMR data, see Table 1; ESIMS m/z 259 [M
+ Na]+
; HRESIMS m/z 259.1676 [M + Na]+(calcd for C15H24O2Na,
259.1674).
8β-Hydroxyprespatane (4): colorless, viscous oil; [R]23 D+9 (c
0.4, CHCl3); IR (KBr)νmax3307, 2947, 1640, 1457, 1384, 1239, 1035,
931, 738 cm-1;1H NMR and13C NMR data, see Table 2; ESIMS m/z
243 [M + Na]+; HRESIMS m/z 243.1726 [M + Na]+ (calcd for C15H24ONa, 243.1725).
8β-Hydroperoxyprespatane (5): colorless, viscous oil; [R]23 D+8
(c 0.2, CHCl3); IR (KBr)νmax3384, 2951, 1640, 1459, 1442, 1374,
1252, 1129, 1048, 945, 892 cm-1;1H NMR and13C NMR data, see
Table 2; ESIMS m/z 433 [M + Na]+; HRESIMS m/z 259.1675 [M + Na]+(calcd for C15H24O2Na, 259.1674).
24-Methylenecholesta-2,4,6-trien-19-ol (7): colorless, viscous oil;
[R]23
D+43 (c 0.7, CHCl3); UV (CHCl3)λmax(logε) 247 (3.97), 279
(3.18); IR (KBr)νmax3385, 2958, 1645, 1463, 1374, 1051, 884, 734
cm;1H NMR and13C NMR data, see Table 3; EIMS m/z 394 [M]+
; HREIMS m/z 394.3230 [M]+(calcd for C28H42O, 394.3235).
24-Methylenecholesta-4,6-dien-3β,19-epoxy-2β-ol (8): colorless, viscous oil; [R]23
D+166 (c 0.2, CHCl3); UV (MeOH)λmax(logε) 240
(3.63); IR (KBr)νmax3406, 2932, 1640, 1463, 1374, 1265, 1040, 884,
749 cm-1;1H NMR and13C NMR data, see Table 3; ESIMS m/z 433
[M + Na]+; HRESIMS m/z 433.3084 [M + Na]+ (calcd for C28H42O2Na, 433.3082).
24-Methylenecholesta-6β,19-epoxy-3β,5r-diol (9): colorless, vis-cous oil; [R]23
D+176 (c 0.2, CHCl3); IR (KBr)νmax3396, 2963, 1462,
1380, 1259, 1098, 1030, 858, 795 cm-1;1H NMR and13C NMR data,
see Table 3; ESIMS m/z 453 [M + Na]+; HRESIMS m/z 453.3343 [M + Na]+
(calcd. for C28H46O3Na, 453.3344).
Cytotoxicity Testing. Cytotoxicity was determined against P-388
(mouse lymphocytic leukemia) and HT-29 (human colon adenocarci-noma) tumor cells using the MTT assay method. The experimental details of this assay were carried out according to a previously described procedure.18
Acknowledgment. We thank J. M. Pezzuto, formerly of the
Department of Medicinal Chemistry and Pharmacognosy, University of Illinois at Chicago, for the provision of the P-388 cell line. The HT-29 cell line was purchased from the American Type Culture Collection. This work was supported by grants from the National Science Council and Ministry of Education (95C030311) of the Republic of China awarded to C.-Y.D.
References and Notes
(1) Blunt, J. W.; Copp, B. R.; Munro, M. H. G.; Northcote, P. T.; Prinsep, M. R. Nat. Prod. Rep. 2006, 23, 26–78, and literature cited in previous reviews.
(2) Coll, J. C.; Bowden, B. F.; Tapiolas, D. M.; Willis, R. H. Tetrahedron
1985, 41, 1085–1092.
(3) Kitagawa, I.; Cui, Z.; Son, B. W.; Kobayashi, M.; Kyogoku, Y. Chem.
Pharm. Bull. 1987, 35, 124–135.
(4) Handayani, D.; Edrada, R. A.; Proksch, P.; Wray, V.; Witte, L. J.
Nat. Prod. 1997, 60, 716–718.
(5) Duh, C.-Y.; Wang, S.-K.; Weng, Y.-L. Tetrahedron Lett. 2000, 41, 1401–1404.
(6) Duh, C.-Y.; Wang, S.-K.; Weng, Y.-L.; Chiang, M.-Y.; Dai, C.-F. J.
Nat. Prod. 1999, 62, 1518–1521.
(7) Rao, M. R.; Venkatesham, U.; Venkateswarlu, Y J. Nat. Prod. 1999,
62, 1584–1585.
(8) El-Gamal, A. A. H.; Wang, S.-K.; Dai, C.-F.; Duh, C.-Y. J. Nat. Prod.
2004, 67, 1455–1458.
(9) Duh, C.-Y.; Wang, S.-K.; Chu, M.-J.; Sheu, J.-H. J. Nat. Prod. 1998,
61, 1022–1024.
(10) König, G. M.; Wright, A. D. J. Org. Chem. 1997, 62, 3837–3840. (11) Warners, U.; Wihstutz, K.; Bülow, N.; Fricke, C.; König, W. A.
Phytochemistry 1998, 49, 1723–1731.
(12) Bortolotto, M.; Braeckman, J. C.; Daloze, D.; Losman, D.; Tursch, B. Steroids 1976, 28, 461–466.
(13) Wang, G.; Li, F.; Zeng, L.; Ma, L.; Tu, G. Chem. J. Chin. UniV. 1992,
13, 623–627.
(14) Nagashima, F.; Murakami, M.; Takaoka, S.; Asakawa, Y. Chem.
Pharm. Bull. 2004, 52, 949–952.
(15) Fischer, N. H.; Oliver, E. J.; Fischer, H. D. Prog. Chem. Org. Nat.
Prod. 1979, 38, 47–390.
(16) Barrero, A. F.; Quilez del Moral, J. F.; Herrador, M. M.; Akssira, M.; Bennamara, A.; Akkad, S.; Aitigri, M. Phytochemistry 2004, 65, 2507– 2516.
(17) Masuyama, A.; Sugawara, T.; Nojima, M.; McCullough, K. J.
Tetrahedron 2003, 59, 353–366.
(18) Hou, R.-S.; Duh, C.-Y.; Chiang, M. Y.; Lin, C.-N. J. Nat. Prod. 1995,
58, 1126–1130.