Synthesis and characterization of alternating fluorene-based copolymers
containing diaryl- and non-substituted bithiophene units
Hong-Cheu Lin*, Hsiao-Hsien Sung, Chien-Min Tsai, Kuang-Chieh Li
Department of Materials Science and Engineering, National Chiao Tung University, Hsinchu, Taiwan, ROCReceived 10 June 2005; received in revised form 22 July 2005; accepted 25 July 2005 Available online 11 August 2005
Abstract
A series of soluble alternating fluorene-based copolymers containing diaryl- and non-substituted bithiophene units are synthesized by palladium-catalyzed Suzuki coupling reaction. All polymers demonstrate green colors of photoluminescence (PL) in chloroform, good thermal stability (with decomposition temperatures above 436 8C), and high glass transition temperatures (in the range of 120–144 8C). Owing to the large steric hindrance of diaryl substituents on bithiophenes in the polymers (P2–P4), the aggregation of solids is reduced as well as the solubility is improved, so the performance of their PLED devices are superior to that of the non-substituted polymer (P1). Compared with P1, the introduction of substitutents at 3,30-position of bithiophene in P2–P4 has significant effects on the photophysical properties of resulting polymers in solution and solid states. Though the PL quantum yield of P1 is much higher than those of diaryl-substituted polymers (P2–P4), the PLED device of P1 has the worst electroluminescence (EL) properties due to the poor solubility of P1. Consequently, among these polymers, the device made of P3 as an emitter has the highest luminance of 2590 cd/m2at 9.5 V. For optimum device performance, a device of P3 blended with PVK can be further enhanced to a brighter luminance of 4284 cd/m2at 18 V.
q2005 Elsevier Ltd. All rights reserved.
Keywords: Alternating fluorene-based copolymers; Bithiophene; Diaryl substituents
1. Introduction
Poly(fluorene) (PF) is a well-known hole transporting blue-emitting material for polymeric light-emitting diodes (PLEDs) application[1,2]. This material displays extremely high photoluminescence (PL) efficiencies in both solution and solid states [3,4]. However, PF has some drawbacks, such as aggregation and excimer formation in the solid state
[5], high energy barrier for hole injection [6–8], and unbalance charge mobilities between holes and electrons
[9]. Fortunately, these unfavorable issues can be overcome by proper molecular design, i.e. fluorene monomers copolymerize with other monomers to form copolymers or introduction of electron-deficient oxadiazoles (OXD) groups into C-9 position of the fluorene units to balance
charge injection and transportation as well as recombination of resulting polymers[10–12].
Polythiophenes and their derivatives have a number of interesting properties [13–15], such as relatively high conductivity and stability under ambient atmosphere along with easy preparation and tailoring of band gaps by adjusting chemical structures of the monomers [14,15]. Nevertheless, the quantum yields of these polymers are far from unity[16]. Therefore, in order to increase the quantum yields of polythiophenes, thiophene monomers can be copolymerized with monomers possessing high quantum yields, such as fluorene monomers. Besides, the p-excessive
thiophene rings [17] can help lowering the oxidation
potentials of the fluorene moieties, and the onset potential values of PFO are reported around 1.4 V[18].
In order to obtain highly efficient PLED devices, it is important to suppress the intermolecular interactions (aggregation between molecules), which results in the formation of excimers in the solid state and consequently lower the quantum yields of the polymers. Hence, the introduction of large substituted groups into conjugated polymers has been utilized to a great extent in controlling their physical and electronic properties [19]. In addition,
www.elsevier.com/locate/polymer
0032-3861/$ - see front matter q 2005 Elsevier Ltd. All rights reserved. doi:10.1016/j.polymer.2005.07.075
* Corresponding author. Tel.: C886 3 5712121x55305; fax: C886 3 5724727.
large steric hindrance of lateral substitution will reduce crystallinity in the resulting polymers and provide the polymer films with amorphous properties [19–22]. From previous studies, it is known that anti-type bithiophene units have a dihedral angle of qZ147.98 between these two thiophene rings [23], and 3,30-diphenyl-2,20-bithiophene lies in an anti conformation with a dihedral angle of qZ1538
[16]. Such a conformation of these two bulky substituents at
3,30-position of bithiophene can provide large steric hindrance to restrain interchain interaction in the solid state. To control the emitting colors of light-emitting polymers, the most effective means is to introduce monomers with different functional groups and flexible parts as side chains to control the conjugation lengths. In addition, the regioregularity is also an important factor to control the conjugation length along the polymer backbone. To our
knowledge, very few literatures regarding copolymers containing fluorene and bithiophene units were investigated, and only related alternating copolymers of fluorenone and dialkylbithiophene have been reported[24,25]. In this work, the synthesis and characterization of four alternating fluorene-based conjugated polymers containing regioregu-lar head-to-head coupling bithiophene with different substituents at 3,30-position of bithiophene in the backbone are reported. The effects of substituents on the electro-chemical, photophysical, and electroluminescent (EL) properties of resulting polymers are also investigated.
2. Experimental 2.1. Measurements
1H NMR spectra were recorded on a Varian unity
300 MHz spectrometer using CDCl3 solvent. Elemental
analyses were performed on a HERAEUS CHN-OS RAPID elemental analyzer. Transition temperatures were deter-mined by differential scanning calorimetry (Perkin–Elmer Diamond) with heating and cooling rates of 10 8C/min. The mesophases were studied by a polarizing optical microscope (Leica DMLP) equipped with a hot stage. Thermogravi-metric analysis (TGA) was conducted on a Du Pont Thermal Analyst 2100 system with a TGA 2950 thermogravimetric analyzer under a heating rate of 20 8C/min. Gel permeation chromatography (GPC) analysis was conducted on a Water 1515 separation module using polystyrene as a standard and tetrahydrofuran (THF) as an eluant. UV-visible absorption spectra were recorded in dilute chloroform solutions (10K5M) on a HP G1103A spectrophotometer, and fluorescence (PL and EL) spectra were obtained on a Hitachi F-4500 spectrophotometer. Polymer thin films were spin-coated on a quartz substrate from chloroform solutions with a concentration of 10 mg/ml (this condition consists with that of EL devices). Fluorescence quantum yields were determined by comparing the integrated PL density of a reference quinine sulfate in 0.1 M H2SO4 with a known quantum yield (ca. 1!10K5M, quantum yieldZ0.54)[26]. Cyclic voltammetry (CV) was performed at a scanning rate of 100 mV/s on a BAS 100 B/W electrochemical analyzer, which was equipped with a three-electrode cell. Pt wire was used as a counter electrode, and an Ag/AgCl was used as a reference electrode in the CV measurement. The polymer thin films were cast onto a Pt disc as a working electrode with ferrocene as a standard in acetonitrile, and 0.1 M
tetrabutylammonium hexafluorophosphate (TBAPF6) was
used as a supporting electrolyte. A series of double-layer EL devices with the configuration of ITO/polymer/TPBI (30 nm)/MgAg (50 nm)/Ag (100 nm), in which 2,20,20 -(1,3,5-phenylene)-tris[1-phenyl-1H-benzimidazole] (TPBI) as an electron transporting layer, were made by spin-coating the polymer solutions (with a concentration of 10 mg/ml in dichloroethane and the spin rate of 3000 rpm for 40 s) onto
ITO glass substrates. The thickness of these films was measured on a Sloan DektakIIA surface profilometer, and the thicknesses of the polymer emitting layers were about 60 nm. The luminance-current-voltage characteristics were recorded on a power source (Keithley 2400) and photometer (MINOLTA CS-100A).
2.2. Materials
Chemicals and solvents were reagent grades and purchased from Aldrich, ARCROS, TCI, and Lancaster Chemical Co. Dichloromathane and THF were distilled to keep anhydrous before use. The other chemicals were used without further purification. The synthetic routes of
monomers (M1–M4) are shown inScheme 1.
2.2.1. 3,30,5,50-Tetrabromo-2,20-bithiophene (1)
Bromine (14.58 g, 91.2 mmol) was added dropwise to a solution of 2,20-bithiophene (7.12 g, 42.8 mmol) in a mixed solvent of glacial acetic acid (30 ml) and chloroform (77 ml) at a temperature of 5 8C. The mixture was stirred at room temperature for 5 h and then refluxed for 24 h. After the reaction was completed, the mixture was cooled to room temperature and 10% KOH aqueous was added. The mixture was extracted with chloroform and water, and the organic solvent was collected and removed by rotavapor. The residue was recrystallized from ethanol afforded yellow crystals in a yield of 85%.1H NMR (CDCl3, 300 MHz, d): 7.06 (s, 2H).
2.2.2. 3,30-Dibromo-2,20-bithiophene (2)
3,30,5,50-Tetrabromo-2,20-bithiophene (9.81 g, 20.3 mmol) was added in portion to a zinc powder (5.33 g, 81.2 mmol) dispersion in 100 ml of ethanol containing 10 ml of water, 24 ml of acetic acid, and 2 ml of 2 M HCl aqueous. After refluxed for 2 h and then cooled to room temperature, the mixture was filtered and washed with ethanol, and filtrate was collected. After removed the solvent, the mixture was extracted with ether and water. The solvent was filtered and removed by rotavapor. Recrystalli-zation from hexane afforded light yellow crystals in a yield of 71%.1H NMR (CDCl3, 300 MHz, d): 7.07 (d, JZ5.4 Hz, 2H), 7.40 (d, JZ5.4 Hz, 2H).
2.2.3. 3,30-Diphenyl-2,20-bithiophene (3)
A mixture of 3,30-dibromo-2,20-bithiophene (1.04 g, 3.2 mmol), benzeneboronic acid (1.20 g, 9.6 mmol), and tetrakis(triphenylphosphine) palladium (0.19 g, 0.16 mmol) were added in a degassed mixture of 1,2-dimethixyethane (DME) ([monomer] Z0.2 M) and aqueous 2 M sodium carbonate (3:2 in volume). The mixture was vigorously stirred at 85 8C for 48 h. After the mixture was cooled to room temperature, it was extracted with CH2Cl2and water. The solvent was removed by rotavapor, the residue was purified by chromatograph (hexane). Yield: 86%.1H NMR
(CDCl3, 300 MHz, d): 7.00–7.05 (m, 6H), 7.08–7.11 (m, 6H), 7.27 (d, JZ5.4 Hz, 2H).
2.2.4. 3,30-Bis-(4-octyloxy-phenyl)-2,20-bithiophene (4) 3,30-Bis-(4-octyloxy-phenyl)-2,20-bithiophene was syn-thesized according to the procedure described for 3,30
-diphenyl-2,20-bithiophene (3). Yield: 85%. 1H NMR
(CDCl3, 300 MHz, d): 0.88 (t, 6H), 1.28–1.43 (m, 20H), 1.72–1.76 (m, 4H), 3.88 (t, 4H), 6.66 (d, JZ8.4 Hz, 4H), 7.00–7.05 (m, 6H), 7.25 (d, JZ5.4 Hz, 2H).
2.2.5. 3,30-Bis-(40-octyloxy-biphenyl-4-yl)-2,20-bithiophene (5)
3,30-Bis-(40-octyloxy-biphenyl-4-yl)-2,20-bithiophenyl was synthesized according to the procedure described for 3,30-diphenyl-2,20-bithiophene (3). Yield: 78%. 1H
NMR (CDCl3, 300 MHz, d): 0.89 (t, 6H), 1.26–1.53
(m, 20H), 1.78–1.83 (m, 4H), 3.99 (t, 4H), 6.92 (d, JZ 9 Hz, 4H), 7.04 (d, JZ8.4 Hz, 4H), 7.11 (d, JZ5.4 Hz, 2H), 7.27 (d, JZ8.4 Hz, 4H), 7.35 (d, JZ5.4 Hz, 2H), 7.43 (d, JZ9 Hz, 4H).
2.2.6. M2 (6). 5,50-Dibromo-3,30-diphenyl-2,20-bithiophene N-Bromosuccinimide (0.98 g, 5.5 mmol) was added dropwise to a solution of 3,30-diphenyl-2,20-bithiophene (0.79 g, 2.5 mmol) in a mixed solvent of glacial acetic acid (3 ml) and chloroform (6 ml) at room temperature. The mixture was refluxed for 4 h. The mixture was extracted
with CH2Cl2 and water, and the organic solvent was
collected and removed by rotavapor. The residue was purified by chromatograph (CH2Cl2:hexaneZ1:20). Yield: 94%. 1H NMR (CDCl3, 300 MHz, d): 6.96–7.00 (m, 4H), 7.11–7.15 (m, 6H). Anal. Calcd for C20H12Br2S2: C, 50.44; H, 2.54. Found: C, 50.22; H, 2.90.
2.2.7. M3 (7). 5,50-Dibromo-3,30-bis-(4-octyloxy-phenyl)-2, 20-bithiophene
5,50-Dibromo-3,30-bis-(4-octyloxy-phenyl)-2,20 -bithio-phene was synthesized according to the procedure described for 5,50-dibromo-3,30-diphenyl-2,20-bithiophene (6). Yield: 95%. 1H NMR (CDCl3, 300 MHz, d): 0.90 (t, 6H), 1.26– 1.44 (m, 20H), 1.74–1.78 (m, 4H), 3.90 (t, 4H), 6.69 (d, JZ 8.4 Hz, 4H), 6.98–7.01 (m, 6H). Anal. Calcd for C36H44Br2O2S2: C, 59.01; H, 6.05. Found: C, 58.79; H, 6.02.
2.2.8. M4 (8). 5,50-Dibromo-3,30-bis-(40 -octyloxy-biphenyl-4-yl)-2,20-bithiophene
5,50-Dibromo-3,30-bis-(40-octyloxy-biphenyl-4-yl)-2,20 -bithiophene was synthesized according to the procedure described for 5,50-dibromo-3,30-diphenyl-2,20-bithiophene (6). Yield: 95%. 1H NMR (CDCl3, 300 MHz, d): 0.89 (t, 6H), 1.25–1.46 (m, 20H), 1.75–1.82 (m, 4H), 3.98 (t, 4H), 6.92 (d, JZ8.4 Hz, 4H), 7.00–7.04 (m, 6H), 7.28 (d, JZ 8.4 Hz, 4H), 7.42 (d, JZ8.4 Hz, 4H). Anal. Calcd for C48H52Br2O2S2: C, 65.15; H, 5.92. Found: C, 65.15; H, 5.93.
2.2.9. M1 (9). 5,50-Dibromo-2,20-bithiophene
In the absence of light, N-bromosuccinimide (2.15 g, 11.9 mmol) was added dropwise to a solution of bithiophene (0.98 g, 5.8 mmol) in N,N-dimethylformamide (DMF) at room temperature. The mixture was stirred for 3 h at room temperature and poured into ice water, and the precipitate was filtered and washed with water and ethanol. Yield: 92%.
1H NMR (CDCl
3, 300 MHz, d): 6.84 (d, JZ3.9 Hz, 2H), 6.95 (d, JZ3.9 Hz, 2H). Anal. Calcd for C8H4Br2S2: C, 29.65; H, 1.24. Found: C, 29.81; H, 1.46.
2.3. Polymerization
The synthetic routes of polymers (P1–P4) are shown in
Scheme 2. A general procedure of polymerization is
proceeded through the Suzuki coupling reaction. A mixture of
2,7-bis-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-9,9-dihexyllfluorene (1 equiv), dibromo compound
(1 equiv), and tetrakis(triphenylphosphine) palladium (1.0 mol%) were added in a degassed mixture of toluene ([monomer]Z0.2 M) and aqueous 2 M potassium carbonate (3:2 in volume). The mixture was vigorously stirred at 87 8C for 72 h. After the mixture was cooled to room temperature, it was poured into 200 ml of methanol. A fibrous solid was obtained by filtration. The solid was washed sequentially with methanol, water, and methanol.
P1.1H NMR (CDCl3, 300 MHz, d): 0.60–0.84 (m, 10H), 0.90–1.22 (m, 12H), 2.01 (br, 4H), 7.05–7.40 (m, 4H), 7.45– 7.75 (m, 6H). Anal. Calcd for C33H36S2: C, 79.79; H, 7.30. Found: C, 78.90; H, 6.98.
P2. Yield: 62%.1H NMR (CDCl3, 300 MHz, d): 0.71– 0.92 (m, 10H), 1.09–1.20 (m, 12H), 2.04 (br, 4H), 7.05–7.21 (m, 10H), 7.38 (s, 2H), 7.59–7.72 (m, 6H). Anal. Calcd for C45H44S2: C, 83.28; H, 6.83. Found: C, 80.01; H, 6.43.
P3. Yield: 70%.1H NMR (CDCl3, 300 MHz, d): 0.70– 1.46 (m, 48H), 1.74–1.80 (m, 4H), 2.03 (br, 4H), 3.93 (t, 4H), 6.72 (d, JZ8.4 Hz, 4H), 7.12 (d, JZ8.4 Hz, 4H), 7.38 (s, 2H), 7.58–7.71 (m, 6H). Anal. Calcd for C61H76O2S2: C, 80.92; H, 8.46. Found: C, 80.34; H, 8.58.
P4. Yield: 83%.1H NMR (CDCl3, 300 MHz, d): 0.75– 1.49 (m, 48H), 1.79–1.84 (m, 4H), 2.06 (br, 4H), 4.01 (t, 4H), 6.95 (d, JZ8.4 Hz, 4H), 7.15 (d, JZ8.1 Hz, 4H), 7.31 (d, JZ8.1 Hz, 4H), 7.42 (s, 2H), 7.46 (d, JZ8.4 Hz, 4H), 7.58–7.71 (m, 6H). Anal. Calcd for C73H84O2S2: C, 82.90; H, 8.01. Found: C, 83.12; H, 8.10.
3. Results and discussion
3.1. Synthesis and characterization
The synthetic routes of monomers (M1–M4) and
polymers (P1–P4) are shown in Schemes 1 and 2. All
these polymers were dissolved in common organic solvents to different degrees (P1!P4!P2!P3), and they form good transparent films on glass substrates. The
number-average molecular weights (Mn) were measured by gel
Table 1
Molecular weights and thermal properties of polymers P1–P4
Polymer Mn Mw/Mn Td a (8C) Tg(8C) Tm b (8C) P1 41,800 1.90 442 129 O290 P2 9400 1.81 452 138 – P3 14,400 1.61 436 120 – P4 99,400 2.75 442 144 –
a Temperature of 5% weight loss measured by TGA in nitrogen. b Melting temperature measured by polarizing optical microscopy (POM).
permeation chromatography (GPC) with polystyrene as a standard and THF as an eluting solvent. The related data
obtained from GPC are given in Table 1. The
number-average molecular weights of the polymers were determined to be 9400–99,400 g/mol with polydispersity indexes of 1.61–2.75.
The thermal stability of the polymers (P1–P4) in nitrogen evaluated by thermogravimetric analysis (TGA) indicates that the degradation temperatures (Td) of 5% weight loss in nitrogen are higher than 430 8C for all polymers (Table 1). The glass transition temperatures (Tg) and melting temperatures (Tm) of these polymers were
determined by differential scanning calorimetry (DSC) in nitrogen atmosphere at a scanning rate of 10 8C/min and by polarizing optical microscopy (POM), correspondingly. Their glass transition temperatures are shown between 120 and 144 8C, which are higher than poly(9,9-dihexylfluorene) (w75–80 8C)[27]and also higher than most other
fluorene-based copolymers [28]. Comparing P4 with P1, when
biphenyl groups are attached to 3,30-position of bithiophene, an increase of 15 8C in Tg is observed in P4. That may originate from the contribution of the rigidity of the biphenyl side groups (due to the steric hindrance of the bulky biphenyl side groups) and/or the higher molecular weight of P4. Besides, P1 exhibits mesomorphism at high temperatures (O290 8C), but P2–P4 do not exhibit any mesophase. This may be because that bulky side sub-stituents of bithiophene interrupt the interactions between rigid cores and prohibit from liquid crystalline packing. Another reason is that maybe the diaryl substituents disperse the dipole moments of the main chains in polymers to reduce the mesogenic interactions. With diaryl substituents on bithiophene units, the relatively high glass transition temperatures of molecular architectures are necessary for PLED applications as an emission layer, and the thermal stabilities will improve operating lifetime of PLED devices. 3.2. Optical properties
The UV–visible absorption spectra of these polymers are shown inFig. 1. The larger lmaxvalues in the region of 370– 500 nm are attributed to the electron transition of p–p* along the conjugated backbones of the polymers. It is noticed that P1 exhibits a much larger absorption lmaxvalue than P2–P4. The blue shift of the absorption maxima in P2– P4may be ascribed to loss coplanarity of backbones in the polymers. The bulky substituents on 3,30-positions of thiophene rings give steric hindrance between the side groups and/or the adjacent thiophene rings thus to result in the distortion of the backbones in P2–P4. Therefore, the conjugation of polymer backbones is interrupted which induces a blue shift of the absorption maximum. Comparing P2with P3, two polymers show almost identical absorption spectra. It is obvious that the alkoxy side chains on the pendants of the thiophene do not have any noticeable influence on the absorption spectra of polymers.
The shorter wavelengths (in the range of 270–350 nm) of absorption spectra may originate from the absorption of the
Fig. 1. UV–vis spectra of polymers (a) in dilute chloroform solutions (10K5M). (b) In films.
Table 2
PL emission spectral data of polymers in chloroform solutions and thin solid films
Polymer lPL,sol(nm) lPL,film(nm) FWHMsol(nm) FWHMfilm(nm) FPL,sola Rel. FPL,filma
P1 506 (537)b 543 (514)b 55 87 0.42 1
P2 507 514 78 81 0.05 0.33
P3 509 514 76 78 0.04 0.41
P4 515 517 83 84 0.04 0.69
a
Excited at the absorption region of polymer backbones.
b
substituents, and the intensities of absorption increase with longer conjugation lengths of the substituents. Comparing UV-visible spectra of thin films with diluted solutions, although the polymer main-chain absorption peaks of the solution and film samples are similar, all polymers show tails in the low energy regions. This result indicates some extents of ground-state aggregation exist in the solid state, especially in P1. Therefore, it is obvious that the pendant groups of bithiophene units have strong influence on the absorption of polymers.
The optical band gap energies obtained from UV-visible absorption spectra in solutions are also given inTable 3. The
band gap energies of polymers were determined by the intersection of the tangent through the turning point of the lower energy side of the spectrum and the lengthened baseline for each polymer. The optical band gap of P1 is measured to be 2.43 eV. Nevertheless, diaryl-substituted polymers (P2–P4) have blue-shifted absorption curves and larger band gaps (2.61–2.63 eV). It is because that the head-to-head configurations of 3,30-disubtituted bithiophene units in these polymers lead to an increase of repulsive interactions between the thiophene rings themselves and/ or between thiophene and adjacent fluorene rings, which force the backbones out of coplanarity. Therefore, P2–P4 have larger band gaps due to the shorter conjugation lengths by the twisting of the backbones which is induced by the side diaryl-substituted groups. Nevertheless, the optical band gaps of P2–P4 are almost indistinguishable, regardless of the differences in polarities and rigidities of phenyl and biphenyl rings among the substituents.
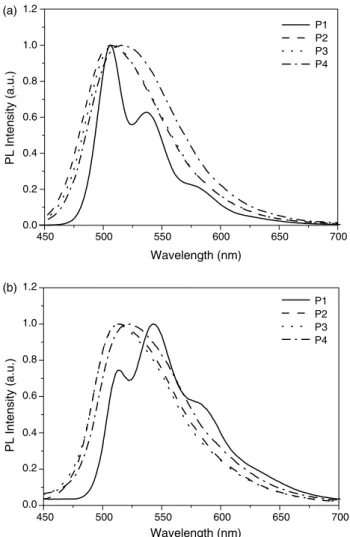
The PL quantum yield (FPL), PL emission maxima (lPL), and full width at half-maximum (FWHM) values of these polymers are exhibited inTable 2. All emission data were obtained by the excitation at the absorption regions of polymer backbones. The PL emission spectra in dilute solutions as well as in thin films are shown inFig. 2, where these polymers exhibit green emissions from the polymer backbones. P2–P4 only show one PL emission peak ranging from 507 to 515 nm in dilute solutions, and they show red shifts of PL emissions ca. 2–7 nm in thin films. On the contrary, P1 shows two well-resolved excimer bands in both of the solution and solid states, and due to strong aggregation it even forms a dominant PL excimer peak (w543 nm) in the solid state. This phenomenon suggests that the bulky substitutents at 3,30-positions of bithiophenes can efficiently reduce aggregation in the solid state and enhance the amorphism of the polymers. Comparing absorption and PL spectra of P1 with those of P2–P4 in solutions, the absorption maxima of P2–P4 are blue-shifted ca. 38–47 nm, but PL emission maxima of P2–P4 are slightly red-shifted ca. 1–9 nm. This result indicates P2–P4 have shorter conjugated lengths than P1 in the ground state, but P2–P4 have longer conjugated lengths similar to P1 in the excited state. As for diaryl-substituted polymers (P2– P4), compared with the PL spectra in solutions, the PL emission peaks in solid films only show small red shifts (the maximum red-shifted value is 7 nm). However, in contrast to diaryl-substituted polymers, the PL peak of P1 in thin
Fig. 2. Spectra of polymers (a) in dilute chloroform solutions (10K5M). (b) In films.
Table 3
HOMO and LUMO energies, and electrochemical properties of polymers P1–P4
Polymer Eox/onset(V) Eox/peak(V) EHOMO(eV) ELUMO(eV)a Optical band gap (eV)
P1 0.72 0.76, 1.24 K5.07 K2.64 2.43
P2 0.84 0.91, 1.20 K5.19 K2.56 2.63
P3 0.79 0.87, 1.07 K5.14 K2.51 2.63
P4 0.77 0.89, 1.38 K5.12 K2.51 2.61
a
LUMO values of polymers are obtained by deduction of optical band gap values from HOMO values due to the lack of reduction peaks in CV measurements.
films is red-shifted 37 nm (from 506 nm in solutions to 543 nm in films) and the emission peak of 543 nm becomes a dominated emission peak, which is due to the aggregation of polymer chains in the solid state of non-substituted P1.
It is interesting to note that the PL quantum yields of diaryl-substituted polymers (P2–P4) are much lower than that of non-substituted polymer (P1) in Table 2. This phenomenon is similar to our previous report and other literatures[29–32]. The PL quantum yields will be decreased when the pendant groups (side chains) are conjugated with the polymer main chains. It may be due to the multifarious conjugated forms in the conjugated substituents of bithiophene units, which cannot confine the excitons efficiently; hence, the PL quantum yields drop considerably in the diaryl-substituted polymers. On the other hand, the PL quantum yields of P2–P4 in solutions are quite similar, indicating that the variation of side-chain diaryl substituents on bithiophene units have little effect on PL quantum yields of the polymers based on the same structures of polymer backbones. However, the quantum yields in films increase in the order of P2!P3!P4 according to the size of pendant groups (Table 2). This result can be explained by the large steric hindrance of the pendant groups to restrain diaryl-substituted polymers from interchain interactions in solids, so the quantum yields of diaryl-substituted polymers with larger pendant groups are enhanced. The effect of large steric hindrance can also be observed when comparing emission peaks and FWHM in solution and solid states, respectively (Table 2). Comparing lPL and FWHM values of solution and solid states, P4 has the least red shift of 2 nm (from 515 nm in solutions to 517 nm in films) in the PL emission peak (lPL) and the smallest change of 1 nm (from 83 nm in solutions to 84 nm in films) in the value of FWHM of the emission peak.
3.3. Electrochemical properties
Cyclic voltammetry (CV) is used to estimate the band gaps, electron affinities, and work functions of conjugated polymers. The potentials were based on the reference energy level of ferrocene (4.8 eV below the vacuum level) according to the following equation[26]: EHOMO/ELUMOZ [K(EonsetK0.45)K4.8] eV. The onset potentials were determined from the intersection of two tangents drawn at the rising and background currents of the cyclic voltammo-gram. Since the onset of the oxidation potentials (i.e. Eox/ onset) for the polymers were obtained from the cyclic voltammetry measurements, the HOMO (i.e. EHOMO) values were calculated from the previous equation, i.e. EHOMOZ [K(Eox/onsetK0.45)K4.8] eV. The reduction potential peaks of the polymers were not observed in the CV measurements, so crude estimations of LUMO values in reduction processes of polymers are made by deduction of optical band gap values from HOMO values. HOMO and LUMO energies, and electrochemical properties (from CV measurements) of polymers (P1–P4) are shown inTable 3.
Fig. 3. Cyclic voltammetry of polymers during the oxidation process.
Table 4
PLED device performance data Polymer lmax(nm) Von a (V) Max. brightness (cd/m2) (V) FWHMEL (nm) P1b 520 4.0 12 (11.0) 90 P2b 516 4.0 1265 (13.5) 84 P3b 515 3.5 2590 (9.5) 78 P4b 528 2.4 474 (11.5) 92 P3c 518 3.5 4284 (18) 82 a
Vonis the turn-on voltage of light. b
Device structure: ITO/polymer/TPBI (30 nm)/MgAg (50 nm)/Ag (100 nm).
c
Device structure: ITO/PVK:P3 (100:30 by weight) (60 nm)/TPBI (30 nm)/MgAg (50 nm)/Ag (100 nm).
These polymers have the same backbone structures of poly(fluorene-co-alt-bithiophene) with different sub-stituents on 3,30-position of bithiophene units. It is interesting to evaluate that how these pendants modulate the electrochemical behavior of these polymers. As shown in Fig. 3, the polymers possess two anodic peaks at potentials ranging from 0.76 to 1.38 V (Table 3) and
onset potentials of oxidation between 0.72 and 0.84 V in anodic scans. Compared with P1, it seems that the onset potentials of oxidation of P2–P4 slightly larger than P1. This result reveals that P1 has a smaller band gap than P2–P4. Compared with P2, the oxidized onset potentials of P4 are not remarkably changed by the replacement of phenyl groups with p-extended biphenyl groups.
Table 5
PLED device performance data measured at 100 mA/cm2
Polymer Voltage (V) Brightness (cd/m2) Luminance efficiency (lm/W) Power efficiency (cd/A) External Q.E. (%) P1a 5.4 2 0c 0c 0c P2a 5.5 110 0.06 0.11 0.04 P3a 6.7 932 0.44 0.94 0.29 P4a 6.5 40 0.02 0.04 0.01 P3b 7.5 1025 0.43 1.03 0.46 a
Device structure: ITO/polymers/TPBI (30 nm)/MgAg (50 nm)/Ag (100 nm).
b
Device structure: ITO/PVK:P3 (100:30 by weight) (60 nm)/TPBI (30 nm)/MgAg (50 nm)/Ag (100 nm).
c
Too small to be detected.
3.4. Electroluminescent (EL) properties of PLED devices The PLED device performance data are summarized in
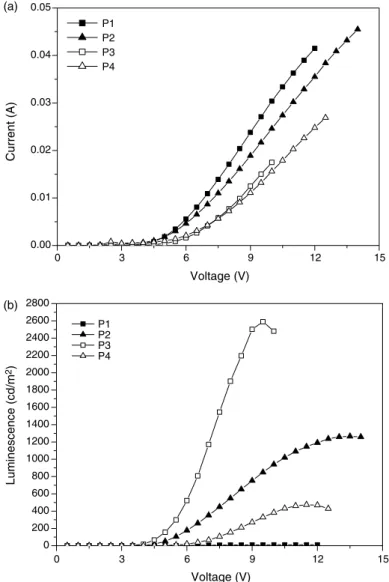
Tables 4 and 5. The current-voltage and
luminescence-voltage characteristics are displayed inFig. 4. These devices show turn on voltages for light at ca. 2.4–4 V, and the device of P4 with device structure: ITO/polymer/TPBI (30 nm)/ MgAg (50 nm)/Ag (100 nm) has the lowest turn-on voltage (2.4 V). Though the non-substituted polymer (P1) has the highest PL quantum yield in solutions (in chloroform), the PLED device of P1 has the worst emission properties due to the poor solubility of P1 in dichloroethane during the spin-coating process. Hence, the device luminance order (P1! P4!P2!P3) is approximately followed by the solubility order of the polymers in dichloroethane. Due to the highest solubility of P3 in dichloroethane, the device with P3 as an emitter has the highest luminance of 2590 cd/m2 at 9.5 V (with a power efficiency of 0.94 cd/A and an external
quantum yield of 0.29% at 100 mA/cm2) among these
polymers (Tables 4 and 5), and shows stable emission with
increasing operating voltages. The device performance can be further improved by blending polymers with poly(N-vinyl carbazole) (PVK), i.e. PVK:P3Z100:30 by weight in device structure: ITO/PVK:P3 (100:30 by weight) (60 nm)/ TPBI (30 nm)/MgAg (50 nm)/Ag (100 nm), a bright luminance of 4284 cd/m2at 18 V (with a power efficiency of 1.03 cd/A and an external quantum yield of 0.46% at 100 mA/cm2) can be reached (Tables 4 and 5). The device performance is probably improved by the film uniformity of PVK or by electron blocking of the interface between PVK and TPBI.
Most EL spectra are well matched with the correspond-ing PL emission spectra of thin films, which indicates that comparable fluorescent emissions of excited states are involved in both EL and PL processes. As shown in
Fig. 5(a), EL emission spectra of these polymers are alike at
8 V, and exhibit similar EL and PL spectra in solid films, except P1. Moreover, emission peaks around 380 nm attributed to TPBI emission were perceptible in PLED devices with the device configuration: ITO/polymer/TPBI (30 nm)/MgAg (50 nm)/Ag (100 nm). The TPBI emission peaks were observed to different degrees, in the order of P1OP4OP2OP3, depending on the emission intensities of the major emission in the polymers. Compared with PL spectra of P1, a dominant PL excimer peak (w543 nm) in the solid state may be suppressed or shifted to 520 nm in EL spectra due to a stronger emission from TPBI. Fig. 5(b) shows that the EL spectra of P3 are almost unchanged and without any TPBI emission bands (due to the bright major emission of P3) as the voltage increases from 6 to 10 V. Thus, this PLED device demonstrates good EL properties without voltage dependent effects.Fig. 6shows EL spectra of P3 blended with PVK in the ratio of PVK:P3Z100:30 by weight, as can be seen that the intensity of 380 nm TPBI emission peak ca. 380 nm will increase as the voltage increases. This result implies that PVK can block electrons in the interface between PVK and TPBI, which results in growing emission intensities of TPBI by increasing voltages.
Fig. 6. Normalized EL spectra of ITO/PVK:P3 (100:30 by weight)/TPBI (30 nm)/MgAg (50 nm)/Ag (100 nm) device operating at different voltages. Fig. 5. (a) Normalized EL spectra of ITO/polymer/TPBI (30 nm)/MgAg
(50 nm)/Ag (100 nm) devices operating at 8 V. (b) Normalized EL spectra of ITO/P3/TPBI (30 nm)/MgAg (50 nm)/Ag (100 nm) device operating at different voltages.
4. Conclusions
† These polymeric bithiophene derivatives incorporating with fluorenes exhibit relatively high Tg and good thermal stabilities.
† Owing to the large steric hindrance of diaryl substituents on bithiophenes, the aggregation of the polymers (P2– P4) in solids is reduced. Therefore, even though the PL quantum yields of diaryl-substituted polymers (P2–P4) are much lower than that of non-substituted polymer (P1), the performance of their PLED devices are superior to that of P1.
† Compared with the non-substituted polymer (P1), the introduction of diaryl substitutents at 3,30-positions of bithiophene units in P2–P4 has significant effects on the photophysic properties of resulting polymers in solution and solid states. However, different diaryl substituents on bihiophene units only affect the electronic properties of the resulting polymers slightly.
† Consequently, the device made of P3 as an emitter has the highest luminance of 2590 cd/m2 at 9.5 V among these polymers. For optimum device performance, a device of P3 blended with PVK can be further enhanced to a brighter luminance of 4284 cd/m2at 18 V.
Acknowledgements
We are grateful for the financial support provided by the National Science Council of Taiwan, ROC through NSC 92-2113-M-009-016.
References
[1] Redecker M, Bradley DDC, Inbasekaran M, Woo EP. Appl Phys Lett 1998;73:1565.
[2] Grell M, Bradley DDC, Ungar G, Hill J, Whitehead KS. Macromol-ecules 1999;32:5810.
[3] Klaerner G, Miller RD. Macromolecules 1998;31:2007.
[4] Chen X, Liao JL, Liang Y, Ahmed MO, Tseng HE, Chen SA. J Am Chem Soc 2003;125:636.
[5] Jenekhe SA, Osaheni JA. Science 1994;265:765.
[6] Weinfurthner KH, Fujikawa H, Tokito S, Taga Y. Appl Phys Lett 2000;76:2502.
[7] Pei Q, Yang Y. J Am Chem Soc 1996;118:7416.
[8] Kim JS, Friend RH, Cacialli F. Appl Phys Lett 1999;74:3084. [9] Ego C, Grimsdale AC, Uckert F, Yu G, Srdanov G, Mullen K. Adv
Mater 2002;14:809.
[10] Wu FI, Reddy S, Shu CF, Liu MS, Jen AKY. Chem Mater 2003;15: 269.
[11] Shu CF, Dodda R, Wu FI, Liu MS, Jen AKY. Macromolecules 2003; 36:6698.
[12] Sung HH, Lin HC. J Polym Sci, Part A: Polym Chem 2005;43:2700. [13] Andersson MR, Berggren M, Inganas O, Gustafsson G,
Gustafsson-Carlberg JC, Selse D, et al. Macromolecules 1995;28:7525. [14] Kham K, Sadki S, Chevrot C. Synth Met 2004;145:135.
[15] Naudin E, Mehdi NE, Soucy C, Breau L, Belanger D. Chem Mater 2001;13:634.
[16] Xu B, Holdcroft S. Macromolecules 1993;26:4457.
[17] Albert IDL, Marks TJ, Ratner MA. J Am Chem Soc 1997;119:6575. [18] Janietz S, Bradley DDC, Grell M, Giebeler C, Inbasekaran M,
Woo EP. Appl Phys Lett 1998;73:2453.
[19] Ruiz JP, Dharia JR, Reynolds JR, Buckley LJ. Macromolecules 1992; 25:849.
[20] Chen ZK, Meng H, Lai YH, Huang W. Macromolecules 1999;32: 4351.
[21] Meng H, Yu WL, Hunang W. Macromolecules 1999;32:8841. [22] Pu YJ, Soma M, Kido J, Nishide H. Chem Mater 2001;13:3817. [23] Aleman C, Julia L. J Phys Lett 1996;100:14661.
[24] (a) Demadrille R, Divisia-Blohorn B, Zagorska M, Quillard S, Rannou P, Traversa JP, et al. New J Chem 2003;27:1479. (b) Demadrille R, Patrice R, Bleuse J, Oddou JL, Pron A,
Zagorska M. Macromolecules 2003;36:7045.
[25] (a) Lim E, Jung BJ, Shim HK. Macromolecules 2003;36:4288. (b) Liu B, Yu WL, Lai YH, Huang W. Macromolecules 2000;33:
8945.
[26] Egbe DAM, Cornelia B, Nowotny J, Gunther W, Klemm E. Macromolecules 2003;36:5459.
[27] (a) Zeng G, Yu WL, Chua SJ, Huang W. Macromolecules 2002;35: 6907.
(b) Marsitzky D, Murray J, Scott JC, Carter KR. Chem Mater 2001; 13:4285.
[28] (a) Chen Y, Araki Y, Doyle J, Strevens A, Ito O, Blau WJ. Chem Mater 2005;17:1661.
(b) Kreyenschmidt M, Klaerner G, Fuhrer T, Ashenhurst J, Karg S, Chen WD, et al. Macromolecules 1998;31:1099.
[29] Sung HH, Lin HC. Macromolecules 2004;37:7945.
[30] Lee YZ, Chen X, Chen SA, Wei PK, Fann WS. J Am Chem Soc 2001; 123:2296.
[31] Xu B, Pan Y, Zhang J, Peng Z. Synth Met 2000;114:337.
[32] (a) Liu MS, Jiang X, Herguth P, Jen AKY. Chem Mater 2001;13: 3820.
(b) Wilson JN, Windscheif PM, Evans U, Myrick ML, Bunz UHF. Macromolecules 2002;35:8681.