Synthesis, Characterization, and Modification
FANG-IY WU, CHING-FONG SHUDepartment of Applied Chemistry, National Chiao Tung University, Hsin-Chu, Taiwan, 30035, Republic of China
Received 4 March 2001; accepted 1 May 2001
ABSTRACT: The synthesis and characterization of hyperbranched aromatic poly(ether imide)s are described. An AB2monomer, which contained a pair of phenolic groups and an aryl fluoro moiety activated toward displacement by the attached imide heterocyclic ring, was prepared. The nucleophilic substitution of the fluoride with the phenolate groups led to the formation of an ether linkage and, subsequently, to the hyperbranched poly(ether imide), which contained terminal phenolic groups. A similar one-step poly-merization involving a monomer that contained silyl-protected phenols yielded a hy-perbranched poly(ether imide) with terminal silylated phenols. The degree of branching of these hyperbranched polymers was approximately 55%, as determined by a combi-nation of model compound studies and1H NMR integration experiments. End-capping reactions of the terminal phenolic groups were readily accomplished with a variety of acid chlorides and acid anhydrides. The nature of the chain-end groups significantly influenced physical properties, such as the glass-transition temperature and the solu-bility of the hyperbranched poly(ether imide)s. As the length of the acyl chain of the terminal ester groups increased, the glass-transition temperature value for the polymer decreased, and the solubility of the polymer in polar solvents was reduced, becoming more soluble in nonpolar solvents.© 2001 John Wiley & Sons, Inc. J Polym Sci Part A: Polym Chem 39: 2536 –2546, 2001
Keywords: hyperbranched; poly(ether imide)s; AR2monomer; degree of branching
INTRODUCTION
Hyperbranched polymers have been the subject of considerable interest in recent years because their unique highly branched structures can be predicted to exhibit some unusual properties.1,2 Such polymers are prepared in a single step via a random one-pot polymerization of ABn-type
monomers. As predicted theoretically by Flory,3 the direct polymerization of ABn-type monomers
is expected to give rise to a highly branched, ir-regular structure that contains a large number of terminal functional groups. Hyperbranched
poly-mers are generally composed of three parts, den-dritic, linear, and terminal units, and may be considered to be irregular analogues of dendritic macromolecules,4 which are built up by
step-by-step synthetic sequences.5,6 Nevertheless, they still maintain many of the properties found in their more perfectly defined dendrimer counter-parts.
One-step synthesis allows hyperbranched poly-mers to be more readily available (as well as large-scale preparation) for potential applica-tions. This significant feature has led to the de-velopment of novel synthetic routes for the prep-aration of such polymers.1,2Aromatic poly(ether imide)s represent a class of polymers that have gained technical interest because of their excel-lent electrical, thermal, and mechanical proper-Correspondence to: C.-F. Shu (E-mail: [email protected])
Journal of Polymer Science: Part A: Polymer Chemistry, Vol. 39, 2536 –2546 (2001) © 2001 John Wiley & Sons, Inc.
ties.7 Two typical synthetic methods have been
employed for their preparation. The first involves the condensation of dianhydride and diamine monomers, thus generating a poly(amic acid), fol-lowed by cyclodehydration to form the imide ring.8 The second method involves aromatic
cleophilic substitution reactions of phenoxide nu-cleophiles with nitro- or fluoro-substituted phtha-limides to form the ether linkage.9,10 Because of
the high reactivity of amine and anhydride groups, no stable AB2-type monomer that
con-tains one amino group and two anhydride groups (or one anhydride group and two amino groups) appears to be available for the preparation of hyperbranched poly(ether imide)s. To solve this problem, Kakimoto et al.11recently reported the
synthesis of a hyperbranched poly(ether imide) derived from a monomer composed of an amino group and a phthalic acid monomethyl ester. Moore et al. reported a modification of the nucleo-philic displacement method for the preparation of hyperbranched poly(ether imide)s with a Krichel-dorf-type reaction involving tert-butyldimethylsi-lyl (TBDMS) protected benzenediol groups and an activated fluoride in the presence of a catalytic amount of CsF.12,13
In a previous study, we reported the synthesis of dendritic poly(ether imide)s from the building block 1-(4-aminophenyl)-1,1-bis(4-hydroxyphenyl)ethane (1),14in which two hydroxy groups are attached to
two separated benzene rings, to prevent the for-mation of complex products due to electron-trans-fer redox reactions between the electron-deficient nitrophthalimide and electron-rich benzenediol dianion.15In this article, we report the synthesis and characterization of hyperbranched poly(ether imide)s containing terminal phenolic groups. The polymers were prepared directly via aromatic nu-cleophilic substitution from an AB2 monomer, which contained a fluoro-substituted phthalim-ide, derived from compound 1. Even though the nitro group was readily displaced, the resulting nitrite ion was reactive and could participate in side reactions during the polymerization at a high temperature.16Because of this, the fluoride was the more desirable leaving group. The phenolic terminal units of the resulting polymers were modified by reaction with several end-capping agents. The influence of the chain-end groups on the solubility and glass-transition temperature (Tg) of the hyperbranched polymers was also
in-vestigated.
EXPERIMENTAL
General Directions
Anhydrous tetrahydrofuran (THF) was distilled from a sodium diphenyl ketyl solution just prior to use. Other starting materials and reagents were used as obtained from the suppliers. NMR spectra were recorded on a Varian Unity 300-MHz spectrometer or a Bruker-DRX 300-300-MHz spectrometer. Differential scanning calorimetry (DSC) was performed on a Seiko SSC 5200 DSC with a heating/cooling rate of 10 °C min⫺1. Ther-mogravimetric analysis (TGA) was performed on a Seiko TG/DTA 200 instrument. The thermal stability of samples was determined in nitrogen on the basis of the weight loss at a heating rate of 10 °C min⫺1. Size exclusion chromatography (SEC) was carried out on a Waters chromatogra-phy unit interfaced with a Waters 410 differential refractometer. Three 5-m Waters styragel col-umns (300 ⫻ 7.8 mm) connected in series in de-creasing order of pore size (105, 104, and 103 Å) were used with dimethylformamide (DMF)/0.05 M LiBr as an eluent, and poly(methyl methacry-late) standard samples were used for calibration. Mass spectra were obtained on a JEOL JMS-SX/SX 102A mass spectrometer. Analytical thin-layer chromatography (TLC) was performed on silica gel GF254 plates. The silica gel used for column chromatography was Merck Kieselgel 60 (70 –230 mesh). 114and N-phenyl-4-fluorophtha-limide (5)9 were prepared according to literature methods.
N-{4-[1,1-Di(4-hydroxyphenyl)ethyl]phenyl}-4-fluorophthalimide (2)
A mixture of 1 (1.01 g, 3.30 mmol) and 4-fluoroph-thalic anhydride (0.55 g, 3.31 mmol) in 1,2-dichlo-robenzene (5.0 mL) was heated at reflux with stirring under nitrogen for 2 h. The resulting mixture was added to hexane (150 mL). The re-sulting precipitate was collected and dried at 60 °C in vacuo to give 2 (1.27g, 84.9%).
1H NMR [dimethyl sulfoxide (DMSO)-d 6, ␦]: 9.29 (s, 2 H) 8.02 (dd, 1 H, J⫽ 4.6, 8.2 Hz), 7.86 (dd, 1 H, J⫽ 2.2, 7.4 Hz), 7.74–7.67 (m, 1 H), 7.32 (d, 2 H, J⫽ 8.4 Hz), 7.15 (d, 2 H, J ⫽ 8.4 Hz), 6.85 (d, 4 H, J⫽ 8.6 Hz), 6.67 (d, 4 H, J ⫽ 8.6 Hz), 2.05 (s, 3 H).13C NMR (DMSO-d 6,␦): 166.1, 165.9 (d, JCOF⫽ 252 Hz), 165.8 (d, JCOF⫽ 3 Hz), 155.3, 149.9, 139.2, 134.6 (d, JCOF ⫽ 10 Hz), 129.3, 129.2, 128.7, 127.7 (d, JCOF⫽ 3 Hz), 126.4, 126.2
(d, JCOF⫽ 10 Hz), 121.5 (d, JCOF⫽ 24 Hz), 114.6, 111.2 (d, JCOF⫽ 25 Hz), 50.6, 30.3. High-resolu-tion mass spectrometry (HRMS) [M⫹]: 453.1373. Calcd. for C28H20FNO4: 453.1376.
N-{4-[1,1-Di(4-tert- butyldimethylsilyloxyphenyl)ethyl]phenyl}-4-fluorophthalimide (3)
A mixture of 2 (1.28 g, 2.81 mmol), tert-butyldim-ethylsilyl chloride (1.01 g, 6.70 mmol), and imida-zole (0.46 g, 6.76 mmol) in CH2Cl2 (8.0 mL) was stirred under nitrogen at 25 °C for 7 h. The imi-dazole salts were removed by filtration, and the solvent was removed in vacuo. The crude product was purified by column chromatography (CH2Cl2) to give 3 (1.64 g, 85.8%). 1H NMR (CDCl 3,␦): 7.94 (dd, 1 H, J ⫽ 4.4, 8.2 Hz), 7.61 (dd, 1 H, J⫽ 2.1, 7.0 Hz), 7.46–7.40 (m, 1 H), 7.29 (d, 2 H, J⫽ 8.7 Hz), 7.19 (d, 2 H, J ⫽ 8.7 Hz), 6.94 (d, 4 H, J⫽ 8.7 Hz), 6.72 (d, 4 H, J ⫽ 8.7 Hz), 2.13 (s, 3 H), 0.97 (s, 18 H), 0.19 (s, 12 H).13C NMR (CDCl3,␦): 166.6 (d, JCOF⫽ 256 Hz), 166.3, 166.0 (d, JCOF⫽ 3 Hz), 153.7, 150.0, 141.5, 134.5 (d, JCOF ⫽ 9 Hz), 129.6, 129.4, 129.1, 127.5 (d, JCOF ⫽ 3 Hz), 126.1 (d, JCOF ⫽ 9 Hz), 121.5, 121.4 (d, JCOF ⫽ 24 Hz), 119.2, 111.4 (d, JCOF ⫽ 25 Hz), 51.2, 30.7, 25.7, 18.2, ⫺4.4. HRMS [M⫹]: 681.3097. Calcd. for C40H48FNO4Si2: 681.3106.
N-{4-[1,1-Di(4-hydroxyphenyl)ethyl]phenyl}phthalimide (4)
A mixture of 1 (0.52 g, 1.69 mmol) and phthalic anhydride (0.25 g, 1.69 mmol) in 1,2-dichloroben-zene (1.2 mL) was heated at reflux with stirring under nitrogen for 1.5 h. The resulting mixture was added to hexane (100 mL). The precipitate was collected and dried at 60 °C in vacuo to give 4 (0.606 g, 82.3%). 1H NMR (DMSO-d 6,␦): 9.28 (s,2 H), 7.97–7.93 (m, 2 H), 7.91–7.87 (m, 2 H), 7.33 (d, 2 H, J⫽ 8.6 Hz), 7.15 (d, 2 H, J⫽ 8.6 Hz), 6.86 (d, 4 H, J ⫽ 8.7 Hz), 6.66 (d, 4 H, J⫽ 8.7 Hz), 2.06 (s,3 H). 13C NMR (DMSO-d6, ␦): 167.1, 155.3, 149.8, 139.2, 134.7, 131.5, 129.4, 129.2, 128.6, 126.4, 123.4, 114.6, 50.6, 30.3. HRMS [M⫹]: 435.1463. Calcd. for C28H21NO4: 435.1470.
Synthesis of Model Compounds 6 and 7
A mixture of 4 (40 mg, 90 mol), 5 (21 mg, 90 mol), CsF (13 mg, 90 mol), and diphenylsulfone
(DPS; 0.50 g) was heated under nitrogen at 235 °C for 15 min. The model compounds were isolated from the reaction mixture by preparative TLC (1/2/2 hexane/EtOAc, ethyl acetate/CH2Cl2). Compound 6 1 H NMR (DMSO-d6, ␦): 9.37 (s, 1 H), 7.97–7.95 (m, 3 H), 7.89 –7.87 (m, 2 H), 7.54 –7.37 (m, 9 H), 7.22 (d, 2 H, J⫽ 8.6 Hz), 7.20 (d, 2 H, J ⫽ 8.9 Hz), 7.13 (d, 2 H, J⫽ 8.9 Hz), 6.92 (d, 2 H, J ⫽ 8.7 Hz), 6.73 (d, 2 H, J⫽ 8.7 Hz), 2.15 (s, 3 H).13C NMR (DMSO-d6, ␦): 167.0, 166.4, 166.2, 162.7, 155.6, 152.7, 149.0, 146.0, 138.4, 134.7, 134.2, 131.9, 131.5, 130.3, 129.6, 129.3, 128.8, 128.7, 128.0, 127.3, 126.6, 125.9, 125.3, 123.4, 122.9, 119.5, 114.8, 111.8, 51.1, 30.3. HRMS [M⫹ ⫹ H]: 657.2017. Calcd. for C42H29FN2O6: 657.2025. Compound 7 1H NMR (DMSO-d 6,␦): 7.97–7.94 (m, 4 H), 7.92– 7.87 (m, 2 H), 7.54 –7.39 (m, 16 H), 7.30 (d, 2 H, J ⫽ 8.7 Hz), 7.26 (d, 4 H, J ⫽ 8.9 Hz), 7.17 (d, 4 H, J⫽ 8.9 Hz), 2.25 (s, 3 H).13C NMR (DMSO-d 6,␦): 167.0 166.3, 166.2, 162.5, 153.0, 148.2, 145.2, 134.7, 134.2, 131.9, 131.5, 130.4, 130.0, 128.8, 128.7, 128.0, 127.3, 126.8, 125.9, 125.4, 123.4, 123.0, 119.6, 111.9, 51.5, 30.2. HRMS [M⫹ ⫹ H]: 878.2501. Calcd. for C56H36N3O8: 878.2502. N-{4-[1,1-Di(4-tert- butyldimethylsilyloxyphenyl)ethyl]phenyl}-4-fluorophthalimide (8)
A mixture of 4 (0.62 g, 1.41 mmol), tert-butyldim-ethylsilyl chloride (0.43 g, 2.83 mmol), and imida-zole (0.19 g, 2.82 mmol) in CH2Cl2 (5.0 mL) was
stirred under nitrogen at 25 °C for 12 h. The imidazole salts were removed by filtration, and the solvent evaporated in vacuo. The crude prod-uct was purified by column chromatography (CH2Cl2) to give 8 (0.48 g, 51.3%). 1 H NMR (CDCl3,␦): 7.95–7.92 (m, 2 H), 7.79– 7.75 (m, 2 H), 7.31 (d, 2 H, J⫽ 8.9 Hz), 7.20 (d, 2 H, J⫽ 8.9 Hz), 6.95 (d, 4 H, J ⫽ 8.8 Hz), 6.73 (d, 4 H, J⫽ 8.8 Hz), 2.13 (s, 3 H), 0.97 (s, 18 H), 0.18 (s, 12 H).13C NMR (CDCl3,␦): 167.4, 153.7, 149.7, 141.6, 134.3, 131.8, 129.7, 129.4, 129.3, 125.6, 123.7, 119.2, 51.2, 30.7, 25.7, 18.2, ⫺4.4. HRMS [M⫹ CH3]: 648.2953. Calcd. for C39H46NO4Si2: 648.2965.
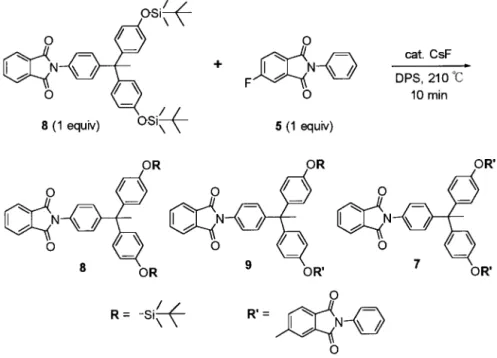
Synthesis of Model Compound 9
A mixture of 8 (60 mg, 90 mol), 5 (23 mg, 90 mol), CsF (1.4 mg, 9 mol), and DPS (0.60 g) was
heated under nitrogen at 210 °C for 10 min. The model compound 9 was isolated from the reaction mixture by preparative TLC (5/1/2 hexane/EtOAc/ CH2Cl2). 1H NMR (CDCl 3,␦): 7.95–7.93 (m, 2 H), 7.88 (d, 1 H, J⫽ 8.2 Hz), 7.79–7.76 (m, 2 H), 7.51–7.32 (m, 9 H), 7.24 (d, 2 H, J⫽ 8.6 Hz), 7.18 (d, 2 H, J ⫽ 8.7 Hz), 7.01–6.98 (m, 4 H), 6.77 (d, 2 H, J ⫽ 8.6 Hz), 2.20 (s, 3 H), 0.96 (s, 9 H), 0.20 (s, 6 H).13C NMR (CDCl3,␦): 167.3, 166.8, 166.7, 163.6, 153.9, 152.9, 148.9, 146.3, 140.8, 134.4, 134.2, 131.7, 130.7, 129.6, 129.5, 129.3, 129.1, 128.0, 126.5, 125.8, 125.7, 125.1, 123.7, 123.0, 119.6, 119.4, 112.1, 51.6, 30.7, 25.6, 18.2, ⫺4.4. HRMS [M⫹ ⫹ H]: 771.2893. Calcd. for C48H43N2O6Si:
771.2890.
Preparation of Hyperbranched Poly(ether imide) P1
A mixture of 2 (0.15 g, 0.33 mmol), CsF (40 mg, 0.26 mmol), and DPS (0.45 g) was heated at 235 °C with stirring under nitrogen for 15 min. The mixture was then dissolved in DMF (3.0 mL) and precipitated into methanol (40 mL). The resulting polymer was collected and purified by precipita-tion from DMF into methanol two times to give P1 (0.123 g, 85.5%).
Preparation of Hyperbranched Poly(ether imide) P2
A mixture of 3 (3.00 g, 4.40 mmol), CsF (6.67 mg, 44mol), and DPS (4.50 g) was heated at 210 °C with stirring under nitrogen for 10 min. The re-action mixture was dissolved in THF (30 mL) and precipitated into a mixture of hexane (50 mL) and methanol (150 mL). The resulting polymer was collected and purified by precipitation from THF into a mixture of hexane, acetone, and methanol (1/1/3 v/v/v) two times to give P2 (2.19 g, 91.0%). Preparation of Hyperbranched
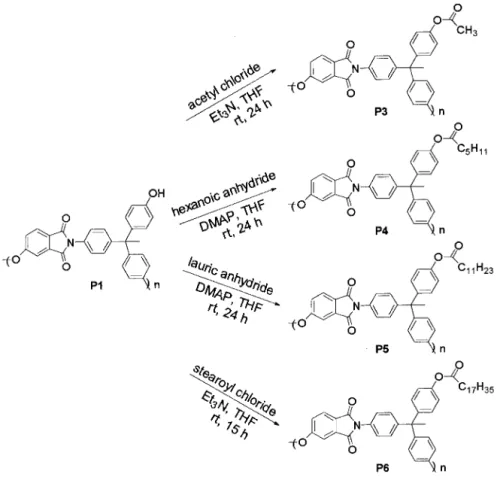
Poly(ether imide) P3
To a solution of P1 (60 mg, 0.14 mmol) and tri-ethylamine (Et3N) (73 mg, 0.72 mmol) in anhy-drous THF (6.0 mL), acetyl chloride (57 mg, 0.73 mmol) was added dropwise under nitrogen. The mixture was stirred at 25 °C for 24 h and then added to a mixture of water (10 mL) and metha-nol (10 mL). The resulting polymer was collected and purified by precipitation from CHCl3 into methanol to give P3 (50 mg, 76%).
Preparation of Hyperbranched Poly(ether imide) P4
To a solution of P1 (60 mg, 0.14 mmol) and 4-(dimethylamino)pyridine (DMAP) (91 mg, 0.74 mmol) in anhydrous THF (3.0 mL), hexanoic an-hydride (154 mg, 0.72 mmol) was added dropwise under nitrogen. The mixture was stirred at 25 °C for 24 h and then added to a mixture of water (10 mL) and methanol (10 mL). The resulting poly-mer was collected and purified by precipitation from CHCl3 into methanol to give P4 (61 mg, 83%).
Preparation of Hyperbranched Poly(ether imide) P5
To a solution of P1 (60 mg, 0.14 mmol) and DMAP (88 mg, 0.72 mmol) in anhydrous THF (3.0 mL), lauric anhydride (275 mg, 0.72 mmol) was added dropwise under nitrogen. The mixture was stirred at 25 °C for 24 h and then added to a mixture of water (10 mL) and methanol (10 mL). The result-ing polymer was collected and purified by precip-itation from CHCl3into methanol to give P5 (70 mg, 83%).
Preparation of Hyperbranched Poly(ether imide) P6
A mixture of stearic acid (204 mg, 0.72 mmol) and thionyl chloride (0.8 mL) was heated at reflux with stirring under nitrogen for 2.5 h. The excess thionyl chloride was then removed in vacuo. To the acid chloride was added a solution of P1 (50 mg, 0.12 mmol) and Et3N (73 mg, 0.72 mmol) in anhydrous THF (6.0 mL). The mixture was stirred at 25 °C for 15 h and then poured into a mixture of water (10 mL) and methanol (10 mL). The collected polymer was purified by precipita-tion from CHCl3 in methanol and then precipi-tated from CHCl3into a solvent mixture of hexane and methanol (1/2 v/v) to give P6 (49 mg, 61%).
RESULTS AND DISCUSSION
Synthesis and Characterization
Two types of AB2-type monomers, 2 and 3, were prepared as shown in Scheme 1. The condensa-tion of the amino group of compound 1 with 4-flu-orophthalic anhydride followed by cyclodehydra-tion gave monomer 2, which contained an acti-vated aryl fluoride and two phenolic groups. The
phenolic groups of 2 were then converted into
t-butyldimethylsilyl ether by stirring with
TB-DMS chloride in methylene chloride in the pres-ence of imidazole to yield monomer 3.
The polymerization of monomer 2 was per-formed in DPS at 235 °C in the presence of CsF (0.8 equiv), as shown in Scheme 2. The nucleo-philic substitution of the fluoride with the phe-nolic groups,9 activated by the imide moiety, led
to the formation of an ether linkage and, subse-quently, to the hyperbranched poly(ether imide) P1 with terminal phenolic groups. For the sake of simplicity, Scheme 2 only shows the overall
com-positional repeat units, even though the hyper-branched polymer contained a combination of dendritic, linear, and terminal units. For the poly-condensation of monomer 3, reaction conditions similar to those reported by Moore et al.13were
employed to give the corresponding hyper-branched poly(ether imide) P2, which contained terminal silylated phenol units. The TBDMS end groups in P2 remained fully intact, as indicated by the 1H NMR integration ratio of the protons
attributed to the methyl (or t-butyl) groups in silyl ether group versus the methyl (or phenyl) protons of the AB2 unit. Both hyperbranched Scheme 1
poly(ether imide)s were obtained in good yields (85–90%).
The molecular weight of P1 and P2 was deter-mined by SEC analysis in a DMF solution cali-brated against linear poly(methyl methacrylate) standards. SEC measurements of P1 and P3 indicated that the weight-average molecular weights were 34,500 and 74,900, respectively, rel-ative to standard poly(methyl methacrylate), and the polydispersity values (weight-average molec-ular weight/number-average molecmolec-ular weight) were determined to be 3.9 and 3.4, respectively. Because of the highly branched nature of the hy-perbranched macromolecules, the result from SEC measurements was used only for a rough estimate. The structures of the polymers were characterized by infrared spectroscopy. A Fourier transform infrared spectrum of P1 showed char-acteristic absorptions corresponding to imide at 1778 (CAO asymmetric stretching), 1718 (CAO symmetric stretching), and 1375 cm⫺1 (CON stretching). The corresponding imide bands of the P2 polymer appeared at 1779, 1727, and 1374 cm⫺1. For P1, an additional broad band appeared at 3470 cm⫺1 and was assigned to the OOH stretching of the terminal phenolic groups. Tgof the hyperbranched poly(ether imide)s was determined by DSC. Tgof P1, which had polar hydroxyl termi-nal groups, was observed at 270 °C. Tgof P2, which had silyl-protected terminal phenols, decreased to
223 °C. TGA was used to examine the thermal sta-bility of these polymers. A 5% weight loss for P1 was observed at 376 °C, followed by an additional 5% weight loss at 402 °C. P2, in which the terminal phenols were protected by TBDMS groups, showed better thermal stability, with a 5 wt % loss at 467 °C followed by a 10 wt % loss at 480 °C.
Degree of Branching (DB)
The DB, which is a typical characteristic often used to evaluate the irregularity of the structure of hyperbranched polymers, was defined as the sum of dendritic and terminal units versus total units (linear, dendritic, and terminal units).17A
combination technique of model compound stud-ies and NMR spectroscopy was used to quantify the different subunits that were present in the hyperbranched polymer and, subsequently, to de-termine its DB.17Scheme 3 shows the model
re-action used in determining the DB of P1. An equimolar mixture of compounds 4 and 5 was reacted under conditions that were similar to those used for the polymerization of monomer 2. The model compounds were separated from the reaction mixture by preparative TLC. Figure 1 shows the1H NMR spectra of 4, which resembles
the terminal unit, the monosubstituted product 6, which resembles the linear unit, and the disub-stituted product 7, which resembles the dendritic Scheme 3
unit. The peak assignments were based on the peak positions of compounds 4 and 5 as well as the auxiliary of two-dimensional (H, H) and (C, H) correlation NMR spectroscopy. Distinct reso-nances for the terminal model compound 4 ap-peared at 9.28 and 2.06 ppm, respectively, and were assigned to the protons of the phenolic groups and the methyl group, whereas the corre-sponding protons for the linear model compound 6 were observed at 9.37 and 2.15 ppm, respectively. The resonance for the methyl protons of the den-dritic model compound 7 appeared at 2.25 ppm. Figure 1 also shows the1H NMR spectrum of the
hyperbranched poly(ether imide) P1. A good cor-relation can be seen in a comparison of the 1H
NMR spectra of these model compounds with that of P1. The resonances at 9.33 and 9.25 ppm were assigned to the phenol protons of the linear and terminal subunits, respectively, whereas the res-onances at 2.21, 2.15, and 2.03 ppm were assigned to the methyl protons of the dendritic, linear, and terminal subunits, respectively.18The integration
of these resonances allowed the relative percent-age of each subunit to be determined. The DB of P1 was determined to be 52% on the basis of the relative integration of the phenolic protons. By calculating the integration ratio of the methyl protons, we also estimated the DB of P1 to be 55%,18which is in good agreement with the value
determined from the phenolic protons. The DB
values calculated for P1 are close to the statistical value of 50%, which would be expected for a ran-dom AB2polycondensation.19
The DB of the hyperbranched poly(ether imide) P2 was also evaluated with a similar rationale. The synthesis of the model compounds is detailed in Scheme 4. Figure 2 shows the1H NMR spectra
of model compounds 7, 8, and 9 and the polymer P2. A comparison of the1H NMR spectra of these
model compounds with that of P2 allowed the resonances corresponding to the dendritic, linear, and terminal subunits of the hyperbranched poly-mer to be identified. The resonances of the AB spin system at 6.75 and 6.70 ppm were assigned to the protons in the ortho position of the si-lylether of the linear and terminal subunits, re-spectively, whereas the resonances at 2.25, 2.18, and 2.11 ppm correspond to the methyl protons of the dendritic, linear, and terminal units, respec-tively.20Based on the integration ratio of the
or-tho protons of the silylether and that of the methyl protons, the DB values of P2 were deter-mined to be 57% and 54%,20values which are in
reasonable agreement with each other and ap-proach the expected statistical value 50%.19
Chemical Modification of Hyperbranched Poly(ether imide) P1
Hyperbranched polymers are characterized by a large number of chain-end groups. The terminal Figure 1. 1H NMR spectra in DMSO-d
6of model compounds (a) 7, (b) 6, and (c) 4 compared with (d) P1.*indicates a signal arising from DMSO.
phenolic groups in P1 could be easily functional-ized to yield hyperbranched polymers with a va-riety of functional chain ends. The nature of the end groups influences the physical and chemical properties of the hyperbranched polymers.21 In
this study, the terminal phenolic groups of P1
were acylated with acid chlorides or anhydrides to give the corresponding ester derivatives, as shown in Scheme 5. The degree of modification could be estimated from the1H NMR integration
ratio of proton signals attributed to the methyl-ene group in the ␣ position of the ester group Scheme 4
Figure 2. 1H NMR spectra in CDCl
3 of model compounds (a) 7, (b) 9, and (c) 8 compared with (d) P2.*indicates a signal arising from CHCl3.
versus the methyl protons of the AB2unit. For all
the modification reactions, the use of excess re-agents resulted in almost complete (95–100%) functionalization.
The Tg and solubility of the hyperbranched
poly(ether imide)s P1–P6 are summarized in Ta-ble I. The influence of Tgthrough variation of the
end groups could be assigned to the translational motion and an intermolecular effect.22,23 It is
known that for hyperbranched polymers, the transition from the polar hydroxy function to non-polar aliphatic end groups results in a decrease in
Tgdue to the reduction in the extent of
intermo-lecular interactions in the polymer molecules.23
Tg of P1, which had polar hydroxyl terminal
groups, was 270 °C, whereas the Tgvalues of P2
and P3, which had fewer polar terminal groups, namely, silylether and ester groups, were 223 and Scheme 5
Table I. Thermal and Solution Properties of Hyperbranched Poly(ether imide)s
Polymer Tg(°C) Solubilitya Toluene CH2Cl2 CHCl3 THF DMF DMSO P1 270 ⫺ ⫺ ⫺ ⫹ ⫹ ⫹ P2 223 ⫹ ⫹ ⫹ ⫹ ⫹ ⫺ P3 227 ⫺ ⫹ ⫹ ⫹ ⫹ ⫹ P4 163 ⫹ ⫹ ⫹ ⫹ ⫹ ⫺ P5 43 ⫹ ⫹ ⫹ ⫹ ⫹ ⫺ P6 ⫺41 ⫹ ⫹ ⫹ ⫹ ⫺ ⫺ a⫹ ⫽ soluble; ⫺ ⫽ insoluble.
227 °C, respectively. The reduction in Tg was caused by a reduction in the extent of hydrogen bonding. The polar hydrogen-bonding interac-tions were replaced by nonpolar hydrophobic in-teractions as the result of esterification with fatty acids. Therefore, the intermolecular interaction was lower, and the mobility was increased. A further decrease in Tgto 163, 43, and⫺41 °C was observed for P3, P4, and P5, respectively, be-cause of the increasing length of the alkoxyl chain of the terminal ester groups. Hult et al.24
inves-tigated the alkyl modification of hyperbranched polyesters based on 2,2-bis(hydroxymethyl)propi-onic acid. They reported that, when a sufficiently long alkyl chain (12–16 carbons) was introduced, the side chain of the polymers tended to crystal-lize, so an increase in the polyester Tg was ob-served. However, the same phenomenon was not observed for the hyperbranched poly(ether imi-de)s, which contained rigid aromatic imide units and could exhibit a lower tendency for side-chain crystallization.
Because of their highly branched structures, these hyperbranched poly(ether imide)s pos-sessed good solubility in organic solvents. How-ever, the different chain ends resulted in differ-ences of solubility in polar vis-a`-vis nonpolar sol-vents. P1, which had polar phenolic terminal groups, was soluble in polar solvents such as DMSO, DMF, and THF. P2, which contained fewer polar silylether terminal groups, was insol-uble in DMSO but solinsol-uble in relatively nonpolar solvents such as toluene, CH2Cl2, and CHCl3, in which P1 was insoluble. The length of the acyl chain of the terminal ester groups also had an influence on the solubility of the hyperbranched poly(ether imide)s. P3, which contained acetyl terminal groups, was soluble in DMSO and insol-uble in toluene, whereas the P4–P6 polymers, which contained longer acyl chains (6 –18 car-bons), were soluble in toluene and insoluble in DMSO.
SUMMARY
A hyperbranched poly(ether imide) that con-tained terminal phenolic groups was prepared by the one-step polymerization of an AB2 monomer containing a pair of phenolic groups and an aryl fluoride. The nucleophilic substitution of the flu-oride by the phenolate groups, which was acti-vated by the imide ring, resulted in the formation of an ether linkage and, subsequently, the
hyper-branched poly(ether imide) P1. A Kricheldorf-type polymerization reaction involving TBDMS protected groups and catalyzed by CsF was also carried out to give the corresponding hyper-branched poly(ether imide) P2, which contained terminal silylated phenols. As determined by a combination of model compound studies and 1H
NMR integration data, the DB of both P1 and P2 was approximately 55%. The terminal phenolic groups in P1 were easily modified, yielding hy-perbranched polymers that contained a variety of functional chain ends. Physical properties, such as the Tg and solubility of the hyperbranched poly(ether imide)s, were dependent on the nature of the chain ends.
The authors thank the National Science Council (Re-public of China; NSC 89-2113-M009-019) for its finan-cial support.
REFERENCES AND NOTES
1. (a) Malmstro¨m, E.; Hult, A. J Macromol Sci Rev Macromol Chem Phys 1997, 37, 555–579; (b) Kim, Y. H. J Polym Sci Part A: Polym Chem 1998, 36, 1685–1698; (c) Voit, B. J Polym Sci Part A: Polym Chem 2000, 38, 2505–2525 and references therein. 2. (a) Drohmann, C.; Mo¨ller, M.; Gorbatsevich, O. B.; Muzafarov, A. M. J Polym Sci Part A: Polym Chem 2000, 38, 741–751; (b) Lin, Q.; Long, T. E. J Polym Sci Part A: Polym Chem 2000, 38, 3736 –3741; (c) Emrick, T.; Chang, H.-T.; Fre´chet, J. M. J. J Polym Sci Part A: Polym Chem 2000, 38, 4850 – 4869; (d) Gong, C.; Fre´chet, J. M. J. J Polym Sci Part A: Polym Chem 2000, 38, 2970 –2978.
3. Flory, P. J. J Am Chem Soc 1952, 74, 2718 –2723. 4. (a) Fre´chet, J. M. J. Science 1994, 263, 1710 –1715;
(b) Advances in Dendritic Molecules; Newkome, G. R., Ed.; JAI: Greenwich, CT, 1994; Vol. 1; (c) Newkome, G. R.; Moorefield, C. N.; Vo¨gtle, F. Den-dritic Molecules: Concepts, Syntheses, Perspec-tives; VCH: Weinheim, 1996; (d) Zeng, F.; Zimmer-man, S. C. Chem Rev 1997, 97, 1681–1712; (e) Frey, H.; Lach, C.; Lorenz, K. Adv Mater 1998, 10, 279 – 293; (f) Crooks, R. M.; Zhao, M. Q.; Sun, L.; Chechik, V.; Yeung, L. K. Acc Chem Res 2001, 34, 181–190; (g) Hecht, S.; Fre´chet, J. M. J. Angew Chem Int Ed Engl 2001, 40, 74 –91.
5. (a) Tomalia, D. A.; Baker, H.; Dewald, J.; Hall, M.; Kallos, G.; Martin, S.; Roeck, J.; Ryder, J.; Smith, P. Polym J 1985, 17, 117–132; (b) Newkome, G. R.; Yao, Z.-Q.; Baker, G. R.; Gupta, V. K. J Org Chem 1985, 50, 2003–2004.
6. (a) Hawker, C. J.; Fre´chet, J. M. J. J Am Chem Soc 1990, 112, 7638 –7647; (b) Miller, T. M.; Neenan,
T. X. Chem Mater 1990, 2, 346 –349; (c) Xu, Z. F.; Moore, J. S. Angew Chem Int Ed Engl 1993, 32, 1354 –1357.
7. Polyimides; Wilson, D.; Stenzenberger, H. D.; Her-genrother, P. M., Eds.; Blackie, Chapman and Hall: New York, 1990.
8. Harris, F. W. In Polyimides; Wilson, D.; Stenzen-berger, H. D.; Hergenrother, P. M., Eds.; Blackie, Chapman and Hall: New York, 1990; Chapter 1, pp 1–37.
9. Williams, F. J.; Donahue, P. E. J Org Chem 1977, 42, 3414 –3419.
10. White, D. M.; Takekoshi, T.; Williams, F. J.; Relles, H. M.; Donahue, P. E.; Klopfer, H. J.; Louks, G. R.; Manello, J. S.; Matthews, R. O.; Schluenz, R. W. J Polym Sci Polym Chem Ed 1981, 19, 1635–1658. 11. Yamanaka, K.; Jikei, M.; Kakimoto, M.
Macromol-ecules 2000, 33, 6937– 6944.
12. (a) Kricheldorf, H. R.; Bier, G. J. J Polym Sci Polym Chem Ed 1983, 21, 2283–2286; (b) Kricheldorf, H. R.; Bier, G. J. Polymer 1984, 25, 1151–1156. 13. (a) Markoski, L. J.; Thompson, D. S.; Moore, J. S.
Macromolecules 2000, 33, 5315–5317; (b) Moore, J. S.; Thompson, D. S.; Markoski, L. J. PCT Int. Appl. WO 26267, 2000.
14. (a) Leu, M.; Chang, Y.-T.; Shu, F.; Teng, C.-F.; Shiea, J. Macromolecules 2000, 33, 2855–2861; (b) Leu, C.-M.; Shu, C.-F.; Teng, C.-F.; Shiea, J. Polymer 2001, 42, 2339 –2348.
15. Takekoshi, T. In Polyimides; Wilson, D.; Stenzen-berger, H. D.; Hergenrother, P. M., Eds.; Blackie,
Chapman and Hall: New York, 1990; Chapter 2, pp 38 –57.
16. Markezich, R. L.; Zamek, C. S. J Org Chem 1977, 42, 3431–3434.
17. Hawker, C. J.; Lee, R.; Fre´chet, J. M. J. J Am Chem Soc 1991, 113, 4583– 4588.
18. The relative integrations of the resonances as-signed to the methyl protons of the dendritic, lin-ear, and terminal subunits were 26, 45, and 29, respectively. The number of terminal units was slightly larger than the number of dendritic units. This may due to the oligomer content. The DB was 55% according to the equation DB⫽ (D ⫹ T)/(D ⫹ L ⫹ T), where D, L, and T represent the fractions of dendritic, linear, and terminal units, respectively. 19. Ho¨lter, D.; Burgath, A.; Frey, H. Acta Polym 1997,
48, 30 –35.
20. The relative integrations of the resonances associ-ated with the methyl protons of the dendritic, lin-ear, and terminal subunits of P2 were 26, 46, and 28, respectively. The DB was 54% according to the equation DB⫽ (D ⫹ T)/(D ⫹ L ⫹ T).
21. Hawker, C. J.; Chu, F. Macromolecules 1996, 29, 4370 – 4380.
22. Kim, Y. H.; Webster, O. W. Macromolecules 1992, 25, 5561–5572.
23. Schmaljohann, D.; Ha¨ußler, L.; Po¨tschke, P.; Voit, B. I.; Loontjens, T. J. A. Macromol Chem Phys 2000, 201, 49 –57.
24. Malmstro¨m, E.; Johansson, M.; Hult, A. Macromol Chem Phys 1996, 197, 3199 –3207.