Molecular elimination in photolysis of fluorobenzene at 193 nm: Internal energy of HF
determined with time-resolved Fourier-transform spectroscopy
Chia-Yan Wu, Yu-Jong Wu, and Yuan-Pern Lee
Citation: The Journal of Chemical Physics 121, 8792 (2004); doi: 10.1063/1.1802537 View online: http://dx.doi.org/10.1063/1.1802537
View Table of Contents: http://scitation.aip.org/content/aip/journal/jcp/121/18?ver=pdfcov
Published by the AIP Publishing Articles you may be interested in
Photodissociation of CH3CHO at 248 nm by time-resolved Fourier-transform infrared emission spectroscopy: Verification of roaming and triple fragmentation
J. Chem. Phys. 140, 064313 (2014); 10.1063/1.4862266
Gas-phase photodissociation of CH3COCN at 308 nm by time-resolved Fourier-transform infrared emission spectroscopy
J. Chem. Phys. 136, 044302 (2012); 10.1063/1.3674166
Molecular elimination in photolysis of o - and p -fluorotoluene at 193 nm: Internal energy of HF determined with time-resolved Fourier transform spectroscopy
J. Chem. Phys. 123, 224304 (2005); 10.1063/1.2131072
Three-center versus four-center elimination of haloethene: Internal energies of HCl and HF on photolysis of CF 2 CHCl at 193 nm determined with time-resolved Fourier-transform spectroscopy
J. Chem. Phys. 117, 9785 (2002); 10.1063/1.1518028
I. Three-center versus four-center HCl-elimination in photolysis of vinyl chloride at 193 nm: Bimodal rotational distribution of HCl (v7) detected with time-resolved Fourier-transform spectroscopy
J. Chem. Phys. 114, 160 (2001); 10.1063/1.1328736
Molecular elimination in photolysis of fluorobenzene at 193 nm:
Internal energy of HF determined with time-resolved
Fourier-transform spectroscopy
Chia-Yan Wu and Yu-Jong WuDepartment of Chemistry, National Tsing Hua University, Hsinchu 30013, Taiwan
Yuan-Pern Leea)
Department of Chemistry, National Tsing Hua University, Hsinchu 30013, Taiwan and Institute of Atomic and Molecular Sciences, Academia Sinica, Taipei, Taiwan
共Received 1 June 2004; accepted 11 August 2004兲
Following photodissociation of fluorobenzene (C6H5F) at 193 nm, rotationally resolved emission
spectra of HF(1⭐v⭐4) in the spectral region 2800–4000 cm⫺1 are detected with a step-scan Fourier-transform spectrometer. In the period 0.1–1.1s after photolysis, HF(v⭐4) shows similar Boltzmann-type rotational distributions corresponding to a temperature ⬃1830 K; a short extrapolation from data in the period 0.1– 4.1 s leads to a nascent rotational temperature of 1920⫾140 K with an average rotational energy of 15⫾3 kJ mol⫺1. The observed vibrational distribution of (v⫽1):(v⫽2):(v⫽3):(v⫽4)⫽(60⫾7):(24⫾3):(10.5⫾1.2):(5.3⫾0.5) corres-ponds to a vibrational temperature of 6400⫾180 K. An average vibrational energy of 33⫾9/3 kJ mol⫺1 is derived based on the observed population of HF(1⭐v⭐4) and an estimate of the population of HF(v⫽0) by extrapolation. The observed internal energy distribution of HF is consistent with that expected for the four-center 共␣,兲 elimination channel. A modified impulse model taking into account geometries and displacement vectors of transition states during bond breaking predicts satisfactorily the rotational excitation of HF. We also compare internal energies of HF observed in this work with those from photolysis of vinyl fluoride (CH2CHF) and 2-chloro-1,1-difluoroethene (CF2CHCl) at 193 nm. © 2004 American Institute of Physics.
关DOI: 10.1063/1.1802537兴
I. INTRODUCTION
Competition between three-center and four-center lecular elimination is the subject of extensive study of mo-lecular dynamics because internal distributions of the prod-ucts are distinct in each process.1–3 Photolysis of vinyl halides (CH2CHX, X⫽F, Cl, or Br兲 at 193 nm is a proto-typical system in which both three-center and four-center HX-elimination channels compete. Although experiments of photofragment translational spectroscopy provide detailed mechanisms of various dissociation channels,4 they provide no information to distinguish between these two molecular elimination channels for production of HX. Step-scan time-resolved Fourier-transform spectroscopy共TR-FTS兲 has been demonstrated to be more powerful than other techniques such as resonance-enhanced multiphoton ionization
共REMPI兲 or laser-induced fluorescence in determining the
internal-energy distributions of reaction products.2–7 With TR-FTS, we observed rotationally resolved emission of HX from photodissociation of vinyl halides at 193 nm; highly vibrationally and rotationally excited HX are attributed to production via three-center 共␣,␣兲 and four-center 共␣,兲 elimination channels.2,3 Extending the investigation to pho-tolysis of 2-chloro-1, 1-difluoroethene (CF2CHCl) at 193
nm, we observed emission of HCl(v⭐3) and HF(v⭐4) both with Boltzmann-type rotational distributions.5 Energy participation via the four-center elimination channel is non-statistical; a modified impulse model based on geometries and displacement vectors of transition states during bond breaking predicts satisfactorily the rotational excitation of HX observed in these investigations. A phenyl halide has a structure partially resembling that of a vinyl halide, hence it is of interest to investigate its molecular dissociation chan-nels and to test the model for dissociation dynamics of four-center elimination.
Photodissociation of phenyl halides in the ultraviolet
共UV兲 region is typically less complicated than that of
ben-zene or toluene,8 –10partly because of the possibility to excite nonbonding electrons of the halogen atom and partly because the bond energy of C– X (X⫽Cl, Br, or I兲 is smaller than that of the C–H bond on the phenyl ring. Excitation of the nonbonding electron of the halogen atom to an antibonding orbital leads to a prompt direct dissociation along the repul-sive surface, whereas excitation of the bonding electron in the phenyl ring leads to an excited state that might dissociate through various direct or indirect processes. Reported inves-tigations on C6H5Cl,11–15 C6H5Br,11,16and C6H5I共Refs. 11
and 17兲 all show that the major channel in photodissociation at 193 nm is fission of the C– X bond, with both direct and indirect channels being observed.
a兲Author to whom correspondence should be addressed. Present address:
Department of Applied Chemistry, National Chiao Tung University, Hsin-chu 30010, Taiwan. Electronic mail: [email protected]
8792
0021-9606/2004/121(18)/8792/8/$22.00 © 2004 American Institute of Physics This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to IP:
In contrast, although the F-elimination channel is ener-getically accessible at 193 nm共619 kJ mol⫺1兲,
C6H5F⫹hv→C6H5⫹F, ⌬Hⴰ⫽534 kJ mol⫺1, 共1兲
Huang et al.18 observed no evidence of this channel. They employed the multimass ion-imaging technique to find that HF-elimination,
C6H5F⫹hv→C6H4⫹HF, ⌬Hⴰ⫽283 kJ mol⫺1, 共2兲
is the major channel and H-elimination,
C6H5F⫹hv→C6H4F⫹H, ⌬Hⴰ⬵467 kJ mol⫺1, 共3兲
is a minor channel. Observed intensities of fragment ions C6H4 and C6H4F exhibit a ratio of ⬃25:1, indicating a
branching ratio likely greater than 0.9 for reaction 共2兲. Ex-perimental enthalpies of reaction are derived from enthalpies of formation for C6H5F,19 HF,20 C6H4 共o-benzyne兲,21,22and
C6H5 共Ref. 23兲; ⫺115.9⫾1.3, ⫺273.3⫾0.7, 440⫾10, and
339⫾8 kJ mol⫺1, respectively. An experimental enthalpy of formation of C6H4F is unknown; a value⬃352 kJ mol⫺1 is
estimated based on theoretical calculations.
We report here investigation of the HF-elimination chan-nel in photolysis of fluorobenzene at 193 nm with time-resolved Fourier-transform spectroscopy and compare the observed internal energy distribution of HF with other simi-lar four-center HF-photoelimination processes.18
II. EXPERIMENTS
The apparatus employed to obtain step-scan time-resolved Fourier-transform spectra has been described previously;5,24,25only a brief summary is given here. A lens mildly focused the photolysis beam from an ArF laser at 193 nm 共Lambda Physik, Optex兲 to ⬃3⫻4 mm2 at the reaction center with a fluence ⬃40 mJ cm⫺1. Filters passing either 2800– 4200 cm⫺1 or 3050–5000 cm⫺1 were employed. We used an InSb detector with a rise time of 0.7 s, and its transient signal was preamplified with a gain factor 105V A⫺1 共EG&G, Judson, PA9-50, 1.5 MHz兲, followed by amplification by a factor of 100共Stanford Research Systems, SRS560, 1 MHz兲 before being digitized with an external data-acquisition board共PAD1232, 12-bit ADC兲 at 50 ns reso-lution. Data were typically averaged over 27 laser pulses at each scan step; 2157 scan steps were performed to yield an interferogram resulting in a spectrum of resolution 2.0 cm⫺1. To improve the ratio of signal to noise (S/N) of the spec-trum, 10–30 consecutive time-resolved spectra were subse-quently summed to yield a satisfactory spectrum representing emission at intervals 0.5–1.5 s.
An IR laser beam at 5958 cm⫺1with a temporal width
⬃10 ns was directed into the spectrometer and temporal
pro-files for the amplified signal of the detector were recorded to yield the instrument response function of the detecting sys-tem. The IR laser beam was generated from the second Stokes shift of a dye laser共Spectra Physics, PDL-3兲 at 700.8 nm that was pumped with a Nd-YAG laser共Spectra Physics, Lab170兲; a single-pass cell filled with H2 at ⬃15 bar was
employed for Raman shifting. A delay of 1.3s was deter-mined for this specific combination of detector, preamplifier, and amplifier.
C6H5F was injected into the vacuum chamber as a dif-fusive beam through a slit-shaped inlet. The vapor pressure of C6H5F is ⬃77 Torr at 298 K and the partial pressure of
C6H5F in the chamber was⬃0.013 Torr. Ar 共Scott Specialty
Gases, 99.999%兲 in a minimal amount was added near the entrance port for the photolysis beam to suppress formation of solid deposit on the quartz window. With Ar purging, the pressure of the system was maintained ⬃35 mTorr. C6H5F 共Acros, 99%兲 was used without purification except for
degas-sing; no impurity was detected in its IR spectrum.
III. RESULTS AND ANALYSIS
We irradiated C6H5F at 0.26 Torr in a small static mul-tipass cell共⬃1.5 L, total path length 6.4 m兲 with an excimer laser at 193 nm 共⬍20 mJ, 20 Hz兲 for 120 s and recorded conventional infrared absorption spectra with the Fourier-transform spectrometer operating in a continuously scanning mode. The loss of C6H5F and formation of acetylene (C2H2,
3282 cm⫺1兲 and 1,3-butadiyne (C4H2, 3329 cm⫺1兲 were
readily identifiable, whereas absorption of HF and o-benzyne (C6H4) was absent.
Experiments on dynamics were performed with the Fourier-transform spectrometer operating in a step-scan mode. To maintain a nearly collisionless condition within 1.0 s period, the partial pressures of C6H5F共0.013 Torr兲 and Ar 共0.022 Torr兲 were reduced as much as possible while
main-taining a satisfactory S/N ratio; the partial pressure of C6H5F
in the photolysis region near the exit of the slit is likely greater than the observed average pressure in the chamber. C6H5F has an absorption cross section⬃1.9⫻10⫺17cm2 at
193 nm.26 An investigation of the dependence of signal in-tensity on the fluence of photolysis laser indicates that the signal intensity deviates from linearity when the laser fluence is greater than 50 mJ cm⫺2. With a fluence greater than 65 mJ cm⫺2, we recorded unresolved emission in the region 4000–10 000 cm⫺1 associated with multiphoton processes. Hence experiments performed only with a photolysis fluence⬍50 mJ cm⫺2were used to determine the internal en-ergy of HF. Although, based on time-resolved emission ex-periments, we are unable to exclude the slight possibility of production of HF from fragmentation of molecular ions that might be produced via a 1⫹1 REMPI process, we believe that such a contribution is negligible based on results from separate experiments using photofragmentation translational spectroscopy共to be published兲; observed translational energy of HF and C6H4 fit well with a single distribution and there
is no evidence of production of HF from fragmentation of the parent ion.
A. Infrared emission of HF
Figure 1 shows partial emission spectra of HF, at a reso-lution of 2.0 cm⫺1, recorded 0.1–0.6, 0.6 –1.1, 1.1–1.6, and 1.6 –2.1 s after photolyses of C6H5F 共0.013 Torr兲 and Ar 共0.022 Torr兲. The small intensities in periods before 1.1 s are partially due to the slow response of the detection sys-tem, the temporal evolution of the signal is described later. Assignments based on spectral parameters reported by Sen-gupta et al.27and Ram et al.28are shown as stick diagrams in
8793 J. Chem. Phys., Vol. 121, No. 18, 8 November 2004 Photolysis of fluorobenzene
Fig. 2; values of J
⬘
are indicated. The spectrum exhibits emission from HF with J⬘
up to 15 and v up to 4. Each vibrational-rotational line in the P branch was normalized with the instrument response function and divided by its re-spective Einstein coefficient29 to yield a relative population Pv(J⬘
). Partially overlapped lines, such as J⬘
⫽4, 6, 9, 11–13 of HF(v⫽1), J⬘
⫽3, 8, 9, 11–13 of HF(v⫽2), J⬘
⫽2, 7–9, 12 of HF(v⫽3), and J
⬘
⫽2, 7, 9 of HF(v⫽4),were deconvoluted to yield their intensities. Semilogarithmic plots of Pv(J
⬘
)/(2J⬘
⫹1) versus J⬘
(J⬘
⫹1) for HF(v⫽1 – 4) produced from C6H5F are shown in Fig. 3. Fitted
Boltzmann-type rotational distributions of HF, derived from the spectrum recorded in the range 0.1–1.1 s, yield rota-tional temperatures of 1840⫾90, 1830⫾110, 1830⫾90, and 1800⫾160 K for v⫽1 – 4, respectively; unless specified,
er-ror limits listed in this paper represent one standard deviation in fitting. Similar procedures were carried out for spectra averaged over 1.1–2.1, 2.1–3.1, and 3.1– 4.1 s. With a short extrapolation, we estimate that the nascent rotational temperature to be 1930⫾20, 1920⫾20, 1920⫾40, and 1910⫾10 K for v⫽1 – 4, respectively; an average rotational temperature of 1920⫾140 K is thus derived; an estimated error is listed.
We assume a Boltzmann distribution and associate an interpolated population with overlapped lines. Relative popu-lations obtained on counting levels up to observed Jmax in FIG. 1. Infrared emission spectra of HF in spectral region 2800– 4000 cm⫺1
recorded at varied intervals after photolysis of C6H5F共0.013 Torr兲 in Ar
共0.022 Torr兲 at 193 nm. 共A兲 0.1–0.6s;共B兲 0.6–1.1s;共C兲 1.1–1.6s;共D兲 1.6 –2.1s. Spectral resolution is 2.0 cm⫺1; 27 laser pulses were averaged at each scan step of the interferometer.
FIG. 2. Infrared emission spectra of HF in spectral region 2950–3950 cm⫺1recorded 0.1–1.1s after photolysis of C6H5F共0.013 Torr兲 in Ar 共0.022 Torr兲
at 193 nm. Spectral resolution is 2.0 cm⫺1; 27 laser pulses were averaged at each scan step of the interferometer. Assignments are shown as stick diagrams. FIG. 3. Semilogarithmic plots of relative rotational populations of HF(v
⫽1 – 4) after photolysis of C6H5F共0.013 Torr兲 in Ar 共0.022 Torr兲 at 193 nm.
Solid lines represent least-squares fits.
each vibrational level共referred as ‘‘observed data’’ hereafter兲 are listed as ⌺JPv(J) in Table I. We normalized values of
⌺JPv(J) associated with each vibrational state to yield a relative vibrational population (v⫽1):(v⫽2):(v⫽3):(v
⫽4)⫽(60⫾7):(24⫾3):(10.5⫾0.9):(5.3⫾0.4), correspond
to a vibrational temperature of 6400⫾180 K. Rotational en-ergies for each vibrational level Er(v) obtained on summing a product of rotational level energy and normalized popula-tion for each rotapopula-tional level, are also listed in Table I. An average rotational energy Er⫽13.7⫾1.6 kJ mol⫺1 for HF(v
⫽1 – 4) observed 0.1–1.1 s after photolysis is derived on summing a product of vibrational population and associated Er(v). After applying a correction factor 1920/1830⫽1.05 for rotational quenching, we derive a nascent rotational en-ergy of 14.4⫾1.7 kJ mol⫺1based on observed data.
The highest level of HF observed, J
⬘
⫽11 of v⫽4 has an energy 17 120 cm⫺1 above the ground vibrational level; this energy corresponds to Jmax(v)⫽26, 22, and 17 for v ⫽1 – 3, respectively. We assume a Boltzmann distributionand associate an extrapolated population with unobserved lines up to Jmax(v) for each vibrational level to derive a
re-vised population distribution, referred to as ‘‘extrapolated data’’ hereafter; the relative rotational population ⌺JPv(J) and rotational energy Er(v) thus derived are listed in paren-theses in Table I. An average rotational energy of 14.8⫾1.8 kJ mol⫺1 and a nascent rotational energy of Er⫽15.5
⫾1.8 kJ mol⫺1estimated with a small correction for
quench-ing are thus derived from extrapolated data. Takquench-ing this up-per limit into account, we report an average rotational en-ergy of HF as 15⫾3 kJ mol⫺1.
Assuming a Boltzmann distribution, we estimate the population of v⫽0 relative to v⫽1 to be 2.14 and 2.17 for observed and extrapolated data, respectively. The vibra-tional distribution of HF normalized for v⫽0 – 4 is thus (v⫽0):(v⫽1):(v⫽2):(v⫽3):(v⫽4)⫽56.2:26.2:10.6:4.6: 2.4 and 56.4:26.0:10.6:4.7:2.3 for observed and extrapolated data, respectively, as shown in Table I; the differences be-tween observed and extrapolated data are negligible, so only the distribution derived from observed data is shown in Fig. 4. The average vibrational energies of HF derived from ob-served and extrapolated data are Ev⫽32.7⫾2.4 kJ mol⫺1.
If the vibrational population has a smooth non-Boltzmann distribution, we may estimate a lower bound of the population of v⫽0 to be 1.3 times that of v⫽1. The average vibrational energy of Ev⫽42 kJ mol⫺1 thus derived might be taken as an upper limit. Taking this upper limit into account, we report an average vibrational energy of HF as 33⫾9/3 kJ mol⫺1.
B. Unresolved emission in the 2800–3500 cmÀ1region
A weak continuous emission in the range 2800–3400 cm⫺1was present at an early stage共t⭐5s兲 after irradiation
共Fig. 1兲 and diminished after t⭓20 s; the lower bound of the spectrum might be limited by the transmission of the IR filter. With the present detectivity and resolution, we are un-able to assign positively the carrier of this broad feature. A possible candidate is o-benzyne (C6H4). The C–H stretching modes of o-benzyne isolated in solid Ne absorb at 3049, 3071, 3086, and 3094 cm⫺1,30 consistent with the observed region of emission. Emission of the internally excited parent C6H5F cannot, however, be positively excluded because
C6H5F also absorbs in the region 3020–3130 cm⫺1.
C. Temporal profiles of emission
The temporal evolution of emission of HF and the broad feature produced from photolysis of C6H5F共0.013 Torr兲 with
Ar 共0.022 Torr兲 at 193 nm is shown in Fig. 5. The total intensity for emission of the broad feature was derived by integration of the spectrum with features associated with HF subtracted at each time interval, whereas that of HF was derived by integration of lines with the broad feature re-moved. Emission of the broad feature reaches a maximum at
⬃1.4 s, whereas that of HF reaches a maximum at⬃2.9 s; the variation is mainly due to a quenching rate for the broad feature共like o-benzyne兲 much greater than that for HF. We deconvoluted these profiles with the instrument response function and a mechanism consisting of formation and quenching in first order to derive rates of formation kf
⫽(1.5⫾0.2)⫻106 and (3.3⫾1.3)⫻106s⫺1 and rates of
quenching kq⫽(2.6⫾0.2)⫻104 and (1.1⫾0.4)⫻105s⫺1for FIG. 4. Relative vibrational distributions of HF(v⫽0 – 4) after photolysis of
C6H5F共0.013 Torr兲 in Ar 共0.022 Torr兲 at 193 nm. 䊉, from observed data; 䉭,
estimate by extrapolation. TABLE I. Fitted rotational temperature, rotational energy, and vibrational
population of HF(v) recorded 0.1–1.1s after photolysis of C6H5F at 193
nm.
v Trot共K) ⌺JPv(J)
a E
r(v) (kJ mol⫺1) Vibrational populationb
0 0.562c共0.564兲d 1 1840⫾90 1154c共1176兲d 14.1c共15.2兲d 0.262共0.260兲 2 1830⫾110 467共477兲 13.9共15.1兲 0.106共0.106兲 3 1830⫾90 204共214兲 12.5共14.5兲 0.046共0.047兲 4 1800⫾160 102共102兲 10.4共10.4兲 0.024共0.023兲 a
Pv(J)⫽共relative integrated emittance兲/关共instrumental response
factor兲共Ein-stein coefficient兲兴; arbitrary unit.
b
Normalized tov⫽0 – 4; populations of v⫽0 are estimated from
extrapo-lation.
c
Observed data. Only levels below observed Jmaxin each vibrational level
are included; see text.
d
Extrapolated data are listed parenthetically. All levels up to E
⫽17 120 cm⫺1above the ground level are included; see text.
8795
J. Chem. Phys., Vol. 121, No. 18, 8 November 2004 Photolysis of fluorobenzene
HF and the broad feature, respectively. Considering the error associated with the intensity of the broad feature, both HF and the broad feature have similar rates of production.
D. Calculations on transition states of C6H5F and branching ratios
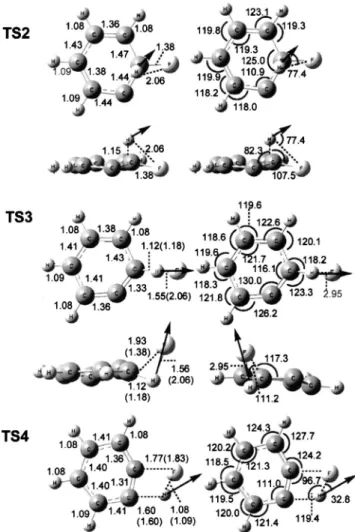
We performed calculations to optimize structures of tran-sition states for H-shift 共TS2兲, three-center 共TS3兲, and four-center 共TS4兲 elimination channels of C6H5F with the B3LYP/6-311G(d, p) density-functional theory31,32using the
GAUSSIAN 03program.33Geometries of transition states TS2, TS3, and TS4 of C6H5F and displacement vectors
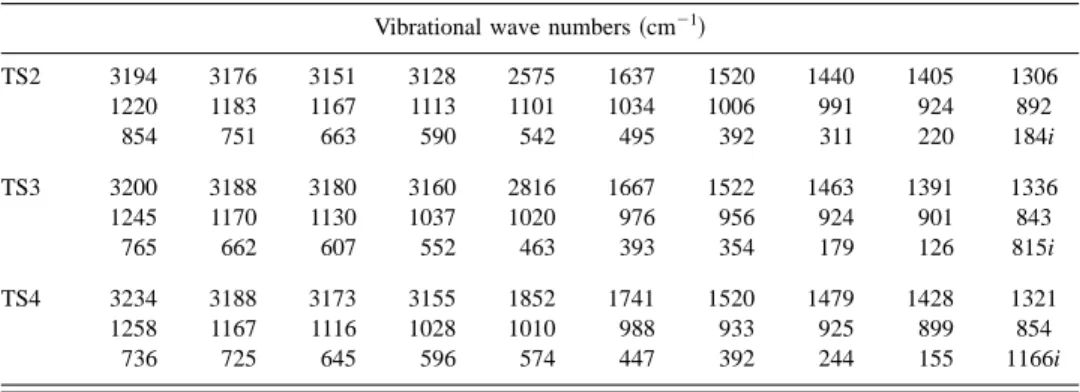
corre-sponding to imaginary vibrational wave numbers predicted with the B3LYP method are shown in Fig. 6; available data derived previously18 with B3LYP/6-31⫹G*are listed in pa-rentheses for comparison. Predicted vibrational wave num-bers for TS2, TS3, and TS4 of C6H5F are listed in Table II.
Relative energies and barriers of the H-shift and both mo-lecular elimination channels are shown in Fig. 7; estimates from a similar figure reported previously18 using CCSD/6-311⫹G*//B3LYP/6-31⫹G* are listed in parenthe-ses for comparison. Calculations using the B3LYP/6-311G(d, p) method yield barriers of 497 and 387 kJ mol⫺1 for three-center and four-center elimination channels, respec-tively, within 17 kJ mol⫺1of those predicted using the CCSD method. The three-center elimination proceeds via a transi-tion state TS2 with a barrier of 389 kJ mol⫺1 to form iso-C6H5F with energy nearly identical to TS2, followed by a
further barrier of 108 kJ mol⫺1to reach TS3.
We estimated rates of dissociation via these two chan-nels on the ground electronic surface of C6H5F with a mi-crocannonical transition-state theory. The Whitten-Rabinovitch equations34 were used to calculate density of states. Using barriers of 497 and 387 kJ mol⫺1 and vibra-tional wave numbers listed in Table II, we calculated rates of dissociation for four-center and three-center elimination channels to be 8.7⫻105 and 89 s⫺1, respectively; the latter was derived on assuming a steady-state of intermediate iso-C6H5F. Accordingly, production of HF via the three-center
elimination path on the ground electronic surface is negli-gible.
IV. DISCUSSION
In experiments with a static cell, only C2H2 and
1,3-butadiyne (C4H2) were observed as end products. These
products are likely produced from decomposition of C6H4
via secondary photolysis,35
C6H4→C2H2⫹C4H2, ⌬H⫽251 kJ mol⫺1. 共4兲 Enthalpies of formation for C6H4, C2H2, and C4H2are 440
⫾10, 226.7⫾0.8, and 464 kJ mol⫺1, respectively.36 The
ab-sence of HF might be due to reactions with the walls of the absorption cell or the multireflection optics in the cell. Ob-servation of time-resolved emission of HF and possibly C6H4 with TR-FTS clearly demonstrates its superiority over
conventional Fourier transform infrared共FTIR兲 spectrometer in investigating photodissociation processes.
Our previous investigation on photolysis of CH2CHF 共0.180 Torr兲 in Ar 共0.270 Torr兲 showed that rotational
quenching of HF(v,J) is small but nonnegligible. In this experiment we were able to use a total pressure as small as 0.035 Torr and employ a data acquisition window of 1.0 s FIG. 5. Temporal profiles of HF共䊐兲 and the continuum in a region 2850–
3450 cm⫺1共䊊兲 after photolysis of C6H5F共0.013 Torr兲 in Ar 共0.022 Torr兲 at
193 nm. IRF: instrument response function.
FIG. 6. Geometries of transition states for 1,2-H-shift 共TS2兲, three-center elimination 共TS3兲 and four-center elimination 共TS4兲 of C6H5F predicted
with the B3LYP/6-311G(d, p) method. TS2 and TS3 are nonplanar. Bond lengths共shown on the left兲 are in Å and bond angles 共shown on the right兲 are in degrees 共°兲. Results reported by Huang et al. using the B3LYP/6-31⫹G*method are shown in parentheses for comparison. Dis-placement vectors corresponding to imaginary wave numbers are shown with solid arrows.
so that observed rotational quenching is nearly negligible; the correction to rotational temperature due to quenching is only ⬃5%. Vibrational quenching is negligible under our experimental conditions. It should be noted that the pressure in the photolysis region might be slightly greater than the pressure determined with the gauge because of the pressure gradient in the system.
According to a previous report18 and RRKM 共Rice-Ramsperger-Kassel-Marcus兲 calculations, HF is produced via the four-center elimination channel of C6H5F. The
internal-energy distribution of HF observed in this work is also consistent with such a model, as discussed below.
A. Rotational energy of HF
Our previous experience2,3 indicates that average rota-tional and vibrarota-tional energies of HF, produced via the four-center elimination channel, do not agree with predictions us-ing phase-space theory37–39 and separate statistical ensemble40 models. A revised impulse model predicts satis-factorily the rotational energy of HX products. Assuming that the H atom receives most available energy, we distribute
available energy between H and C atoms during bond break-ing, followed by calculations of translational and rotational energy of HF according to classical mechanics. Instead of assuming that the H atom moves along the direction of the breaking bond as in the standard impulse model, we consider motions associated with the reaction coordinates described by displacement vectors associated with the imaginary vibra-tional frequency of transition state TS4 of C6H5F. The
direc-tion of the H atom is assumed to follow the displacement vector shown in Fig. 6. Rotational energies are predicted with this modified impulse model according to the equation Erot⫽关mFmC/共mH⫹mF兲共mH⫹mC兲兴Eavailsin2•␣, 共5兲
in which Eavailis the available energy共exit barrier兲 and␣is the torque angle between the direction of motion of H and that of the H–F bond. With available energies of 56 and 166 kJ mol⫺1and␣⫽32.8° and 3.0° predicted for four-center and three-center elimination channels, respectively, rotational en-ergies of 14.4 and 0.4 kJ mol⫺1are predicted. The former is nearly identical to the experimental value of 15⫾3 kJ mol⫺1. Considering possible errors associated with the estimated exit barrier and the relatively simply impulse model, the agreement is satisfactory. The average rotational energy of 15⫾3 kJ mol⫺1for HF implies that a small fraction of total available energy is partitioned into HF, with fr⬵0.045
⫾0.009.
B. Vibrational energy of HF
The average vibrational energy of 33⫾9/3 kJ mol⫺1for HF implies that a moderate fraction of available energy is partitioned into HF, with fv⬵0.10⫾0.03/0.01. The partition of vibrational energy depends on the deviation of the dis-tance between two bond-forming atoms from the equilibrium bond length. Predicted distances between H and F for TS3 and TS4 in dissociation of C6H5F are 1.56 and 1.08 Å, re-spectively. Because the equilibrium bond distance of HF is 0.9168 Å 共Ref. 41兲, one would expect that the vibrational distribution of HF produced via the three-center elimination be highly inverted, in contrast to the vibrational distribution observed in this work. As a comparison, the distance be-tween H and F in TS4 for four-center elimination of CH2CHF is predicted to be 1.281 Å and the observed
vibra-tional population of HF has a maximum near v⫽1 or 2.5 FIG. 7. Energies共in kJ mol⫺1兲 of transition states 共TS2, TS3, and TS4兲 and
dissociation products relative to C6H5F. Energies are predicted with the
B3LYP/6-311G(d, p) method; those from Ref. 18 using CCSD/6-311⫹G*//B3LYP/6-31⫹G*are shown in parentheses for comparison. The experimental value is in brackets.
TABLE II. Vibrational wave numbers共cm⫺1兲 of transition states TS2, TS3, and TS4 of C6H5F predicted with
the B3LYP/6-311G(d, p) method.
Vibrational wave numbers共cm⫺1兲
TS2 3194 3176 3151 3128 2575 1637 1520 1440 1405 1306 1220 1183 1167 1113 1101 1034 1006 991 924 892 854 751 663 590 542 495 392 311 220 184i TS3 3200 3188 3180 3160 2816 1667 1522 1463 1391 1336 1245 1170 1130 1037 1020 976 956 924 901 843 765 662 607 552 463 393 354 179 126 815i TS4 3234 3188 3173 3155 1852 1741 1520 1479 1428 1321 1258 1167 1116 1028 1010 988 933 925 899 854 736 725 645 596 574 447 392 244 155 1166i 8797
J. Chem. Phys., Vol. 121, No. 18, 8 November 2004 Photolysis of fluorobenzene
With a distance of 1.08 Å for TS4, one would expect the v
⫽0 or 1 level of HF product to have the greatest population.
Hence, the observed vibrational distribution of HF fits satis-factorily with the four-center elimination mechanism.
C. Energy balance
In previous work using multimass imaging detection, the distribution of translational energy release of reaction 共2兲 upon photolysis at 193 nm is reported to show a maximum at 155 kJ mol⫺1 and a decreasing population extending to the maximum available energy ⬃336 kJ mol⫺1.18 We estimated an average translational energy of⬃146 kJ mol⫺1from their distribution plot; this value implies that the fraction of en-ergy partitioned into translation is ft⬵0.43.
With an available energy of 336 kJ mol⫺1 and an ob-served average translational release of 146 kJ mol⫺1, 190 kJ mol⫺1is distributed between internal energies of HF and C6H4. Our observation of ⬃48 kJ mol⫺1 in internal energy of HF implies that C6H4 has an average internal energy of 142 kJ mol⫺1. If available energy above the barrier were dis-tributed statistically, one would expect that most energy would be partitioned to C6H4 because of its high degree of
freedom relative to the other fragment HF. Our observation clearly indicates that the statistical model fails in this case. The dissociation energy of C6H4 to form C2H2 and C4H4,
C6H4→C2H2⫹C4H2, 共4兲
is ⬃251 kJ mol⫺1; hence most C6H4 undergoes no further
dissociation unless it absorbs a second photon.
We noticed that the previously reported translational en-ergy of 146 kJ mol⫺1for the C6H4⫹HF channel, determined
from the velocity distribution of C6H4, is atypically large; the exit barriers predicted with B3LYP/6-311G(d, p) in this work and CCSD/6-311⫹G*//B3LYP/6-31⫹G* are only 56 and 63 kJ mol⫺1, respectively. The reason for this discrep-ancy is unclear. Possibilities include interference from mul-tiphoton processes, decomposition of some C6H4 with a
large internal energy 共hence a small kinetic energy兲, signifi-cant access of regions well above TS4, and an unexpectedly large error in calculations of the exit barrier for the four-center elimination channel. Further study using a molecular beam system utilizing synchrotron radiation for ionization is scheduled. Although the exit barrier of the three-center elimi-nation, 166 kJ mol⫺1, fits satisfactorily with the observed
av-erage translational energy, this possibility is excluded based on the internal-energy distribution of HF observed in this work and RRKM calculations.
D. Rate of production and quenching
Huang et al. reported a rate of (1.4⫾0.8)⫻106s⫺1 for
production of C6H4.18After deconvolution of temporal pro-files of HF with the instrument response function, we ob-tained a rate of formation of (1.5⫾0.2)⫻106s⫺1 for HF, nearly identical to their results.
According to Huang et al.,18 absorption of 193 nm pho-tons corresponds to excitation of C6H5F to the S3 state,
which internally converts to the ground S0state, followed by
four-center HF-elimination共major channel兲 and C–H fission
共minor channel兲.18They performed RRKM calculations and
obtained rates of formation of HF via three-center and four-center elimination channels to be 2.2⫻103 and 1.1
⫻106s⫺1, respectively; a steady-state approximation was
employed for the intermediate formed after a 1,2-H shift and before the three-center elimination. The rate of formation of HF based on energies predicted with B3LYP/6-311G(d, p) for four-center elimination is 8.7⫻105s⫺1, consistent with experimental and previous calculation results.
The observed rate of quenching for HF, (2.6⫾0.2)
⫻104s⫺1, at关C
6H5F兴⫽4.2⫻1014molecule cm⫺3implies an
apparent bimolecular rate coefficient for vibrational quench-ing kqII⫽(6.3⫾0.5)⫻10⫺11cm3molecule⫺1s⫺1 by C6H5F;
vibrational quenching of HF by Ar is much smaller than that by C6H5F.
42
This rate represents mainly quenching from the v⫽1 state of HF. We did not study detailed rates of quench-ing for each individual vibrational state. A rate of quenchquench-ing for the broad feature greater than that of HF is conceivable, as the possible carrier C6H4 for the broad feature is more
complex than HF and is similar to C6H5F; hence quenching of C6H4 is expected to proceed more readily.
E. Comparison with photolysis of other fluoro-compounds
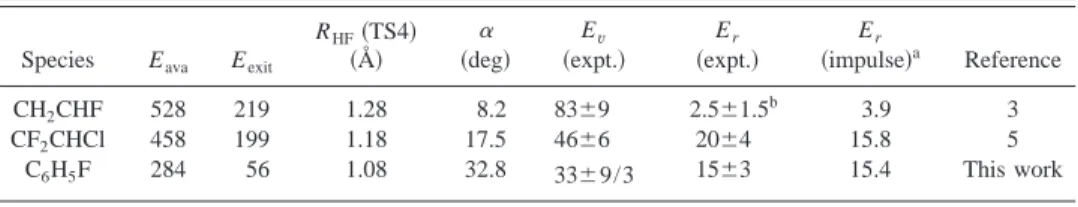
The observed rotational energies of HF from four-center elimination channels of C6H5F and CF2CHCl are compared
with those predicted according to the modified impulse model in Table III. Although the total available energies and the exit barriers for these two systems vary substantially,
TABLE III. Comparison of total available energies, geometry of transition states, observed average internal energies, and predicted rotational energies of HF using a revised impulse model. All energies are in unit of kJ mol⫺1. Eava, available energy; Eexit, exit barrier; ␣, torque angle of TS4; Ev, vibrational energy; Er,
rotational energy.
Species Eava Eexit
RHF共TS4兲 共Å兲 共deg兲␣ Ev 共expt.兲 Er 共expt.兲 Er 共impulse兲a Reference CH2CHF 528 219 1.28 8.2 83⫾9 2.5⫾1.5 b 3.9 3 CF2CHCl 458 199 1.18 17.5 46⫾6 20⫾4 15.8 5 C6H5F 284 56 1.08 32.8 33⫾9/3 15⫾3 15.4 This work aAssuming that the motion of H atom is along the displacement vector corresponding to the imaginary mode of
the transition state; see text.
bRecent trajectory calculations共Ref. 43兲 indicate that the four-center elimination channel produces high-J
components; see text.
observed rotational energies of HF agree satisfactorily with those predicted with the modified impulse model. Rotational excitation of HF produced from four-center elimination of CH2CHF is not compared because recent trajectory
calcula-tions indicate that the four-center elimination produces more rotationally excited HF and the low-J component of HF ob-served previously3might be due to quenching.43 Further ex-periments are needed to clarify this discrepancy. That HF produced from CF2CHCl and C6H5F, both via four-center
elimination, has greater rotational energy is consistent with a significant torque angle in their TS4. The torque angle of 32.8° for TS4 of C6H5F is nearly twice that of CF2CHCl, but its exit barrier is only 56/199⫽0.28 that for CF2CHCl; the resultant rotational energies of HF in these two systems are consequently similar.
Likewise, average vibrational energies of HF via four-center elimination of these molecules are compared in Table III. HF produced via four-center elimination of CH2CHF has
more vibrational energy 共83⫾9 kJ mol⫺1兲 than that 共48⫾6 kJ mol⫺1兲 from CF2CHCl because predicted bond distances
of TS4 are 1.28 and 1.18 Å, respectively. Calculated wave functions also indicate that the H-F bond is ‘‘formed’’ in TS4 of CF2CHCl and C6H5F, whereas the H-F bond in TS4 of
CH2CHF are not yet formed. The vibrational energy of HF,
33⫾9/3 kJ mol⫺1, from photolysis of C6H5F is the smallest,
consistent with the shortest H-F distance 1.08 Å calculated for TS4; it is nearest the equilibrium distance of HF.
V. CONCLUSION
Rotationally resolved emission of HF up tov⫽4 is ob-served after photolysis of C6H5F at 193 nm; HF is likely
produced from the four-center elimination channel on the ground electronic surface. The average rotational energy of 15⫾3 kJ mol⫺1 and vibrational energy of 33⫾9/3 kJ mol⫺1 for HF implies that a moderate fraction of available energy is partitioned into the internal energy of HF, with fv⬵0.10
⫾0.03/0.01 and fr⬵0.045⫾0.009. A modified impulse model considering displacement vectors of transition states during bond breaking predicts the average rotational energy of HF satisfactorily for four-center elimination channels of CF2CHCl and C6H5F. Partition of vibrational energy into
HF upon photolysis is also consistent with distances of H-F predicted for transition states of four-center elimination for these systems.
ACKNOWLEDGMENTS
The authors thank the National Science Council of Tai-wan 共Grant No. NSC92-2113-M-007-034兲 and the Ministry of Education of Taiwan 共Program for Promoting Academic Excellence of Universities, Grant No. 89-FA-04-AA兲 for support and the National Center for High Performance Com-puting for computer time.
1
Y.-P. Lee, Annu. Rev. Phys. Chem. 54, 215共2003兲.
2S.-R. Lin, S.-C. Lin, Y.-C. Lee, Y.-C. Chou, I-C. Chen, and Y.-P. Lee, J.
Chem. Phys. 114, 160共2001兲.
3S.-R. Lin, S.-C. Lin, Y.-C. Lee, Y.-C. Chou, I-C. Chen, and Y.-P. Lee, J.
Chem. Phys. 114, 7396共2001兲.
4D. A. Blank, W. Sun, A. G. Suits, Y. T. Lee, S. W. North, and G. E. Hall,
J. Chem. Phys. 108, 5414共1998兲.
5C.-Y. Wu, C.-Y. Chung, Y.-C. Lee, and Y.-P. Lee, J. Chem. Phys. 117,
9785共2002兲.
6C.-Y. Wu, Y.-P. Lee, J. F. Ogilvie, and N. S. Wang, J. Phys. Chem. A 107,
2389共2003兲.
7C.-Y. Wu, Y.-P. Lee, and N. S. Wang, J. Chem. Phys. 120, 6957共2004兲. 8A. M. Mebel, M. C. Lin, D. Chakraborty, J. Park, S. H. Lin, and Y. T. Lee,
J. Chem. Phys. 114, 8421共2001兲, and references therein.
9S.-T. Tsai, C.-L. Huang, Y. T. Lee, and C.-K. Ni, J. Chem. Phys. 115, 2449
共2001兲.
10C.-K. Lin, C.-L. Huang, J.-C. Jiang, H. H. Chang, Y. T. Lee, S. H. Lin, and
C.-K. Ni, J. Am. Chem. Soc. 124, 4068共2002兲.
11
A. Freeman, S. C. Yang, M. Kawasaki, and R. Bersohn, J. Chem. Phys. 72, 1028共1980兲.
12T. Ichimura, Y. Mori, H. Shinohara, and N. Nishi, Chem. Phys. 189, 117
共1994兲.
13
T. Ichimura, Y. Mori, H. Shinohara, and N. Nishi, Chem. Phys. Lett. 122, 55共1985兲.
14K.-L. Han, G.-Z. He, and N.-Q. Lou, Chem. Phys. Lett. 203, 509共1993兲. 15G.-J. Wang, R.-S. Zhu, H. Zhang, K.-L. Han, G.-Z. He, and N.-Q. Lou,
Chem. Phys. Lett. 288, 429共1998兲.
16
H. Zhang, R.-S. Zhu, G.-J. Wang, K.-L. Han, G.-Z. He, and N.-Q. Lou, J. Chem. Phys. 110, 2922共1999兲.
17M. Dzvonik, S. Yang, and R. Bersohn, J. Chem. Phys. 61, 4408共1974兲. 18C.-L. Huang, J.-C. Jiang, A. M. Mebel, Y. T. Lee, and C.-K. Ni, J. Am.
Chem. Soc. 125, 9814共2003兲.
19CRC Handbook of Chemistry and Physics, 78th ed., edited by D. R. Lide
共RC, New York, 1997兲.
20M. W. Chase, Jr., J. Phys. Chem. Ref. Data Monogr. 9,共1998兲. 21J. M. Riveros, S. Ingeman, and N. M. M. Nibbering, J. Am. Chem. Soc.
113, 1053共1991兲.
22P. G. Wenthold and R. R. Squires, J. Am. Chem. Soc. 113, 7414共1991兲;
116, 6401共1994兲.
23W. Tsang, in Energetics of Organic Free Radicals, edited by J. A.
Mar-tinho Simoes, A. Greenberg, and J. F. Liebman共Blackie Academic and Professional, London, 1996兲, Vol 4, pp. 22–58.
24P.-S. Yeh, G.-H. Leu, Y.-P. Lee, and I.-C. Chen, J. Chem. Phys. 103, 4879
共1995兲.
25S.-R. Lin and Y.-P. Lee, J. Chem. Phys. 111, 9233共1999兲. 26
K. Bowden and E. A. Braude, J. Chem. Soc. 1068共1952兲.
27U. K. Sengupta, P. K. Das, and K. N. Rao, J. Mol. Spectrosc. 74, 322
共1979兲.
28R. S. Ram, Z. Morbi, B. Guo, K.-Q. Zhang, P. F. Bernath, J. V. Auwera, J.
W. C. Johns, and S. P. Davis, Astrophys. J., Suppl. Ser. 103, 247共1996兲.
29
E. Arunan, D. W. Setser, and J. F. Ogilvie, J. Chem. Phys. 97, 1734
共1992兲.
30J. G. Radziszewski, B. A. Hess, Jr., and R. Zahradnik, J. Am. Chem. Soc.
114, 52共1992兲.
31
A. D. Becke, J. Chem. Phys. 98, 5648共1993兲.
32C. Lee, W. Yang, and R. G. Parr, Phys. Rev. B 37, 785共1988兲. 33M. J. Frisch, G. W. Trucks, H. B. Schlegel et al.,
GAUSSIAN03, Revision A.1
共Gaussian Inc., Pittsburgh, PA, 2003兲.
34
K. A. Holbrook, M. J. Pilling, and S. H. Robertson, Unimolecular
Reac-tions, 2nd ed.共Chichester, New York, 1996兲.
35W.-Q. Deng, K.-L. Han, J.-P. Zhan, and G.-Z. He, Chem. Phys. Lett. 288,
33共1998兲.
36H. Y. Afeefy, J. F. Liebman, and S. E. Stein, in Neutral Thermochemical
Data, NIST Chemistry WebBook, NIST Standard Reference Database
Number 69, edited by P. J. Linstrom and W. G. Mallard共National Institute of Standards and Technology, Gaithersburg, MD, 2003兲, p. 20899; http:// webbook.nist.gov
37P. Pechukas and J. C. Light, J. Chem. Phys. 42, 3281共1965兲. 38
C. E. Klots, J. Phys. Chem. 75, 1526共1971兲.
39M. Hunter, S. A. Reid, D. C. Robie, and H. Reisler, J. Chem. Phys. 99,
1093共1993兲.
40C. Wittig, I. Nadler, H. Reisler, M. Noble, J. Catanzarite, and G.
Radhakrishnan, J. Chem. Phys. 83, 5581共1985兲.
41
K. P. Huber and G. Herzberg, Constants of Diatomic Molecules 共Van Nostrand, Princeton, 1979兲.
42S. R. Leone, J. Phys. Chem. Ref. Data 11, 953 共1982兲, and references
therein.
43
E. Martı´nez-Nu´n˜ez and S. A. Va´zqucz, J. Chem. Phys. 121, 5179共2004兲.
8799 J. Chem. Phys., Vol. 121, No. 18, 8 November 2004 Photolysis of fluorobenzene