CASE REPORT
Pure partial monosomy 3p (3p25.3pter): prenatal diagnosis and array
comparative genomic hybridization characterization
Chih-Ping Chen a,b,c,d,e,f,g *, Yi-Ning Su h, Chen-Yu Chen b, Jun-Wei Su b,i, Schu-Rern Chern c, Dai-Dyi Town b and Wayseen Wang c,j
a Department of Medicine, Mackay Medical College, New Taipei City, Taiwan
b Department of Obstetrics and Gynecology, Mackay Memorial Hospital, Taipei, Taiwan c Department of Medical Research, Mackay Memorial Hospital, Taipei, Taiwan
d Department of Biotechnology, Asia University, Taichung, Taiwan
e School of Chinese Medicine, College of Chinese Medicine, China Medical University, Taichung, Taiwan f Institute of Clinical and Community Health Nursing, National Yang-Ming University, Taipei, Taiwan g Department of Obstetrics and Gynecology, School of Medicine, National Yang-Ming University, Taipei,
Taiwan
h Department of Medical Genetics, National Taiwan University Hospital, Taipei, Taiwan
i Department of Obstetrics and Gynecology, China Medical University Hospital, Taichung, Taiwan j Department of Bioengineering, Tatung University, Taipei, Taiwan
* Correspondence to: Chih-Ping Chen, MD
Department of Obstetrics and Gynecology, Mackay Memorial Hospital 92, Section 2, Chung-Shan North Road, Taipei, Taiwan
Tel: +886-2-25433535; Fax: +886-2-25433642, +886-2-25232448 E-mail: [email protected]
Abstract
Objective: To present prenatal diagnosis and molecular cytogenetic characterization of pure partial monosomy 3p (3p25.3pter) by array comparative genomic hybridization (aCGH) and quantitative fluorescent polymerase chain reaction (QF-PCR) on uncultured amniocytes.
Case Report: A 35-year-old, gravida 2, para 0, woman underwent amniocentesis at 19 weeks of gestation because of advanced maternal age. Her husband was 37 years old. She had experienced one intrauterine fetal death. Amniocentesis during this pregnancy revealed a distal deletion of chromosome 3p. The parental karyotypes were normal. Prenatal ultrasound findings were unremarkable. At 22 weeks of gestation, she underwent repeated amniocentesis, and aCGH investigation using CytoChip Oligo Array (BlueGnome, Cambridge, UK) on uncultured amniocytes revealed a 9.29-Mb deletion of 3p26.3p25.3 [arr 3p26.3p25.3 (64,096 – 9,357,258 bp)1] encompassing the genes of CHL1, CNTN4, CRBN, LRRN1, ITPR1 and SRGAP3 but not involving the markers D3S1263 and D3S3594. Polymorphic DNA marker analysis on uncultured amniocytes showed a paternal origin of the deletion. Cytogenetic analysis of cultured amniocytes revealed a karyotype of 46,XX,del(3)(p25.3). At 24 weeks of gestation, prenatal ultrasound findings of the brain, heart and other internal organs were unremarkable. The pregnancy was subsequently terminated, and an 886-g female fetus was delivered with brachycephaly, hypertelorism, a short and thick nose, micrognathia and low-set ears.
Conclusion: In this case, aCGH has characterized a 3p deleted region with haploinsufficiency of the neuro-developmental genes associated with cognitive deficit and mental retardation but without involvement of the CHD susceptibility locus, and QF-PCR has determined a paternal origin of the deletion. aCGH and QF-PCR help to delineate the genomic imbalance in prenatally detected de novo chromosome aberration, and the information acquired is useful for genetic counseling.
Key words: 3p deletion syndrome, CHL1, CNTN4, CRBN, ITPR1, LRRN1, mental retardation, monosomy 3p, prenatal diagnosis, SRGAP3
Introduction
The 3p deletion syndrome (OMIM 613792) is a contiguous gene syndrome associated with partial monosomy 3p (3p25pter) and the characteristic phenotypic features of mental retardation, developmental delay, intrauterine growth restriction, micro- and brachycephaly, a triangular face, hypertelorism, epicanthus, upturned palpebral fissures, palpebral ptosis, frontal bossing, a short and thick nose, micrognathia, low-set ears, hypertrichosis, synophrys, long philtrum, and variable associated abnormalities such as pectus excavatum, scoliosis, hypogenitalia, polydactyly, syndactyly, clinodactyly, atrioventricular septal defects, hiatal hernia, optic atrophy, polycystic renal dysplasia and hypoplastic clavicles [Chen et al, 1996, 2005, 2011; Shuib et al, 2009]. Here, we present our experience of prenatal diagnosis and array comparative genomic hybridization (aCGH) characterization of pure partial monosomy 3p (3p25.3pter) in a fetus.
Case Report
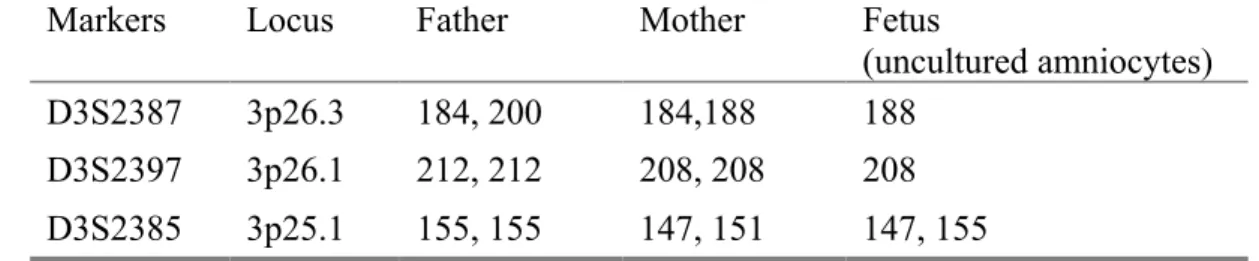
A 35-year-old, gravida 2, para 0, woman underwent amniocentesis at 19 weeks of gestation because of advanced maternal age. Her husband was 37 years old. She had experienced one intrauterine fetal death. Amniocentesis during this pregnancy revealed a distal deletion of chromosome 3p. The parental karyotypes were normal. Prenatal ultrasound findings were unremarkable. At 22 weeks of gestation, she underwent repeated amniocentesis, and aCGH investigation using CytoChip Oligo Array (BlueGnome, Cambridge, UK) on uncultured amniocytes revealed a 9.29-Mb deletion of 3p26.3p25.3 [arr 3p26.3p25.3 (64,096 – 9,357,258 bp)1] encompassing the genes of CHL1, CNTN4, CRBN, LRRN1, ITPR1 and SRGAP3 (Fig. 1). Polymorphic DNA marker analysis on uncultured amniocytes showed a paternal origin of the deletion (Fig. 2, Table 1). Cytogenetic analysis of cultured amniocytes revealed a karyotype of 46,XX,del(3)(p25.3) (Fig. 3). At 24 weeks of gestation, prenatal ultrasound findings of the brain, heart and other internal organs were unremarkable. The pregnancy was subsequently terminated, and an 886-g female fetus was delivered with brachycephaly, hypertelorism, a short and thick nose, micrognathia and low-set ears (Fig. 4).
Discussion
The present case was not associated with congenital heart disease (CHD). The deleted segment of chromosome 3p in this case did not involve the candidate CHD critical region associated with the 3p deletion syndrome. Green et al [2000] mapped the 3p25 CHD susceptibility locus to a 3.7 cM distance between D3S1263 (11,492,252 – 11,492,482 bp) and D3S3594 (10,626,638 – 10,626,908 bp). Shuib et al [2009] additionally mapped the 3p25 CHD susceptibility locus to an ~ 200 kb interval between the deletion size of 11.35 Mb (with CHD) and the deletion size of 11.15 Mb (without CHD) by analysis of cases with a deletion of 3p25.3-pter. The present case had a deletion size about 9.35 Mb, and it is likely that a CHD susceptibility gene was not involved within this deletion.
The present case had haploinsufficiency of the neuro-developmental genes of CHL1, CNTN4, CRBN, LRRN1, ITPR1 and SRGAP3. CHL1 (OMIM 607416) encodes cell adhesion molecule L1-like (CALL) protein. CALL is highly expressed in the central nervous system and the peripheral nervous system, and interruption or loss of CALL may cause cognitive deficit [Frints et al, 2003; Shrimpton et al, 2006]. In a study of genotype-phenotype correlation of terminal 3p deletions in two families, Pohjola et al [2010] concluded that a small terminal 3p deletion encompassing only the CHL1 gene may cause only mild mental deficit, mild learning difficulty, microcephaly and growth retardation, but may not be related to profound mental retardation and dysmorphisms.
CNTN4 (OMIM 607280) encodes contactin 4 that belongs to an axon-associated cell adhesion molecule of immunoglobulin superfamily and plays a role in the formation, maintenance and plasticity of functional neuronal networks [Yoshihara et al, 1995; Saito et al, 1998]. Dijkhuizen et al [2006] suggested that loss of CNTN4 and CRBN contributes to mental retardation in the 3p deletion syndrome. Fernandez et al [2004, 2008] suggested that disruption or haploinsufficiency of CNTN4 causes characteristic physical features of the 3p deletion syndrome including developmental delay, dysmorphic features and growth retardation.
CRBN (OMIM 609262) encodes cereblon that is a binding protein for large-conductance calcium-activated potassium channel in the brain [Jo et al, 2005]. Autosomal recessive non-syndromic mental retardation-2 (MRT2) (OMIM 607417) can be caused by homozygous
mutation in CRBN [Higgins et al, 2000, 2004, 2008]. LRRN1 is required for formation of the midbrain-hindbrain boundary during neuronal development [Andreae et al, 2007; Tossell et al, 2011]. ITPR1 (OMIM 147265) encodes inositol 1,4,5-trisphosphate receptor type 1 which releases calcium ions from intracellular stores and modulates intracellular calcium signaling [Berridge, 1993]. Cargile et al [2002] suggested that ITPR1 is a candidate gene for mental retardation in the 3p deletion syndrome.
SRGAP3 (OMIM 606525) encodes Slit-Robo Rho GTPase-activating protein 3 which is involved in the Slit-Robo pathway regulating neuronal migration and axonal branching. Endris et al [2002] reported a patient with a balanced de novo translocation t(X;3)(p11.2;p25) with hypotonia and severe mental retardation. Endris et al [2002] found that the chromosome breakpoint interrupted the SRGAP3 gene in their patient with the 3p deletion syndrome and suggested that haploinsufficiency of SRGAP3 may lead to misregeneration of neuronal signal transduction and abnormal development of neuronal structures. Shuib et al [2009] analyzed 14 patients with cytogenetically detectable deletions of 3p25 and mapped a candidate critical region for mental retardation to an ~ 1 Mb interval containing only SRGAP3 rather than CHL1, CNTN4, CRBN and ITPR1. Shuib et al [2009] suggested that SRGAP3 is a major determinant of mental retardation in the 3p deletion syndrome.
In conclusion, we present prenatal diagnosis and aCGH characterization of pure partial monosomy 3p (3p25.3pter) in a fetus with haploinsufficiency of a candidate critical region for cognitive deficit and mental retardation in the 3p deletion syndrome. In this case, aCGH has characterized a 3p deleted region with involvement of the neuro-developmental genes of CHL1, CNTN4, CRBN, LRRN1, ITPR1 and SRGAP3 but without involvement of the CHD susceptibility locus, and QF-PCR has determined a paternal origin of the deletion. aCGH and QF-PCR help to delineate the genomic imbalance in prenatally detected de novo chromosome aberration, and the information acquired is useful for genetic counseling.
Acknowledgements
This work was supported by research grants NSC-99-2628-B-195-001-MY3 from the National Science Council and MMH-E-100-04 from Mackay Memorial Hospital, Taipei, Taiwan.
References
1. Chen C-P, Liu F-F, Jan S-W, Lin S-P, Lan C-C. Prenatal diagnosis of partial monosomy 3p and partial trisomy 2p in a fetus associated with shortening of long bones and a single umbilical artery. Prenat
Diagn 1996; 16: 270-5.
2. Chen C-P, Lin S-P, Ho C-S, Chern S-R, Lee C-C, Chen W-L, et al. Distal 3p monosomy associated with epilepsy in a boy. Genet Counsel 2005; 16: 429-32.
3. Chen C-P, Su Y-N, Hsu C-Y, Chern S-R, Lee C-C, Chen Y-T, et al. Mosaic deletion-duplication syndrome of chromosome 3: prenatal molecular cytogenetic diagnosis using cultured and uncultured amniocytes and association with fetoplacental discrepancy. Taiwan J Obstet Gynecol 2011; 50: 485-91.
4. Shuib S, Mcmullan D, Rattenberry E, Barber RM, Rahman F, Zatyka M, et al. Microarray based analysis of 3p25-p26 deletions (3p- syndrome). Am J Med Genet 2009; 149A: 2099-105.
5. Green EK, Priestly MD, Waters J, Maliszewska C, Latif F, Maher ER. Detailed mapping of a congenital heart disease gene in chromosome 3p25. J Med Genet 2000; 37: 581-7.
6. Frints S, Marynen P, Hartmann D, Fryns J, Steyaert J, Schachner M, et al. CALL interrupted in a patient with non-specific mental retardation: Gene dosage-dependent alteration of murine brain development and behavior. Hum Mol Genet 2003; 12: 1463-74.
7. Shrimpton A, Jensen K, Hoo J. Karyotype-phenotype analysis and molecular delineation of a 3p26 deletion/8q24.3 duplication case with a virtually normal phenotype and mild cognitive deficit. Am J
Med Genet 2006; 140A: 388-91.
8. Pohjola P, De Leeuw N, Penttinen M, Kääriäinen H. Terminal 3p deletions in two families – correlation between molecular karyotype and phenotype. Am J Med Genet 2010; 152A: 441-6.
9. Yoshihara Y, Kawasaki M, Tamada A, Nagata S, Kagamiyama H, Mori K. Overlapping and differential expression of BIG-2, BIG-1, TAG-1, and F3: four members of an axon-associated cell adhesion molecule subgroup of the immunoglobulin superfamily. J Neurobiol 1995; 28: 51-69. 10. Saito H, Mimmack M, Kishimoto J, Keverne EB, Emson PC. Expression of olfactory receptors,
G-proteins and AxCAMs during the development and maturation of olfactory sensory neurons in the mouse. Dev Brain Res 1998; 110: 69-81.
11. Dijkhuizen T, Van Essen T, Van Der Vlies P, Verheij J, Sikkema-Raddatz B, Van Der Veen A, et al. FISH and array-CGH analysis of a complex chromosome 3 aberration suggests that loss of CNTN4 and CRBN contributes to mental retardation in 3pter deletions. Am J Med Genet 2006; 140A: 2482-7.
12. Fernandez T, Morgan T, Davis N, Klin A, Morris A, Farhi A, et al. Disruption of contactin 4 (CNTN4) results in developmental delay and other features of 3p deletion syndrome. Am J Hum Genet 2004; 74: 1286-93.
13. Fernandez TV, García-González IJ, Mason CE, Hernández-Zaragoza G, Ledezma-Rodríguez VC, Anguiano-Alvarez VM, et al. Molecular characterization of a patient with 3p deletion syndrome and a review of the literature. Am J Med Genet 2008; 146A: 2746-52.
14. Jo S, Lee K-H, Song S, Jung Y-K, Park C-S. Identification and functional characterization of cereblon as a binding protein for large-conductance calcium-activated potassium channel in rat brain. J
Neurochem 2005; 94: 1212-24.
15. Higgins JJ, Rosen DR, Loveless JM, Clyman JC, Grau MJ. A gene for nonsyndromic mental retardation maps to chromosome 3p25-pter. Neurology 2000; 55: 335-40.
16. Higgins JJ, Pucilowska J, Lombardi RQ, Rooney JP. A mutation in a novel ATP-dependent Lon protease gene in a kindred with mild mental retardation. Neurology 2004; 63: 1927-31.
17. Higgins JJ, Hao J, Kosofsky BE, Rajadhyaksha AM. Dysregulation of large-conductance Ca2+
-activated K+ channel expression in nonsyndromal mental retardation due to a cereblon p.R419X
mutation. Neurogenetics 2008; 9: 219-23.
18. Andreae LC, Peukert D, Lumsden A, Gilthorpe JD. Analysis of Lrrn1 expression and its relationship to neuromeric boundaries during chick neural development. Neural Dev 2007; 2: 22.
19. Tossell K, Andreae LC, Cudmore C, Lang E, Muthukrishnan U, Lumsden A, et al. Lrrn1 is required for formation of the midbrain-hindbrain boundary and organiser through regulation of affinity differences between midbrain and hindbrain cells in chick. Dev Biol 2011; 352: 341-52.
20. Berridge MJ. Inositol trisphosphate and calcium signalling. Nature 1993; 361: 315-25.
21. Cargile CB, Goh DL-M, Goodman BK, Chen XN, Korenberg JR, Semenza GL, et al. Molecular cytogenetic characterization of a subtle interstitial del(3)(p25.3p26.2) in a patient with deletion 3p syndrome. Am J Med Genet 2002; 109: 133-8.
22. Endris V, Wogatzky B, Leimer U, Bartsch D, Zatyka M, Latif F, et al. A. The novel Rho-GTPase activating gene MEGAP/srGAP3 has a putative role in severe mental retardation. Proc Nat Acad Sci
Figure Legends
Fig. 1. Array comparative genomic hybridization investigation shows an ~ 9.29-Mb deletion of 3p25.3pter encompassing the genes of CHL1, CNTN4, CRBN, LRRN1, ITPR1 and SRGAP3 but not involving the markers D3S1263 and D3S3594.
Fig. 2. Representative electrophoretograms of quantitative fluorescent polymerase chain reaction assays at short tandem repeat markers specific for chromosome 3p using uncultured amniocytes and parental DNAs. With the marker D3S2387 (3p26.3), only the allele of 188 bp (maternal) is present in the fetus, and with the marker D3S2397 (3p26.1), only the allele of 208 bp (maternal) is present in the fetus. The results indicate a paternal origin of the deletion.
Fig. 3. A karyotype of 46,XX,del(3)(p25.3). The arrow indicates the breakpoint. Fig. 4. The fetus at birth.
Table 1. Molecular results using polymorphic DNA markers specific for chromosome 3p*
Markers Locus Father Mother Fetus
(uncultured amniocytes)
D3S2387 3p26.3 184, 200 184,188 188
D3S2397 3p26.1 212, 212 208, 208 208
D3S2385 3p25.1 155, 155 147, 151 147, 155