Dynamics of the F 2 + C H 3 S C H 3 reaction: A molecule-molecule reaction without
entrance barrier
Yu-Ju Lu, Lance Lee, Jun-Wei Pan, Henryk A. Witek, and Jim J. Lin
Citation: The Journal of Chemical Physics 127, 101101 (2007); doi: 10.1063/1.2780145 View online: http://dx.doi.org/10.1063/1.2780145
View Table of Contents: http://scitation.aip.org/content/aip/journal/jcp/127/10?ver=pdfcov Published by the AIP Publishing
Articles you may be interested in
The problematic C 2 H 4 + F 2 reaction barrier
J. Chem. Phys. 132, 094304 (2010); 10.1063/1.3316088
Barrierless reactions between two closed-shell molecules. II. Dynamics of F 2 + CH 3 SSCH 3 reaction J. Chem. Phys. 130, 014301 (2009); 10.1063/1.3049782
Crossed jet reactive scattering dynamics of F + H 2 O H F ( v , J ) + O H : H F ( v , J ) product quantum state distributions under single-collision conditions
J. Chem. Phys. 129, 184305 (2008); 10.1063/1.2998524
Barrierless reactions between two closed-shell molecules. I. Dynamics of F 2 + C H 3 S C H 3 reaction J. Chem. Phys. 128, 104317 (2008); 10.1063/1.2837801
Infrared laser spectroscopy of C H 3 H F in helium nanodroplets: The exit-channel complex of the F + C H 4 reaction
J. Chem. Phys. 124, 084301 (2006); 10.1063/1.2168450
Dynamics of the F
2+ CH
3SCH
3reaction: A molecule-molecule reaction
without entrance barrier
Yu-Ju Lu
Institute of Atomic and Molecular Sciences, Academia Sinica, Taipei 10617, Taiwan Lance Lee
Institute of Atomic and Molecular Sciences, Academia Sinica, Taipei 10617, Taiwan and Department of Chemistry, National Taiwan University, Taipei 10617, Taiwan Jun-Wei Pan
Institute of Atomic and Molecular Sciences, Academia Sinica, Taipei 10617, Taiwan Henryk A. Witek
Department of Applied Chemistry, National Chiao Tung University, Hsinchu 30010, Taiwan Jim J. Lina兲
Institute of Atomic and Molecular Sciences, Academia Sinica, Taipei 10617, Taiwan
and Department of Applied Chemistry, National Chiao Tung University, Hsinchu 30010, Taiwan 共Received 23 July 2007; accepted 14 August 2007; published online 13 September 2007兲
The F2+ CH3SCH3reaction was studied with crossed molecular beam techniques and high level ab
initio calculations. Significant reactivity was observed even at low collision energies, consistent
with the negligible barrier height obtained from the ab initio calculations. All experimental findings are consistent with a weakly bound reaction intermediate of F – F – S共CH3兲2 structure, which
possesses a special type of three-center four-electron bonding. Analogous intermediates can also explain the reactions of F2 with CH3SH and CH3SSCH3. © 2007 American Institute of Physics.
关DOI:10.1063/1.2780145兴
A molecule with a closed-shell electronic structure is usually believed to be more stable than a radical with an open-shell electronic structure. This common knowledge is often referred to as the octet rule; it has been used with great success to predict the stability of many chemical species over decades. In comparison with radical reactions, much weaker reactivity and higher activation energies are expected for re-actions between two closed-shell molecules. Here, we report a type of reaction that violates this general rule: we found that the reaction between F2and CH3SCH3 has a negligible
activation barrier despite the closed-shell nature of both re-actants. In all previous investigations of interactions between F2and closed-shell molecules共e.g., I2, ICl, HI, CH3I, C2H4,
and C6H6兲 in crossed molecular beams, a considerable colli-sion energy was required to promote a reaction.1–8Of many reactions of F2 with closed-shell molecules, the rate con-stants were found to be much smaller than those of corre-sponding F2reactions with radical species.9However, an
ex-traordinarily large rate constant of 共1.6±0.5兲 ⫻10−11cm3molecule−1s−1 was observed at room
tempera-ture for the F2+ CH3SCH3 reaction.10 It has been speculated10 that this atypical reactivity correlates with the stability of the ion pair关CH3SCH3兴+关F
2兴−. Although several
products have been investigated in bulk by mass spectrom-etry, infrared emission spectroscopy,10 and photoelectron spectroscopy,11 it is hard to deduce the reaction mechanism
due to difficulty of identifying the primary products in the multiple-collision experiments. Moreover, in the absence of high level quantum calculations, the nature of the unusual reactivity remains elusive.
In this work, the crossed molecular beam technique was applied for the first time to investigate the F2+ CH3SCH3 reaction. Such single-collision experiments enable one to identify the primary reaction products at well defined colli-sion energies. Two product channels共channels I and II兲 were directly observed:
I: F2+ CH3SCH3→ HF + CH2S共F兲CH3; II: → F + CH3S共F兲CH3.
Structures of the products were identified by comparing their photoionization thresholds with the values from ab initio cal-culations. The collision energy dependences of the reaction cross section and branching ratio were studied. Energetics and structures on the reaction paths were calculated with high level ab initio methods. A comprehensive and unam-biguous reaction mechanism was proposed based on the ex-perimental and computational results.
The experiments were performed with two crossed mo-lecular beam apparatuses: One employed electron impact ionization12 and the other photoionization.13 Two reactant beams crossed each other at a 90° angle. The short pulse 共⬍20s兲 of the F2 beam produced from a fast solenoid
valve 共Even-Lavie valve, high repetition rate model, 艋1000 Hz兲 共Ref.14兲 was used to define the starting time of a兲Author to whom correspondence should be addressed. Electronic mail:
jimlin@gate.sinica.edu.tw
0021-9606/2007/127共10兲/101101/4/$23.00 127, 101101-1 © 2007 American Institute of Physics
the collision events. Time-of-flight spectra of the scattered products were measured with a time-resolved quadrupole mass spectrometer. Angular distributions of the products were measured on varying the angle between the detector and molecular beams. Synchrotron radiation from the 9 cm undulator beamline15 of the Taiwan Light Source provided the bright tunable vacuum UV photon beam with intensity of ⬃1016photons/ s. Photoionization efficiency spectra of the
products were recorded on varying the wavelength of the synchrotron radiation. To transform the data measured in the laboratory共lab兲 frame to the center-of-mass 共c.m.兲 frame, we used a forward convolution method. The instrument func-tions, such as velocity spreads of the molecular beams, width of the flight length, etc., and the Jacobian factor of the lab-c.m. transformation are included in the computer program. In some experiments, CD3SCD3 共99% D, Aldrich, Inc.兲 was
used in order to shift the masses of the products, and thus to avoid the background from impurities of the commercial CH3SCH3 sample 共99%, Aldrich, Inc.兲. We did not observe
any significant isotope effect in this study, including the re-action thresholds, angular distributions, product photoioniza-tion spectra, etc. Although in many cases it is unnecessary, the impurity contribution from the CH3SCH3sample can be
subtracted on performing Ar+ CH3SCH3 scattering
experi-ments under similar conditions. For the determination of the relative reaction cross section at various collision energies, the CH3SCH3beam intensity was measured with a fast
ion-ization gauge共Beam Dynamics, Inc.兲; the F2beam intensity
was deduced using the attenuation method, in which a reduc-tion of the CH3SCH3 beam intensity was caused by colli-sions with the F2 molecular beam.
The reaction paths were searched mainly using the com-plete active space self-consistent field共CASSCF兲 calculation with the second-order perturbation corrections 共CAS-PT2兲, in which the active spaces were carefully selected and tested. The minimum energy paths were constructed by scanning the reaction coordinate 共or a geometry parameter close to the reaction coordinate兲 from the reactant state to the product state, in which other degrees of freedom were optimized. The zero-point energy was calculated with the CAS-PT2 method. For singlet structures, the geometry optimization and zero-point energies can be checked with quadratic configuration interaction 关QCISD共T兲兴 calculations. Single-point energies were obtained with CCSD共T兲 共couple cluster兲 calculations with a complete basis set extrapolation scheme, in which Dunning’s cc-pVTZ共triple zeta兲, cc-pVQZ 共quadruple zeta兲, and cc-pV5Z共quintuple zeta兲, basis sets were used on the S and F atoms. The quality of basis sets was found to be im-portant for the S and F atoms, but not crucial for the C and H atoms共the results from cc-pVDZ and cc-pVTZ basis sets are very similar; cc-pVDZ basis sets were used on the C and H atoms in most cases兲. The basis set superposition error was checked with the counterpoise method. This error becomes insignificant when using cc-pVQZ or larger basis sets. The calculations were performed with the MOLPRO 2006.1 quan-tum chemistry package.16
Two sulfur-containing species with masses of 80 关iden-tified as CH2vS共F兲–CH3兴 and 81 关identified as
CH3– S共F兲–CH3兴 were detected as nascent products. The
photoionization efficiency spectra were recorded with the synchrotron radiation facilities for both products to discrimi-nate between possible isomers. The ionization thresholds of the products with masses of 80 and 81 were determined to be 8.7± 0.1 and 7.8± 0.1 eV, respectively. The computed adia-batic ionization energy共IE兲 for two most likely isomer prod-ucts with the mass of 80, CH2vS共F兲–CH3 and
CH2F – S – CH3, were found to be 8.59 and 9.09 eV,
respec-tively, with the CCSD共T兲 method. We noticed that fluorine substitution in the methyl group of CH3SCH3 共IE
= 8.70 eV兲 increases its ionization energy by about +0.4 eV. A similar effect has been observed previously for chlorine substitution 共CH2Cl– S – CH3, IE= 9.08 eV兲.17 The isomer
CH2F – S – CH3 has clearly too high ionization energy to
match the observed photoionization threshold, hence the ma-jor product with mass of 80 can be assigned as CH2vS共F兲–CH3, supported by agreement between the
ob-served and calculated IEs. For product with mass of 81, its ionization threshold is 0.9 eV less than that of CH3SCH3. A
structure corresponding to CH3– S共F兲–CH3 is the most
rea-sonable assignment for this mass with the vertical IE calcu-lated to be 7.89 eV. This finding is consistent with an inter-mediate species observed in a previous flow-tube experiment,11 in which the vertical IE was measured to be 8.03 eV with photoelectron spectroscopy and tentatively as-signed to the above structure.
Channel I is highly exothermic, with calculated⌬H0 K° equal to −80.6 kcal/ mol. Its products, HF + CH2S共F兲CH3,
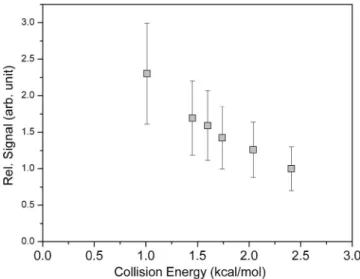
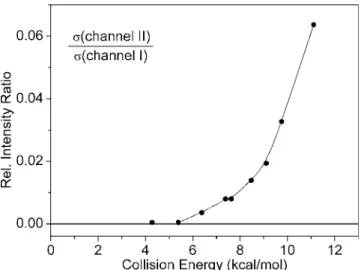
were observed to have high translational energy 共⬃25% of the available energy is deposited into the translational de-grees of freedom兲. The experimental cross section of channel I did not show any significant decrease when we gradually tuned down the collision energy from 11 to 1 kcal/ mol. In fact, it increases for lower collision energies, as shown in Fig. 1. Thus, the barrier of this reaction is expected to be much lower than 1 kcal/ mol, otherwise, we should see a decline in signal when reducing the collision energy. In con-trast, the signal of channel II vanished for low collision en-ergies and increased rapidly for high enen-ergies 共Fig. 2兲. To
FIG. 1. The relative reaction cross section of channel I as a function of collision energy. The relative signal intensity has been normalized with re-spect to the F2and CH3SCH3beam intensities.
101101-2 Lu et al. J. Chem. Phys. 127, 101101共2007兲
determine the threshold of channel II more precisely, we measured the velocity spreads of the molecular beams and calculated the spread of the collision energy to be ±0.7 kcal/ mol 共⫾ half width at half maximum兲. Then, we can perform the deconvolution and determine the threshold for channel II to be 6.0± 0.7 kcal/ mol.
Neither reaction channel shows a forward-backward symmetric angular distribution, a typical signature of a reac-tion with a long-lived intermediate.18 Both sulfur-containing products exhibit significantly forward angular distributions 共with respect to the velocity of the CH3SCH3reactant兲. This
finding indicates that the time scale of the reaction is sub-stantially shorter than one rotational period of the collision complex.
To understand the reaction mechanism, high level ab
ini-tio calculaini-tions were performed. In the entrance region, a
transition state共TS1 in Fig.3兲 was found and characterized
using the CAS-PT2 and CCSD共T兲 calculations. From analy-sis of the CASSCF molecular orbitals near the transition state region, we found that the interaction originates mostly
from the coupling between the 3pz orbital of sulfur and the
2pzorbital of fluorine. Based on both experimental and
com-putational results, we propose the following reaction mecha-nism. In the entrance region, the preferred geometry leading to reaction has a roughly linear F–F–S structure; the axis connecting these atoms共the z axis兲 is approximately perpen-dicular to the C–S–C molecular plane. After passing a neg-ligible barrier 共TS1兲, an intermediate 共INT兲 may be formed. Being weakly bound, the outer F atom is quite floppy. If the collision energy is sufficiently large, this atom can dissociate directly on breaking the F – FS共CH3兲2 bond. Another pathway—with the outer F atom bending toward the methyl group and eventually forming HF product—predominates, however, driven by its lower barrier共TS2兲 and a large reac-tion exothermicity. At the CCSD共T兲 and CAS-PT2 levels of theory, the energies of TS1 and TS2 are lower than the en-ergy of the reactants and the zero-point enen-ergy corrections are found to be insignificant共⬍0.6 kcal/mol兲. That is, both high level ab initio methods predict that there is no activa-tion barrier for channel I, an unusual finding for reacactiva-tions between two closed-shell molecules.
For the reaction to occur, INT is the key structure, in which the F–F–S bonding has a special three-center four-electron character.19 From analysis of the CASSCF molecu-lar orbitals, we found that two electrons from the S lone pair and two electrons from the F2 bond are distributed in the
three active orbitals of the F–F–S bonding共consisting mainly of the 2pz-2pz-3pz orbitals, respectively兲 at the structure of
INT. Structures other than INT can also be imagined and calculated. If both F atoms are directly attached to the S atom of CH3SCH3, a structure with nearly collinear F–S–F
bonds can be formed关SF2共CH3兲2in Fig.4兴. This structure is
similar to SF4 molecule and is, in fact, much more stable
than INT. However, migration of the outer F atom of INT to the opposite side is kinetically unfavorable. If this very stable structure is formed, its lifetime should be much longer than that of INT and thus the product angular distribution should be symmetric. Besides HF formation, the decomposi-tion of SF2共CH3兲2 probably produces CH3+ SF2共CH3兲, a
quite exothermic channel. In fact, these evidences of forming SF2共CH3兲2 cannot be found in the experiments, suggesting
that it hardly competes with the two reaction paths shown in Fig.3.
CH3S共F兲CH3is an intriguing open-shell species; the
cal-culated dissociation energy of the F – S共CH3兲2 bond is
33.4 kcal/ mol, which is only slightly weaker than the disso-ciation energy of F2 共D0,calc= 36.7 kcal/ mol, D0,expt
= 36.9 kcal/ mol兲. Hence, channel II is slightly endothermic. Because the preferred impact direction of F2 does not
coin-cide with the center of mass of the CH3SCH3 molecule, the
FIG. 2. Relative intensity 共branching兲 ratio of product channel II/I as a function of collision energy. These data were obtained by analyzing the time-of-flight spectra of products CD3SFCD3and CD2SFCD3in the crossed beam reaction of F2+ CD3SCD3. The corresponding results obtained from
the F2+ CH3SCH3 reaction yield a similar curve but with a higher
background.
FIG. 3. Optimized structures for the van der Waals entrance well共VDW兲, transition states共TS1, TS2兲, intermediate 共INT兲, and products are shown on schematic reaction paths. Relative energies共in kcal/mol兲, calculated using the CCSD共T兲 method with complete basis set extrapolation, are given in parentheses for each structure. Numbers denoted with *include zero-point
energy corrections.
FIG. 4. Optimized structure of SF2共CH3兲2 关by QCISD共T兲/cc-pVDZ共H,C兲
and cc-pVTZ共S,F兲兴.
experimental threshold of channel II should be corrected in order to compare with the calculated enthalpy of reaction. The direction of reactive F2collisions is roughly
perpendicu-lar to both C–S bonds. The two methyl groups can thus be regarded as spectators along the reaction path of channel II. The effective collision energy for the F2– S interaction can
be expressed approximately as
Eeff=
共F2– S兲
共F2– CD3SCD3兲
Ec= 0.71Ec, 共1兲
where is the reduced mass of the relevant collision part-ners and Ecis the conventional collision energy. The
remain-ing 29% of Ecis transferred mostly into the centrifugal
en-ergy of the collision complex. The experimental threshold of 6.0± 0.7 kcal/ mol should be scaled down with Eq.共1兲, yield-ing the value of 4.3± 0.5 kcal/ mol. Then, the experimental bond dissociation energy of F – S共CH3兲2 can be deduced as
36.9− 4.3= 32.6 kcal/ mol.
Crossed molecular beam reactions of F2with other
orga-nosulfur compounds共CH3SH , CH3SSCH3兲 are under
inves-tigation. As expected, similar results were observed for the F2+ CH3SH reaction. More interesting results were obtained
for the F2+ CH3SSCH3 reaction. The major product channel
was observed to be CH3SF + CH3SF, whereas a minor
chan-nel, F + CH3SS共F兲CH3 共an analog to channel II兲, could be
detected for collision energies greater than 4 kcal/ mol. Simi-lar to the F2+ CH3SCH3reaction, high reactivity at low
col-lision energies 共⬃1.5 kcal/mol兲 was observed. We consider that the reaction mechanism described above is also appli-cable to explain these results. We surmise that, after forming a similar intermediate, the outer F atom reacts preferentially with the other S atom forming a four-member ring structure, eventually leading to breaking of both S–S and F–F bonds. On the other hand, we have found that the reactions of F2
with simple alkenes共C2H4 and C3H6兲 revealed quite
differ-ent features:共a兲 much higher collision energy is required to observe the reaction products; 共b兲 only one product channel producing F + CnH2nF was observed. The HF + CnH2n−1F
product channel was not found;共c兲 the corresponding angu-lar distributions are backward biased, indicating that small impact parameter collisions are required to overcome the
ac-tivation barriers.18 The interaction of F2 with the CvC
double bond shows typical behaviors of a direct reaction with a significant barrier—an interesting comparison to the above F2+ organosulfur reactions that feature a negligible barrier and a short-lived intermediate. Finally, we contend that the above proposed mechanism is generally applicable for reactions of F2 with molecules having loosely bound lone-pair electrons.
National Science Council共Grant Nos. NSC95-2113-M-001-041-MY3 and NSC95-2113-M-009-016兲 and Academia Sinica, Taiwan, supported this work. We thank Professor Yuan T. Lee and Yuan-Pern Lee for valuable comments and National Synchrotron Radiation Research Center, Taiwan for use of facilities.
1J. M. Farrar and Y. T. Lee, J. Am. Chem. Soc. 96, 7570共1974兲. 2J. M. Farrar and Y. T. Lee, J. Chem. Phys. 63, 3639共1975兲.
3M. J. Coggiola, J. J. Valentini, and Y. T. Lee, Int. J. Chem. Kinet. 8, 605
共1976兲.
4J. J. Valentini, M. J. Coggiola, and Y. T. Lee, J. Am. Chem. Soc. 98, 853
共1976兲.
5J. J. Valentini, M. J. Coggiola, and Y. T. Lee, Faraday Discuss. Chem.
Soc. 62, 232共1977兲.
6C. C. Kahler and Y. T. Lee, J. Chem. Phys. 73, 5122共1980兲. 7J. M. Farrar and Y. T. Lee, J. Chem. Phys. 65, 1414共1976兲.
8J. R. Grover, Y. Wen, Y. T. Lee, and K. Shobatake, J. Chem. Phys. 89,
938共1988兲.
9http://kinetics.nist.gov
10A. A. Turnipseed and J. W. Birks, J. Phys. Chem. 95, 6569共1991兲. 11J. Baker, V. A. Butcher, J. M. Dyke, and E. P. F. Lee, J. Phys. Chem. 99,
10147共1995兲.
12J. J. Lin, D. W. Hwang, S. Harich, Y. T. Lee, and X. Yang, Rev. Sci.
Instrum. 69, 1642共1998兲.
13J. J. Lin, Y. Chen, Y. Y. Lee, Y. T. Lee, and X. Yang, Chem. Phys. Lett.
361, 374共2002兲.
14U. Even, J. Jortner, D. Noy, and N. Lavie, J. Chem. Phys. 112, 8068
共2000兲.
15http://140.110.203.42/bldoc/21AU9WL.htm 16H.-J. Werner, P. J. Knowles, R. Lindh et al.,
MOLPRO, version 2006.1, a package of ab initio programs共see http://www.molpro.net兲.
17B.-M. Cheng, E. P. Chew, J. K. Yu, and C.-H. Yu, J. Chem. Phys. 114,
4817共2001兲.
18R. D. Levine and R. B. Bernstein, Reaction Dynamics and Chemical
Reactivity共Oxford, New York, 1987兲, pp. 411–417 and 104–113.
19C. A. Ramsden, Chem. Soc. Rev. 23, 111共1994兲.
101101-4 Lu et al. J. Chem. Phys. 127, 101101共2007兲