行政院國家科學委員會專題研究計畫 成果報告
第二型糖尿病胰小島細胞功能之分子機制研究(3/3)
計畫類別: 個別型計畫 計畫編號: NSC93-2314-B-002-137- 執行期間: 93 年 08 月 01 日至 94 年 07 月 31 日 執行單位: 國立臺灣大學醫學院內科 計畫主持人: 莊立民 計畫參與人員: 張恬君 報告類型: 完整報告 處理方式: 本計畫可公開查詢中 華 民 國 94 年 10 月 28 日
行政院國家科學委員會補助專題研究計畫完整報告
(計畫名稱)
Molecular Studies on Mechanism of Beta Cell Dysfunction Relating to Type 2 Diabetes
計畫類別:□
v個別型計畫 □ 整合型計畫
計畫編號:NSC 93-2314-B -002 - 137 -
執行期間: 93 年 8 月 1 日至 94 年 7 月 31 日
計畫主持人:莊立民
共同主持人:蘇銘嘉
計畫參與人員: 張恬君
成果報告類型(依經費核定清單規定繳交):□精簡報告 □
v完整報告
本成果報告包括以下應繳交之附件:
□赴國外出差或研習心得報告一份
□赴大陸地區出差或研習心得報告一份
□出席國際學術會議心得報告及發表之論文各一份
□國際合作研究計畫國外研究報告書一份
處理方式:除產學合作研究計畫、提升產業技術及人才培育研究計畫、列
管計畫及下列情形者外,得立即公開查詢
□涉及專利或其他智慧財產權,□一年□
v二年後可公開查詢
執行單位:臺大醫學院、內科
中 華 民 國 94 年 10 月 27 日
中文摘要 關鍵字:胰島素分泌、胰島貝它細胞、glucosamine、PPARγ、calpain 10、蛋白質交互作用 我們已經將研究的焦點放在探討第二型糖尿病的致病原因,其中包括胰島素周邊作用及 胰島素分泌功能的缺陷。在第二型糖尿病發生的早期,第一期的胰島素分泌功能已有障礙。 在先前的研究中,我們已報告一種胰島素敏感劑(rosiglitazone)能促進葡萄糖所刺激的胰島素 分泌。在大鼠的胰臟灌流系統中,第一期與第二期的胰島素分泌均會受rosiglitazone 之影響,
而此影響乃是經由PI-3 kinase pathway 所造成的。在此計畫書中,我們將探討 rosiglitazone 能 促進由葡萄糖所刺激的胰島素分泌的分子機。我們已取得並能培養對葡萄糖有反應的小鼠胰 臟貝它細胞株,Min 6。同時我們也以能利用 patch-clamp 技術進行貝它細胞上一些離子通道 的研究。基本上,對ATP 敏感的鉀離子通道(ATP-sensitive K+-channel)參與了由葡萄糖所刺激
的胰島素分泌,但是其他的離子通道也可能參與rosiglitazone 促進胰島素分泌的作用。此外,
利用細胞株我們將探討 PI-3 kinase 如何調控離子通道的電位變化,希望能回答 rosiglitazone
促進胰島素分泌的細胞機轉。
第二型糖尿病是個進行性的慢性疾病,而長期的高血糖可能導致很多的細胞缺陷,此現 象就是所謂的“葡萄糖毒性”。先前的研究已認為透過hexosamine biosynthetic pathway 所增
加的葡萄糖代謝產物,可能是高血糖導致胰島素抗性的機轉之一。通常,只有2-3%被細胞所 吸收的葡萄糖會經由此代謝路徑,最後產生 UDP-N-Acetylglucosamine,此產物可當作 glycoprotein,glycolipids 與 proteoglycanes 之基質。胰島素之標的細胞,如肌肉與脂肪細胞, 及分泌胰島素的胰臟貝它細胞均受其影響。我們與其他研究者均發現glucosamine 可能是長期 高血糖導致“葡萄糖毒性”的元凶之一。在與中興大學楊博士的合作實驗中,我們發現將大 鼠先予以 glucosamine 灌流能減少由葡萄糖所刺激的胰島素分泌,更進一步地,再給予 rosiglitazone 灌流可顯著地回復胰島素的分泌功能。這令人鼓舞的結果暗示我們 rosiglitazone 可能具有恢復受“葡萄糖毒性”抑制的胰島素分泌能力。 因此我們將進一步研究詳細的細胞機轉,包括glucosamine 與 rosiglitazone 如何影響細胞內 ATP 濃度,鈣離子濃度與粒線體膜電位等,並研究可能候選基因之蛋白質交互作用。
英文摘要
Keywords: insulin secretion, beta cells, glucosamine, PPARγ, calpain10, protein-protein interaction
We have focused studies on the pathogenesis of type 2 diabetes that involves both defects in the insulin action and insulin secretion. It is striking that insulin stimulated first phase insulin secretion is impaired in the early stage of development of type 2 diabetes. Previously, we demonstrated that an insulin sensitizer of thiazolidinedione class can potentiate glucose-stimulated insulin secretion. In rat pancreas perfusion system, both first- and second-phase of insulin secretion are affected by rosiglitazone via a PI-3 kinase dependent pathway. In the present proposal, we plan to find out molecular mechanisms by which rosiglitazone potentiate glucose-stimulated insulin secretion. We have obtained and established the culture of a glucose-responsive rat pancreatic beta cell line, MIN6 in our laboratory. We can study the ion channels that affect insulin secretion by the patch-clamp techniques. Basically, ATP-sensitive K+-channel is involved in glucose-stimulated insulin secretion. However, other ion channels might also possibly involve the effect of rosiglitazone on potentiation of insulin secretion. How ion channel regulated by PI-3 kinase could be studied in the cultured cells. In this way, we will answer in cellular mechanisms that might explain how rosiglitazone potentiate insulin secretion.
Type 2 diabetes is a slowly progressive disorder in which long-standing hyperglycemia can cause multiple cellular defects, a process termed glucose toxicity. Previous studies have suggested that an increase flux of the glucose metabolites through the hexosamine biosynthetic pathway maybe the mechanism by which hyperglycemia leads to insulin resistance. Usually, only 2-3% of the total glucose taken up by the cell is metabolized by this pathway that ultimately produces UDP-N-acetylglucosamine, which serves as a substrate in the formation of glycoprotein, glycolipids, and proteoglycans. Both insulin target cells like muscle and adipocytes, and insulin secreting pancreatic beta cells are all affected. We and other have noticed that glucosamine, increased after long-standing hyperglycemia, might contribute to glucose toxicity of the islet cells. In collaboration with Dr. Yang at Chung-Hsin University, we have found that pretreatment with glucosamine could down-regulate glucose-stimulated insulin secretion. Moreover, treatment with rosiglitazone markedly reversed the inhibition. This encouraging data suggest to us that rosiglitazone might reverse the glucose toxicity of pancreatic beta cells. Detailed cellular mechanisms, including how glucosamine and rosiglitazone affect intracellular ATP, calcium ion concentration, mitochondrial membrane potential, and the interacting proteins for some
前言及研究目的
Defects in insulin secretion from the pancreatic β-cells and insulin action on the target
cells are two major causes in pathogenesis of type 2 diabetes (1). Thiazolidinedione (TZD) is a novel class of antidiabetic agent that can produce a potent insulin-sensitizing activity (2). In addition to the insulin-sensitizing effect, TZD also enhances insulin secretory capacity through the amelioration of glucose toxicity on β-cells (3,4). Similarly, a few studies showing that TZDs protect β-cells from lipoapoptosis in rodents implies that part of the therapeutic action of TZDs in human type 2 diabetes may be the result of the prevention of β-cell loss and the restoration of the insulin secretory capacity (3-8). More recently, Lupi et al (9) also reported that both rosiglitazone (RSG) and PGJ2 prevent the lipotoxic effect on the β- cell exerted by increasing the fatty acid
concentration in culture medium of isolated human islet.
Few studies explored the direct effect of TZDs on glucose-induced insulin secretion. Troglitazone, one of TZD derivatives has been reported to cause a dose-dependent increase in insulin secretion after both 10 and 60 min incubation, and the stimulatory effect was associated with an immediate increase of cytoplasmic free Ca2+ (10). In our previous study, we have shown that RSG, a TZD, has a potentiation effect on glucose-stimulated insulin secretion in an isolated pancreatic perfusion system (12). Interestingly, this effect was mediated through a phosphatidylinositol 3-kinase (PI3K)-dependent pathway (10).
ATP-sensitive K+ channels (KATP channels) are characterized by an inhibition of channel
opening when the ATP/ADP ratio at the cytoplasmic cell surface is increased, then depolarized the membrane and open voltage-dependent Ca2+ channels, and stimulate insulin secretion (12). The TZD derivatives troglitazone has been reported to be able to directly stimulate insulin secretion from pancreatic β cells by inhibiting ATP-sensitive K+ channels (KATP channels)(13). .In this study,
we will investigate the effect of RSG on inhibiting the opening of KATP channel to further elucidate
the mechanism of RSG stimulating insulin secretion. 研究方法
Cell line and cell culture. MIN6 cells, obtained from Dr. Seino (Chiba University, Japan), were
used between passage 20 and 30 and grown in DMEM containing 10% (vol/vol) heat-inactivated fetal calf serum, 25mM glucose, 2 mM L-glutamine, 100 mM 2-mercaptoethanol, 100 units/ml penicillin, and 100 µg/ml streptomycin in a humidified atmosphere at 37°C with 5% CO2 unless
specified otherwise.
MIN6 cells were seeded and incubated for about 24 hrs prior to the experiment. The medium was removed and changed to DMEM containing 0.2% BSA at 37°C for 3 hrs. The medium was then removed and replaced with KRB buffer [109 mM NaCl, 4.6 mM KCl, 1 mM MgCl2, 20 mM
HEPES (pH 7.4)], 5 mM sodium bicarbonate, and 0.2% (w/v) BSA containing either 3.3mM or 16.7 mM glucose for another 1 hr at 37°C. Thereafter, test substance was added to the medium for various times at 37°C.
Potassium currents recording in beta cells:
z Whole-cell voltage clamp
z Internal solution (mM): NaCl 10,K+ aspartate 130, KCl 10, MgCl2 2, HEPES 10, pH=7.2
For IKATP recroding: ATP 0.3, ADP 0.3 For IKV recording: ATP 5, ADP 0.3
z External solution (mM): NaCl 135, KCl 5, CaCl2 2, MgCl2 1, HEPES 10, glucose 3.3, pH=7.4
z Two bath glucose conditions: High glucose: 16.7 mM glucose Low glucose: 3.3 mM glucose
Yeast two-hybrid screening using calpain 10 as bait:
To find out the NRP interacting proteins, yeast two-hybrid system will be employed.
MATCHMAKER Two-Hybrid System 3 (from Clontech) is an advanced GAL4-based two-hybrid system that provides a transcriptional assay for detecting protein interactions in vivo in yeast. (a) Preparing for a yeast two-hybrid screen:
Constructing fusion genes:
z Generate the gene fragment of calpain 10 and its mutant by PCR method with useful restriction sites incorporated into the primers. Purify the gene fragment which is generated by PCR method.
z Digest the DNA-BD vector with the appropriate restriction enzyme(s), treat with phosphatase, and purify it. Ligate the DNA-BD vector with the PCR fragments of calpain 10 and its mutant. z Transform ligation mixtures into E. coli. Identify insert-containing plasmids by restriction
analysis or PCR method.
z Use the sequencing primers to check the orientation and reading frame of the junctions. Verify that constructs do not activate reporter genes autonomously:
z Independently transform DNA-BD and AD fusion constructs into yeast strain AH109. z Assay the transformants for MEL1 activation by selecting for transformants on
SD/–Trp/X-a-Gal and SD/–Leu/X-a-Gal, respectively. z Perform positive and negative controls in parallel.
Verify Protein Expression:
z Independently transform the DNA-BD and AD fusion constructs into strain AH109.
z Prepare Western blots from the transformants and probe the blots with antibodies to the GAL4 DNA-BD and AD antibodies.
z Use untransformed yeast as a control.
Transform the library and screen for candidates of calpain 10 and its mutant interacting proteins: This time, we will use the sequential transformation method to deliver pAS2-1 NRP first into AH109, and then perform the second transformation to deliver MIN6 pVP16 cDNA library into AH109 carrying pAS2-1 calpain 10 and its mutant. The methods of first and second transformation differ only with their scale.
Isolate plasmids from putative positive clones carrying high β-galactosidase activity by commercial kits or manual protocols.
Eliminate colonies bearing the same AD/library plasmid by PCR and restriction enzyme digestion methods.
Transform plasmids into E. coli and purify DNA with commercial kits.
Confirm interaction in yeast by cotransformation of DNA-BD/ bait and AD/ library plasmids into AH109 and yeast mating methods.
Perform additional two-hybrid tests such as vectors switch, frameshift mutations and site-specific mutation/deletions to confirm the true interaction.
結果
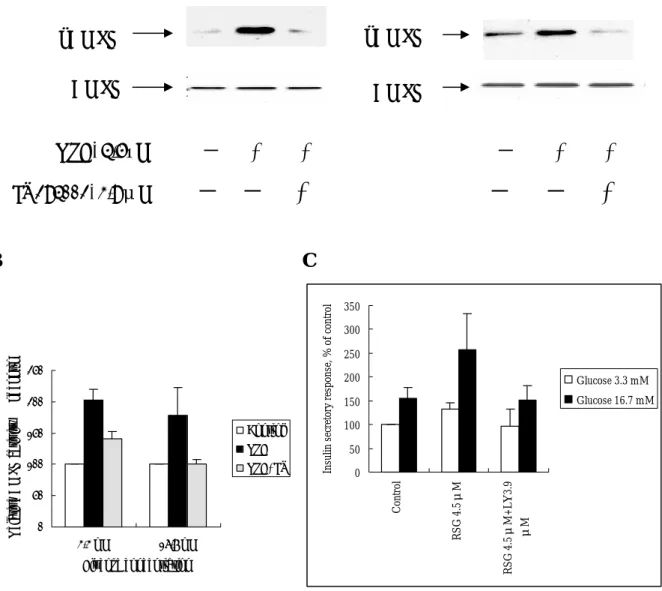
1. RSG enhanced-AMPK phosphorylation and glucose-stimulated insulin secretion (GSIS) is dependent on PI3K in MIN6 beta cells.
Fig. 1. AMPK is activated by RSG treatment either in low (A) and high (B) glucose culture of the MIN6 cells. A Glucose 3.3 mM Glucose 16.7 mM pAMPK RSG, μM 0 0.5 1.5 4.5 AMPK AMPK pAMPK B Glucose 16.7 mM Glucose 16.7 mM RSG 4.5 µM, min AMPK pAMPK AMPK pAMPK 0 10 30 60
Fig. 2. RSG-induced AMPK activation is dependent on PI3K pathway. Both AMPK phosphorylation (A), its enzymatic activity (B) and insulin secretory response (C) is activated by RSG but is blocked by treatment of a PI3K inhibitor LY294002 in either low or high glucose concentrations. A pAMPK pAMPK AMPK AMPK RSG, 4.5µM - + + - + + LY294002, 3.9μM
- - +
- - +
B C 0 50 100 150 200 250 3.3 mM 16.7 mM Glucose concentration Re la tiv e A M PK a ctiv ity , % o f c on tr ol Control RSG RSG+LY 0 50 100 150 200 250 300 350 Cont rol RS G 4.5μ M RS G 4.5μ M + L Y 3.9 μM Ins ul in s ec re to ry re spons e, % of c ont rol Glucose 3.3 mM Glucose 16.7 mM2. Pharmacological activator of AMPK potentiates GSIS in rat islets and beta cells.
Fig. 3. AICAR, an activator AMPK, stimulates insulin secretion in basal glucose concentration (A) and potentiates both first- and second-phase insulin secretion (B) in the isolated pancreatic perfusion system. The activation of AICAR on AMPK activity (C) and the insulin secretion (D) is not affected by PI3K inhibitor as studied in the MIN6 cells. These results indicate AMPK activation by AICAR might be down-stream of PI3K or independent of PI3K pathway.
A. B. C. D. 0 50 100 150 200 250 300 3.3mM 16.7 mM Glucose concentration R el at ive A M PK act iv ity, % of cont ro Control AICAR AICAR+LY 0 50 100 150 200 250 300 350 Control AICA R50μM AIC AR50 μM +LY3 .9μ M In su lin s ec re tor y re spons e, % of c ont ro l Glucose 3.3 mM Glucose 16.7 mM *
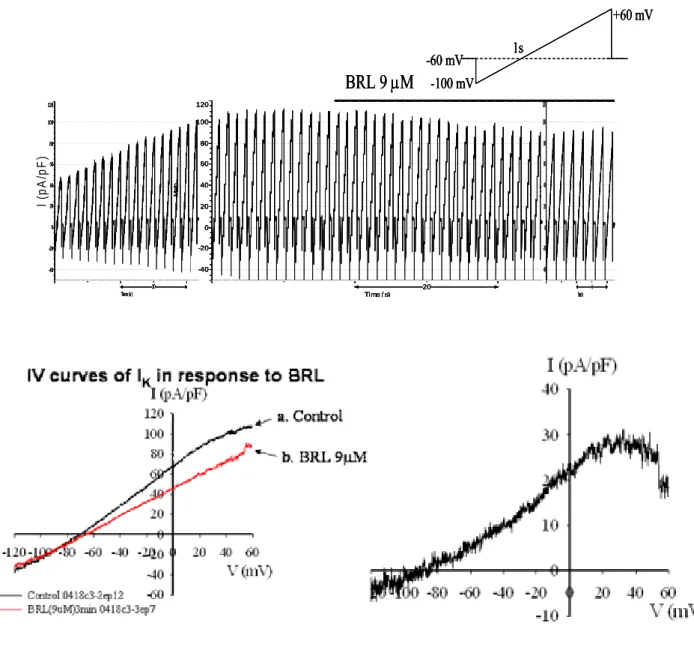
3. Rosiglitazone inhibits KATP channel activity in rat pancreatic islets through the PI3 kinase
dependent pathway.
As shown in Fig. 4A, RSG significantly inhibits K+ currents when the cell membrane voltage of primary islet cells kept above -70mV. In order to confirm that the reduction in the K+ seen in whole cell recordings in the presence of RSG was due to block of KATP channels, primary islets were
incubated in the presence of 30 µM glibenclamide (one of KATP channels inhibitors) and then 9 µM
RSG were added. Initially glibenclamide significantly inhibited the K+ current, and the inhibition was not further enhanced by the addition of RSG (Fig. 4B). It inferred that the reduction in the K+ current seen in whole cell recordings in the presence of RSG was due to block of KATP channels. In
order to verify the inhibition of KATP channel currents by RSG was through the PI3K dependent
pathway, PI3K inhibitor, 10 µM LY294002 was pretreated 10 min before the incubation with RSG. The decreased K+ current at -40 mV in response to RSG was restored by the pretreatment of LY294002 (Fig. 4C).
Fig. 4A 20 Time ( s) Ma th -40 -20 0 20 40 60 80 100 120 10 Time (s) Ma th -40 -20 0 20 40 60 80 100 120 4 Time(s) Ma th -40 -20 0 20 40 60 80 100 120 BRL 9 µM -60 mV-100 mV +60 mV 1s I ( p A /p F ) 20 Time ( s) Ma th -40 -20 0 20 40 60 80 100 120 10 Time (s) Ma th -40 -20 0 20 40 60 80 100 120 4 Time(s) Ma th -40 -20 0 20 40 60 80 100 120 BRL 9 µM -60 mV-100 mV +60 mV 1s 20 Time ( s) Ma th -40 -20 0 20 40 60 80 100 120 10 Time (s) Ma th -40 -20 0 20 40 60 80 100 120 4 Time(s) Ma th -40 -20 0 20 40 60 80 100 120 20 Time ( s) Ma th -40 -20 0 20 40 60 80 100 120 10 Time (s) Ma th -40 -20 0 20 40 60 80 100 120 4 Time(s) Ma th -40 -20 0 20 40 60 80 100 120 BRL 9 µM -60 mV-100 mV +60 mV 1s -60 mV -100 mV +60 mV 1s I ( p A /p F ) Fig. 4B Ma th ) Ma th Ma th Ma th ) I (p A/ I (p A/ glibenclamide 30µM 20 Time (s) -40 -20 0 20 40 60 80 100 20 Time (s) Ma th -40 -20 0 20 40 60 80 100 20 Time (s) Ma th -40 -20 0 20 40 60 80 100 BRL 9µM p F glibenclamide 30µM 20 Time (s) -40 -20 0 20 40 60 80 100 20 Time (s) Ma th -40 -20 0 20 40 60 80 100 20 Time (s) Ma th -40 -20 0 20 40 60 80 100 BRL 9µM glibenclamide 30µM 20 Time (s) -40 -20 0 20 40 60 80 100 20 Time (s) Ma th -40 -20 0 20 40 60 80 100 20 Time (s) Ma th -40 -20 0 20 40 60 80 100 20 Time (s) -40 -20 0 20 40 60 80 100 20 Time (s) Ma th -40 -20 0 20 40 60 80 100 20 Time (s) Ma th -40 -20 0 20 40 60 80 100 BRL 9µM p F
V (mV) -120-100 -80 -60 -40 -20 0 20 40 60 I (pA/pF) -40 -20 0 20 40 60 80 100 Control0418k05-2 ep21 0418k05-3 ep33 glib30uM 0418K05-4 ep10 BRL 2min C. Before BRL After BRL K + cu rr e n t a t -4 0 mV (p A /p F ) 0 10 20 30 40 50 60 70 Control (n=5) LY 10 µM pretreated cells (n=5)
*
9 µM4. Interacting proteins from yeast two hybrid screening for Calpain 10.
a. screening for 3x106 clones ↓AH109 (pAS2-1 Capn 10)
↓50 µg MING6 cDNA library screening ↓SD/-Leu/-Trp/-His +25 mM 3-AT selection ↓30 ℃ incubation for 15 days
↓collect 282 independent colonies
↓restreak onto SD/-Leu/-Trp/-His/-Ade + 25 mM 3-AT
→ test for α-galactosidase activity
→ test for β-galactosidase activity → yeast glycerol stock
α β β β β α α α Clone 1- Clone 32- -ga -ga -ga -ga Clone Clone 102-
However, in the confirmation test with cotransformation assay as depicted below, all clones
o further screening for the interacting proteins for calpain 10 gene, we mutated the catalytic site reveal no significant results.
Cotransformation assay:
↓deliver two plasmids simultaneously into AH109 ivity
Results:
ferences.
↓test for α-galactosidase and β-galactosidase act
No significant dif
T
and rescreen the libraries with this catalytic-defective mutant of calpain 10. This second attempts isolated some clones that remained to be further confirmation (table).
Table. Clones isolated with catalytic-defective mutant of calpain 10 in MIN6 cells. Clone number Gene product Clone number Gene product 5 Translocase of Inner mit.
membrane (Timm 17a)
196 Coatomer protein
6 Amino-terminal enhancer of split (AES)
199 Ubiquitin protein ligase
49 Muf1 protein 202 Tumor protein translationally controlled 1 (Tpt1)
61 Protein tyrosine
phosphatase receptor type Z polypeptide 1 (Ptprz1)
214 Enolase 1
65 kinesin 217 Pyrroline-5-carboxylate synthetase (glutamate γ -semialdehyde synthetase)
70 Insulin II (Ins 2) 233 Zinc metalloproteinase (STE24 homolog)
73 Type 2 proinsulin
processing endopeptidase (subtilisin homolog)
246 dystroglycan
83 Secretogranin III 257 Islet amyloid polypeptide 137 ATPase, H+transporting,
lysosomal 50/57 kD, V1 subunit H (ATP6V1h)
277 Low-density lipoprotein receptor-related protein associated protein 1 (Lrpap1)
194 Syntaxin binding protein 278/280 Lrpap1
討論
this study, we report unequivocal evidence for AMPK in the role of RSG-induced and PI3
e reports for the effect of thiazolidinediones (TZDs) on pancreatic beta cells are not con
In
K-dependent insulin secretion in pancreatic beta cells via negative modulation of KATP channel
activities. So far, th
sistent because of different experimental conditions. In the in vivo studies, treatment with TZDs is often accompanied by an improvement in GSIS in type 2 diabetic patients and various animal models of the disease (Kim HI, 2004). However, the in vitro study results are contradictory in various reports. It has been reported that both troglitazone and rosiglitazone can induce GSIS in the HIT beta cells, rat islets, and isolated rat pancreas (Masuda K, 1995; Ohtani KI, 1998; Yang C, 2001). The effect on beta cells were variably attributed to the binding to SUR1 without closure of the KATP channel, direct activation of Ca2+ channels, and stimulation of phosphatidylinositol (PI)
3-kinase (Masuda K,1995; Ohtani KI,1998; Yang C, 2001). On the other hands, two studies failed to demonstrate the stimulatory effect of TZDs on insulin secretion as studied in perifused rat islet system and in isolated human or rat islets (Zawalich WS, 2003; Dubois M 2000). Interestingly, in
mice with a selective knockout of PPARγ in β cells, treatment of RSG can improve insulin resistance and glucose tolerance when these mice were placed on a high-fat diet (Rosen ED 2003). It is therefore highly speculative that TZDs can potentiate GSIS through a non-genomic and PPARγ-independent pathway.
To explore the mechanism by which RSG acutely stimulates insulin secretion from the β cells, we nd four s to be estab ier G Z, , ent looked for the possible involvement of AMPK. As demonstrated previously, AMPK has been shown to be activated by TZDs in many tissue cells such as the muscle, liver as well as adipose tissue (Fryer, 2002; Saha AK, 2004). For the first time we found that RSG acutely activated the Thr172 phosphorylation and enzyme activity of AMPK in pancreatic beta cells as well. How AMPK is regulated is not clear in the beta cells. In this study, we found activation of AMPK by RSG was blocked by a specific PI3K inhibitor, indicating PI3K is an upstream regulator of AMPK. In supporting our findings, other report in the bovine aortic endothelial cells has elucidated a PI3K-dependent activation of AMPK (Zoo MH, 2003). However, this relationship is not reproduced in the rat skeletal muscles where AMPK-activated glucose transport is mediated by a PI3K-independent pathway (Bergeron R, 1999). Further study to define the detailed signaling pathway(s) that lead to activation of AMPK in the pancreatic beta cells is warranted.
KATP channels consist of a hetero-octamer of four sulfonylurea receptors (SUR1) a
inwardly rectifying K+ channel subunits (Kir6.2) (Seino S, 1999). So far, KATP channel activity has
been known to be regulated by the increased intracellular ATP/ADP ratio in the physiological response to glucose stimulation (Straub SG, 2002), and by certain pharmacological agents that bind to SUR1 directly (Lawrence CL 2001, Meyer M 1999). Although TZDs have ever been shown to potentiate GSIS in insulin-secretory cell lines through blocking KATP channel activity (Rowe, et al
1997; Lee K,1996), the molecular mechanism is not well understood. In present study, we show that RSG-induced acute blockage of KATP channel activity is dependent on PI3 kinase activity.
Whether AMPK serves as a link between RSG-PI3K signaling and KATP channel remain
lished. We found that short-term treatment with AMPK activator, AICAR not only stimulated basal insulin secretion but also potentiated GSIS in both the first- and second phase insulin
secretions. Different reports showed very different relation between activation of AMPK and glucose-stimulated insulin secretion (Akkan AG 1994; Malaisse WJ 1994,34,35; da Silva Xav 2003, da Silva Xavier G 2000). The controversy between different reports may be explained in part by different glucose concentrations and the systems employed in the respective studies. When glucose concentration was in a range of near physiologic conditions between 5.6 and 11.1 mM, AICAR is capable in potentiating insulin secretion (Akkan AG 1994; Malaisse WJ 1994; Wang C 2005). This potentiation effect was abolished in the presence of a very high glucose concentration (20-30 mM) in other reports (da Silva Xavier G 2003, da Silva Xavier G 2000). However, we had reported that PI3K signaling is not involved in the physiologic glucose-stimulated insulin secretion but plays an important role of the potentiation effect of RSG (Yang 2001). It is difficult to conclude how important of the activation of AMPK in physiologic glucose-stimulated insulin secretion. In conclusion, we reported that RSG potentiated insulin secretion via a PI3 kinase-depend activation of AMPK and subsequently inhibited the KATP channel currents (see model in Figure 7).
Due to the acute effect of RSG on signaling molecules, the KATP channel activity, and insulin
secretion, the potentiation effect on insulin secretion by RSG is probably through a non-genomic PPARγ-independent pathway. However, so far, we could not yet confirm the function of calpain 10 and its interacting proteins in the pancreatic beta cells.
Reference
C, Chuang LM: Genetics of type 2 diabetes. J Gen Mol Biol 9:37-68, 1998
ac
3. , Zhou YT, Lee Y, Unger RH: Troglitazone lowers islet fat and
4. Higa M, Zhou YT, Ravazzola M, Baetens D, Orci L, Unger RH: Troglitazone
5. S litazone-induced
6. U in the pathogenesis of obesity-dependent NIDDM: genetics
7. ry JD, Unger RH: Beta-cell
8. L , Esser V, McGarry JD, Unger RH: Increased lipogenic
9. TJ, Chang JC, Liu MW, Tai TY, Hsu WH, Chuang LM
10. litazone and
11. rotein kinase. Fuel gauge of the
12. tassium channels, sulphonylurea
13 but not
14.
1. Chiu K
2. Lebovitz HE: Insulin mimetic and insulin-sensitizing drugs. Diabetes Res Clin Pr 20:89-91, 1993
Shimabukuro M
restores beta cell function of Zucker diabetic fatty rats. J Biol Chem 273:3547-3550, 1998
prevents mitochondrial alternations, β cell destruction, and diabetes in obese prediabetic rats. Pro Natl Acad Sci U S A 96:11513-11518, 1999
himabukuro M, Koyama K, Lee Y, Unger RH: Leptin- or trog
lipopenia protects islets from interleukin 1 beta cytotoxicity. J Clin Invest 100:1750-1754, 1997
nger RH: Lipotoxicity
and clinical implications. Diabetes 44:863-870, 1995 Lee Y, Hirose H, Ohneda M, Johnson JH, McGar
lipotoxicity in the pathogenesis of non-insulin-dependent diabetes mellitus of obese rats: impairment in adipocyte-beta-cell relationship. Proc Natl Acad Sci U S A 91:10878-10882, 1994
ee Y, Hirose H, Zhou YT
capacity of the islets of obese rats: a role in the pathogenesis of NIDDM. Diabetes 46:408-413, 1997
Yang C, Chang
Rosiglitazone (BRL 49653) enhances insulin secretory response via phosphatidylinositol 3-kinase pathway. Diabetes 50:2598-2602, 2001
Fryer LG, Parbu-Patel A, Carling D: The anti-diabetic drugs rosig
metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem 277:25226-25232, 2002
Hardie DG, Carling D: The AMP-activated p mammalian cell? Eur J Biochem 246:259-273, 1997
Dunne MJ, Cosgrove KE, Shepaherd RM, Ammala C: Po
receptors and control of insulin release. Trend Endocrionol Metab 1999;10:146-152. . Sunaga Y, Inagaki N, Gonoi T, Yamada Y, Ishida H, Seino Y, Seino S: Troglitazone
pioglitazone affects ATP-sensitive K+ channel activity. Eur J Pharmacol 1999;381:71-76. Hayward RA, Manning WG, Kaplan SH, Wagner EH, Greenfield S: Starting insulin
therapy in patients with type 2 diabetes: effectiveness, complications, and resource utilization. J Am Med Assoc 1997;278:1663-1669.
Dukes ID, Philipson LH: K+ channels: generating
15. excitement in pancreatic beta-cells.
16. ang J, Smukler SR, Sun AM, Gaisano HY, Salapatek AM,
17. M ang J,
Diabetes 1996;45:845-853. MacDonald PE, Ha XF, W
Backx PH , Wheeler MB: Members of the Kv1 and Kv2 voltage-dependent K(+) channel families regulate insulin secretion. Mol Endocrinol 2001;15:1423-1435.
acDonald PE, Sewing S, Wang J, Joseph JW, Smukler SR, Sakellaropoulos G, W Saleh MC, Chan CB, Tsushima RG, Salapatek AM, Wheeler MB: Inhibition of Kv2.1
voltage-dependent K+ channels in pancreatic β cells enhances glucose-dependent insulin secretion. J Biol Chem 2002;277:44938-44945.