Ab Initio Chemical Kinetics for the Hydrolysis of N
2
O
4
Isomers in the
Gas Phase
R. S. Zhu,
†Ke-Yu Lai,
‡and M. C. Lin*
,†,‡†Department of Chemistry, Emory University, Atlanta, Georgia 30322, United States
‡Center for Interdisciplinary Molecular Science, National Chiao Tung University, Hsinchu 300, Taiwan
*
S Supporting InformationABSTRACT: The mechanism and kinetics for the gas-phase hydrolysis of N2O4 isomers have been investigated at the CCSD(T)/6-311++G(3df,2p)//B3LYP/6-311++G(3df,2p) level of theory in conjunction with statistical rate constant calculations. Calculated results show that the contribution from the commonly assumed redox reaction of sym-N2O4 to the homogeneous gas-phase hydrolysis of NO2can be unequivocally ruled out due to the high barrier (37.6 kcal/mol) involved; instead, t-ONONO2 directly formed by the association of 2NO2, was found to play the key role in the hydrolysis process.
The kinetics for the hydrolysis reaction, 2NO2+ H2O↔ HONO + HNO3(A) can be quatitatively interpreted by the two step mechanism: 2NO2→ t-ONONO2, t-ONONO2+ H2O→ HONO + HNO3. The predicted total forward and reverse rate constants for reaction (A), ktf = 5.36 × 10−50T3.95exp(1825/T) cm6 molecule−2 s−1 and k
tr = 3.31 × 10−19T2.478exp(−3199/T) cm3 molecule−1s−1, respectively, in the temperature range 200−2500 K, are in good agreement with the available experimental data.
I. INTRODUCTION
Nitrogen oxides play important roles in a wide variety of upper and lower atmospheric systems.1Hydrolysis of NO2is of parti-cular interest in atmospheric chemistry because it can generate nitrous acid (HONO), a major source of OH in polluted urban atmospheres; the reaction can be represented by reaction A
+ → +
2NO2 H O2 HONO HNO3 (A)
Reaction A, expressed in terms of the two-step mechanism: =
2NO2 N O2 4 (B)
+ = +
N O2 4 H O2 HNO2 HNO3 (C) has been commonly proposed to describe the conversion of nitrogen dioxide to nitric acid in aqueous solution. Experi-mentally, the heterogeneous formation of HONO in the hydrolysis of NO2 in water has been extensively studied.
2−11
Most studies show that the rate of absorption of nitrogen dioxide is proportional to the concentration of the dimer of nitrogen dioxide, N2O4. Finlayson-Pitts and co-workers proposed a mechanism7 in which the symmetric N2O4 (sym-N2O4) is formedfirst, followed by isomerization to ONONO2; the latter reacts with H2O to form HONO and HNO3. The kinetics for the homogeneous gas-phase reaction A has been studied by England and Corcoran12in the temperature range 298−323 K in N2 (760 Torr) under highly diluted condi-tions. The forward and reverse rate constants for reaction A have been measured and reported. Theoretically, several authors13−16have investigated N2O4isomeration and its interac-tion with H2O from various aspects using density functional
theory (DFT) and“on the fly” molecular dynamics calculations at the MP2 level. Chou et al.13studied the reaction of sym-metric N2O4 with water vapor using the DFT method, their results show that the direct production of HONO and HNO3 needs to overcome more than 30 kcal/mol barrier and the energy barrier is unafected by the presence of multiple water molecules. Pimentel et al.14studied the isomerization of sym-N2O4 to ONONO2 with the combined DFT/B3LYP/13s8p-(2d,1f) level of theory, the isomerization barrier was found to be 31 kcal/mol at 298 K. Miller et al.15 investigated the ionization of N2O4in contact with water using the on-the-fly molecular dynamics simulation method at the MP2 level; they concluded that ionization of N2O4in and on thin water film surface is a key step in the hydrolysis of NO2to form HONO. In the most recent paper of Medeiros and Pimentel,16 their studies show that NO2 dimerization and N2O4 isomerization in the water-film surface are believed to be the key steps in the hydrolysis of NO2. However, the initiation mechanism occurring in the atmosphere involving N2O4and H2O is still poorly understood; no theoretical kinetics calculations have been performed and reported in the literature.
The major objective of this work is to elucidate the gas-phase mechanism for the redox reaction of NO2in the presence of H2O; the roles of the symmetric and asymmetric isomers of N2O4 have been studied by careful mapping of the poten-tial energy surface of the system and the rate constants for the Received: March 7, 2012
Revised: April 16, 2012
Published: April 16, 2012
low-lying energy channels are predicted and compared with the available experimental data. Our result can unequivocally rule out the contribution from the redox reaction of sym-N2O4to the homogeneous gas-phase hydrolysis of NO2.
II. COMPUTATIONAL METHODS
The structures and frequencies of the species involved in the reaction have been fully optimized by using the hybrid density functional (B3LYP) method (i.e., Becke’s three-parameter nonlocal exchange functional17−19with the correlation functional of Lee, Yang, and Parr20) using the standard 6-311++G(3df,2p) basis set. Intrinsic reaction coordinate (IRC) calculations21have been performed to confirm the connection of each transi-tion state with designated intermediates and the single point energies were refined at the CCSD(T)/6-311++G(3df,2p) level of theory. All calculations have been carried out using the Gaussian 03 program package.22
The third-order rate constant for the hydrolysis reaction, 2NO2+ H2O ↔ HONO + HNO3is interpreted by the two-step mechanism:
= K
2NO2 ONONO isomers2 ( eq)
+ ↔ + k
ONONO isomers2 H O2 HONO HNO3 ( )f
The second-order rate constant kf has been predicted by the transition state theory (TST)24 implemented in the Variflex code.23 Microcanonical Rice−Ramsperger−Kassel−Marcus (RRKM) theory,24,25 has been used to test the deactivation effect of the complexes. In the RRKM calculations, the L-J parameters,σ = 3.74 Å, ε/κ = 82 K for N2are taken from the ref 26; the L-J values for H2O−ONONO2complexes,σ = 3.67 andε/κ = 419 K are derived from those of H2O (σ = 2.71 Å, ε/κ = 506 K)26
and N2O4(σ = 4.621 Å, ε/κ = 347 K)27using approximation equations of σ(H2O−N2O4) = [σ(H2O) + σ(N2O4)]/2 and, ε (H2O−N2O4) = [ε(H2O)ε(N2O4)]1/2.
The minimum energy path (MEP) representing the barrierless association processes of H2O + t-ONONO2 is obtained by calculating the potential energy curve along the reaction coordinate O−N bond in the complex LM1 (Figure 1) where the O−N bond length is stretched from the equilibrium value 2.616 to 5.016 Å with an interval step size of 0.2 Å and
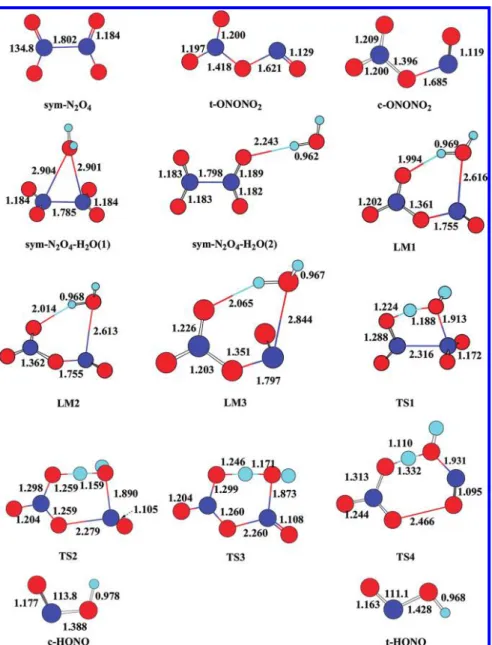
Figure 1.Optimized structures of the reactants, intermediates, and transition states for the reactions of H2O + sym-N2O4and t-ONONO2at the
other geometric parameters are fully optimized. The dissociation curve (see Supporting Information SI-1) can befitted to the Morse potential function, E(R) = De[1− exp(−β(R − Re))]2, which is employed to approximate the MEP path for the variational transition state in the rate calculation. In the above equation, R is the reaction coordinate (i.e., the distance between the two bonding atoms; the O−N in this work), Deis the bond energy excluding zero-point energy, and Re is the equilibrium value of R (2.616 Å). The computed potential energies can be fitted reasonably to the Morse potential function with the parameterβ = 1.763 Å−1. The numbers of states for the tight transition states are evaluated according to the rigid-rotor harmonic-oscillator approximation. The lower vibrational modes in the transition states are treated as classical one-dimensional free rotors in our rate constant calculations. For those paths involving hydrogen atom transfer, Eckart tunneling effects28,29 were taken into account in the rate constant calculations.
Equilibrium constants were calculated on the basis of Kp = exp(−ΔG/RT), where R is the ideal gas law constant with 0.0821 dm3 atm mol−1 K−1; ΔG is the standard Gibbs free energy change, determined byΔH − TΔS, ΔH = ∑Hf,products− ∑Hf,reactants, which is calculated at the CCSD(T)/6-311+ +G(3df,2p) level of theory. ΔS = ∑Sproducts − ∑Sreactants, which is calculated on the basis of the structural parameters calculated at the B3LYP/6-311++G(3df,2p) level.ΔG values at different temperatures are calculated using the ChemRate program.30
III. RESULTS AND DISCUSSION
Several experimental studies31−34 revealed the existence of asymmetric N2O4isomers besides sym-N2O4(D2h). Previous ab initio calculations35−39 predicted the possible geometries of N2O4 isomers. The most stable isomers are sym-N2O4 and trans-ONONO2.36,39 In this work, the reactions of H2O with sym-N2O4, trans-ONONO2, and cis-ONONO2 have been considered. The structures of the key stationary points and the PES diagram are displayed in Figures 1 and 2, respectively.
The vibrational frequencies and rotational constants are summarized in the Supporting Information (SI-2). To judge the reliability of the coupled clusters approach for this system, the T1diagnostics values for the reactants, intermediates, transi-tion states, and products in CCSD(T) were calculated using the 6-311++G(3df,2p) basis set. The data are summarized in Supporting Information SI-3. It can be seen from Supporting Information SI-3 that besides H2O (T1= 0.009), the T1values for all other species lie in 0.02± 0.004, which are close to the proposed maximum value of 0.02.40
For determination of the rate constants from the different mechanisms involving sym-N2O4 and t-ONONO2 given in Figure 2A,B, respectively, we have attempted to scan the MEPs from their intermediates, sym-N2O4−H2O (1) and t-ONONO2−H2O (LM1 and LM2), back to their reactants, 2NO2 + H2O. The most favorable paths led to sym-N2O4 + H2O and t-ONONO2+ H2O because of the much stronger bind-ing between the 2 NO2molecules; accordingly, we interpret the termolcular rate constants for 2NO2+ H2O on the basis of the two-step mechanism as described by reactions B and C given above for the three isomers as discussed below.
Heats of Reaction and Dissociation Energy. To confirm the reliability of the methods employed, we compared the heats of reaction for this reaction and the dissociation energy for sym-N2O4 with available experimental values in Table 1. Our
calculated heats of reaction for the reaction 2NO2 + H2O → HNO3+ c-HONO/t-HONO, give−6.8 and −7.1 kcal/mol, for the formation of cis- and trans-isomers, respectively; these values are in excellent agreement with the experimental values,−6.7 ± 0.6 and−7.2 ± 0.6 kcal/mol, based on the experimental heats of formation:41NO2(8.59 ± 0.19 kcal/mo), H2O (−57.10 ± 0.01 kcal/mol), HNO3 (−29.75 ± 0.1 kcal/mol), c-HONO (−16.85 ± 0.32 kcal/mol), t-HONO (−17.36 ± 0.32 kcal/ mol). The calculated dissociation energies (D0) for sym-N2O4 and t-ONONO2to 2NO2 are 12.0 and 5.7 kcal/mol, respec-tively, which are also consistent with the values, 12.4 and 6.2 kcal/mol calculated at the QCISD/6-31+G(2df)//B3LYP/ 6-31G(d) level.36 The former is in good agreement with the experimental value, 12.7± 0.8 kcal/mol, based on the heats of formation of NO2and sym-N2O4.41
H2O + sym-N2O4. Two NO2 can barrierlessly form sym-N2O4; then the interaction of H2O with sym-N2O4 further forms two complexes, sym-N2O4−H2O (1) and sym-N2O4− H2O (2), with 5.0 and 1.0 kcal/mol association energies at the CCSD(T)/6-311++G(3df,2p) level, which are consistent with the values, 4.3 and 1.3 kcal/mol, reported by Chou et al.13 at the B3LYP/6-311+(2d,p) level. Theoretically, the com-plexes can be formed directly from 2NO2+ H2O (Figure 2).
Figure 2.Schematic energy diagrams (in kcal/mol) of H2O +
sym-N2O4(A) and t-ONONO2 (B), computed at the CCSD(T)/6-311+
+G(3df,2p)//B3LYP/6- 311++G(3df,2p) level.
Table 1. Comparison of Calculated Heats of Reaction (kcal/ mol) for 2NO2+ H2O andD0ofsym-N2O4with the Available Experimental Data CCSD(T)/6-311++G(3df,2p)a calc (0 K) expt (0 K)b 2NO2+ H2O 0.0 t-HONO + HNO3 −7.1 −7.2 ± 0.6 c-HONO + HNO3 −6.8 −6.7 ± 0.6 NO2+ NO2 0.0 sym-N2O4 −12.1 −12.7 ± 0.8 t-ONONO2 −5.7
aBased on the optimized structures at the B3LYP/6-311++G(3df,2p)
sym-N2O4−H2O (1) dissociates to HNO3+ trans-HONO via a five-membered ring transition state TS1 with 37.6 kcal/mol energy above H2O + sym-N2O4. At B3LYP/6-311+(2d,p), this barrier was reported to be 32.1 kcal/mol;13we believe our value is more reliable because of the larger basis set and the higher level calculation. sym-N2O4−H2O (2) has a very shallow well (−1.0 kcal/mol) and the H2O molecule is just a“spectator” in N2O4−H2O (2), which is not involved in the formation of HONO. The rate constant for the formation of HONO + HNO3via sym-N2O4−H2O (1) can be represented by
‐ + = × − × − − − k T T (sym N O H O) 7.61 10 exp( 15011/ ) cm molecule s 2 4 2 26 4.53 3 1 1
in the temperature range 200−2500 K. At 298 K, k = 5.7 × 10−37 cm3molecule−1s −1. Apparently, the rate is too low for the hydrolysis of N2O4producing HONO + HNO3in the gas phase as will be discussed later.
H2O + t-ONONO2. Again, 2NO2 can also directly form t-ONONO2with 5.7 kcal/mol association energy (Figure 2B). The interaction of H2O with t-ONONO2 forms two weakly bonded complexes LM1 and LM2 with 3.3 and 3.5 kcal/mol association energies. LM1 and LM2 can dissociate to HNO3+ t-HONO and c-HONO products via TS2 and TS3 with 6.4 and 8.0 kcal/mol barriers, respectively, above H2O + t-ONONO2. As one can see, these transition state barriers are about 30.0 kcal/mol lower than that of sym-N2O4−H2O system. The fact that t-ONONO2is more reactive than sym-N2O4has also been illustrated recently in the hypergolic reaction of N2H4 with N2O4isomers.39
Rate constants for the following processes are calculated at 760 Torr N2pressure in the temperature range 200−2500 K.
+ ‐ → ‐ ‐ → ‐ + → ‐ + t t k t k c k H O ONONO ONONO H O ( ) HONO HNO ( ) HONO HNO ( ) 2 2 2 2 as 3 2 3 3
The predicted results show that at low temperatures (<280 K), the association process is dominant; with the temperature increasing, the formation of t-HONO + HNO3becomes com-petitive and dominant. The calculated individual rate constants are plotted in Figure 3; they can be represented in units of cm3 molecule−1s−1by the following equations:
= × − = × − = × − + = × − − − − − k T T k T T k T T k k T T 6.49 10 exp( 641/ ) 3.61 10 exp( 2121/ ) 6.96 10 exp( 2775/ ) 3.2 10 exp( 2045/ ) as. 20 14.63 2 17 1.489 3 18 1.695 2 3 18 1.88
It should be mentioned that the mechanism involving the isomerization of sym-N2O4 to ONONO2 is not considered in our calculation due to the high isomeization barrier. For example, at the B3LYP/6-311++G(3df,2p) level, the isomeiza-tion barrier is 40.28 kcal/mol; when one H2O molecule is involved in the isomerization reaction, the barrier is essentially the same, 40.33 kcal/mol, attributable to the factor that the H2O−N2O4complex and the transition state both are stabilized by the same solvation energy of the added water.
H2O +c-ONONO2. Similar calculations have been made for the reaction of c-ONONO2 formed by 2NO2 with only 3.1 kcal/mol association energy; the reaction with H2O forms a weak complex (LM3) with a 2.9 kcal/mol binding energy. LM3 can decompose to t-HONO + HNO3via TS4 with a noticeably higher barrier of 10.2 kcal/mol. The third-order rate constant for the c-ONONO2reaction is shown as a dash-dot-dotted line 4 in Figure 6. As one can see, the lower stability of c-ONONO2 and the higher exit barrier of TS4 result in a significantly less contribution to the third-order redox reaction of NO2 with H2O, <1% at 298−800 K, comparing with that from the t-ONONO2reaction presented above.
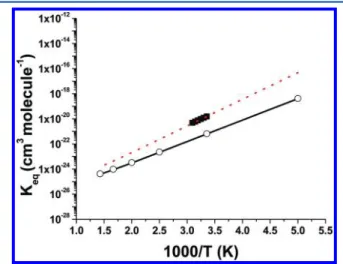
Equilibrium ConstantKA. The predicted KAis plotted as a solid line in Figure 4, which is in good agreement with the
results (given by circles) based on theΔG values reported in NIST-JANAF Thermochemical Tables.41 However, our cal-culated values are lower than those given by England and Corcoran;12in their work, the KA= kf/krwhere kfis the experi-mental third-order rate constant for 2NO2+ H2O and the kris the second-order rate constant for the reverse HONO + HNO3
Figure 3.Rate constants for the reaction of H2O + t-ONONO2. The
dashed line is the association rate constant for the formation of t-ONONO2−H2O complex; the dash-dotted and dotted lines represent
the individual rate constants for the formation of c-HONO + HNO3,
t-HONO + HNO3.
Figure 4.Equilibrium constant for the reaction of 2NO2 + H2O =
HONO + HNO3.Solid and dotted lines are calculated on the basis of
the heat of reactionΔHr=−7.1 and −9.0 kcal/mol, respectively.○,
calculated on the basis of theΔG values taken from ref 41;■, taken from ref 12.
reaction. Based on their KA, the heat of reaction at 298 K is 1.9 kcal/mol lower than ours; using their heat of reaction and our predicted geometric parameters, the calculated values (given by dotted line in Figure 4) can reproduce their reported data.
Rate Constant for HONO + HNO3. On the ground that the bimolecular reaction HONO + HNO3via TS1 producing sym-N2O4has a significantly higher barrier, only the rate con-stants for the low-energy channels via TS2, TS3, and TS4 have been calculated. In these reverse reactions, the deactiva-tion rates for the excited intermediates LM1−LM3 can be rea-sonably ignored as they can readily dissociate to H2O + t-ONONO2 (or H2O + 2NO2) and H2O + c-ONONO2 (or H2O + 2NO2). Accordingly, the conventional TST24was used to calculate the rate constants for these reverse reaction channels with Eckart tunneling corrections.28,29ktris the sum of the individual rates via TS2, TS3, and TS4. The negligible deactivation effects have been confirmed by our RRKM cal-culation. At 760 Torr N2, 100% of activated complexes barrier-lessly dissociate to H2O + t-ONONO2 in the range of 200− 2500 K, followed by the rapid fragmentation of t-ONONO2to 2NO2with only 5.7 kcal/mol dissociation energy.
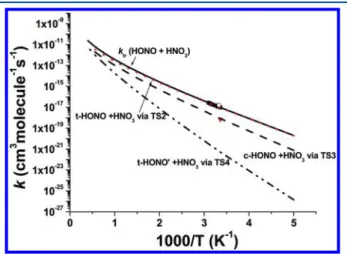
The predicted result is plotted and compared with avail-able experimental values in Figure 5. Our calculated
indivi-dual and total rate constants can be represented in units of cm3 molecule−1s−1by ‐ + = × − ‐ + = × − ‐ ′ + = × − + = × − − − − − k c T T k t T T k t T T k T T ( HONO HNO ) 1.28 10 exp( 3816/ ) via TS3 ( HONO HNO ) 3.32 10 exp( 3253/ ) via TS2 ( HONO HNO ) 2.78 10 exp( 5987/ ) via TS4 (HONO HNO ) 3.31 10 exp( 3199/ ) 3 18 2.186 3 18 2.092 3 19 2.465 tr 3 19 2.478
Figure 5 shows that in the whole temperature range, the reac-tion of t-HONO + HNO3 via TS2 is dominant. At 298 K,
ktr(HONO + HNO3) = 1.04× 10−17cm3molecule−1s−1, which is in good agreement with the experimental values, 1.18× 10−17 (ref 12) and 1.55× 10−17cm3molecule−1s−1(ref 42). However, the value measured by Wallington and Japar,43 7.01 × 10−19 cm3molecule−1 s−1, although in agreement with our predicted result for the c-HONO + HNO3 reaction, 9.34 × 10−19 cm3 molecule−1s−1, is lower than the predicted total rate.
Third-Order Rate Constant for 2NO2+ H2O. In the work of England and Corcoran,12the experimentally measured third-order rate constant for the homogeneous gas-phase hydrolysis reaction, 2NO2 + H2O → HONO + HNO3 (A), was inter-preted according to the two-step mechanism: 2NO2 → sym-N2O4 (B) and sym-N2O4+ H2O→ HONO + HNO3 (C) as alluded to above. On the basis of this mechanism, we can theoretically calculate the equilibrium constant and the second-order rate constant for the above reactions, denoted as Keq (sym-N2O4) and k(sym-N2O4 + H2O), employing our quantum chemically predicted energetics and molecular parameters. The third-order rate constant can be given by kf= Keq(sym-N2O4)× k(H2O + sym-N2O4); the computed result is presented by the dotted line in Figure 6. From thefigure, one can see that in the
experimental temperature range, the calculated kfis more than 10 orders of magnitude lower than the experimental data reported by England and Corcoran,12attributable to the high barrier at TS1 (37.6 kcal/mol) for the formation of HONO + HNO3 from the sym-N2O4 + H2O reaction. Therefore, the role of sym-N2O4in the formation of HONO + HNO3can be ignored.
On the other hand, if we assume that the third-order redox reaction of NO2with H2O involves primarily the reaction of t-ONONO2 with H2O, we can use the following reaction mechanism as depicted by the PES in Figure 2B to interpret England and Corcoran’s experimental data:
↔ ‐t
2NO2 ONONO2
‐ + → +
t ONONO2 H O2 HONO HNO3
Figure 5.Rate constants for the reaction of HONO + HNO3. Dashed,
dotted, and dash-dot-dotted lines represent the calculated values of k(c-HONO + HNO3) via TS3, k(t-HONO + HNO3) via TS2, and
k(t-HONO′ + HNO3) via TS4; the solid line is the total rate, ktr(HONO +
HNO3); symbols are the experimental values:■, ref 12;○, ref 42;▼,
ref 43.
Figure 6.Third-order rate constants for the reaction of 2NO2+ H2O
→ HONO + HNO3, calculated on the basis of the mechanism of
2NO2= t-ONONO2; H2O + t- ONONO2= HONO + HNO3.
Dash-dotted line 1, dashed line 2, and solid line 3 represent the individual rate constants for the formation of c-HONO + HNO3, t- HONO +
HNO3, and the total rate ktf. Dash-dot-dotted line 4 is calculated on
the basis of the mechanism of 2NO2 = c-ONONO2; H2O +
c-ONONO2= t-HONO + HNO3. The dotted line is calculated on the
basis of the mechanism of 2NO2= sym-N2O4; H2O + sym-N2O4=
Similar to the above calculation, the forward third-order rate constant kf for 2NO2 + H2O can be computed by Keq (t-ONONO2)× k(H2O + t-ONONO2), where Keq(t-ONONO2) is the equilibrium constant for 2NO2↔ t-ONONO2; the values for k(H2O + t-ONONO2) have been calculated and presented in Figure 3 as k2and k3. The individual forward rate constants [k(t-HONO) and k(c-HONO)] for 2NO2 + H2O producing t-HONO + HNO3and c-HONO + HNO3 are calculated by k(t-HONO) = Keq(t-ONONO2)× k2and k(c-HONO) = Keq (t-ONONO2) × k3, respectively, and ktf = k(t-HONO) + k(c-HONO).
The calculated total ktfis summarized in Table 2 to compare with available experimental data. The individual and total rate constants given in units of cm6molecule−2s−1are also shown in Figure 6; they can be represented by
‐ = × ‐ = × = × − − − k c T T k t T T k T T ( HONO) 2.72 10 exp(951/ ) ( HONO) 6.80 10 exp(1654/ ) 5.36 10 exp(1825/ ) 49 3.670 47 3.565 tf 50 3.95
in the temperature range 300−2500 K. Calculated results in Figure 6 show that the formation of t-HONO + HNO3is domi-nant in the whole temperature range computed; the predicted total ktf values are in good agreement with the experimental data of England and Corcoran.12In the temperature of range 300−500 K, ktfexhibits a small negative T-dependent because the decrease in the value of Keq(t-ONONO2) with temperature is larger than the increase rate of k(H2O + t-ONONO2), fully consistent with the experimental observation.
IV. CONCLUSIONS
The gas-phase mechanism and kinetics for the reaction of 2NO2+ H2O have been elucidated at the CCSD(T)/6-311+ +G(3df,2p)//B3LYP/6-311++GH(3df,2p) level aided by stat-istical theory calculations. Our predicted results show that the termolecular reaction of 2NO2with H2O can directly form sym-N2O4−H2O, t-ONONO2−H2O, and c-ONONO2−H2O com-plexes with 17.0, 9.0/9.2, and 6.0 kcal/mol association energies, respectively. Further transformation of these complexes by undergoing intramolecular redox reactions giving rise to HONO + HNO3requires 42.6 kcal/mol activation energy at TS1 for the sym-N2O4−H2O complex and only 9.7, 11.5, and
13.1 kcal/mol at TS2, TS3, and TS4 for LM1, LM2 (t-ONONO2−H2O complexes), and LM3 (c-ONONO2−H2O complex). The existing experimental results for the hydrolysis reaction, 2NO2 + H2O → HONO + HNO3, can be quanti-tatively accounted for primarily by the two-step mechanism: 2NO2→ t-ONONO2, t-ONONO2+ H2O→ HONO + HNO3. Our finding may help understand the mechanism for the absorption of nitrogen dioxide into aqueous solution. A comparison of the gas phase with the aqueous solution kinetics for this important system will be investigated in the near future.
■
ASSOCIATED CONTENT*
S Supporting InformationSI-1, the dissociation curve of t-ONONO2−H2O complex; SI-2, the vibrational frequencies and rotational constants for the intermediates, transition states, and products in the reactions of 2NO2+ H2O, computed at the B3LYP/6-311+G(3df,2p) level of theory; SI-3, the T1 diagnostics for the reactants, intermediates, transition states, and products in the reactions of 2NO2 + H2O, computed at the CCSD(T)/6-311++G-(3df,2p) level of theory. Complete citation for ref 22. This material is available free of charge via the Internet at http:// pubs.acs.org
■
AUTHOR INFORMATIONCorresponding Author
*E-mail: [email protected].
Notes
The authors declare no competingfinancial interest.
■
ACKNOWLEDGMENTSWe are grateful to Taiwan’s National Center for High-performance Computing for the CPU and to Taiwan’s National Science Council for a research stipend to K.Y.L. and to M.C.L. for a Distinguished Visiting Professorship at the National Chiao Tung University in Hsinchu, Taiwan. R.S.Z. thanks ONR (N00014-08-1-0106) for the support of this work at Emory University.
■
REFERENCES(1) Finlayson-Pitts, B. J.; Pitts, J. N., Jr. Chemistry of upper and Lower Atmosphere: Theory, Experiments and Application; Academic Press: New York, 2000.
Table 2. Summary of Calculated and Experimental Forward and Reverse Rate Constants for the Reaction of 2NO2+ H2O↔ HNO2+ HNO3(A)
T (K) calc, ktfa calc, ktrb T (K) expt,cktf expt,cktr
300 1.49× 10−37 1.04× 10−17 298 1.52× 10−37 9.71× 10−18 (7.01× 10−19)d 400 9.63× 10−38 3.09× 10−16 303 1.44× 10−37 1.18× 10−17 [1.55× 10−17]e 500 9.62× 10−38 2.68× 10−15 308 1.41× 10−37 1.47× 10−17 600 1.08× 10−37 1.23× 10−14 313 1.36× 10−37 1.79× 10−17 700 1.28× 10−37 3.85× 10−14 318 1.33× 10−37 2.21× 10−17 800 1.57× 10−37 9.51× 10−14 323 1.30× 10−37 2.69× 10−17 1000 2.41× 10−37 3.69× 10−13 1200 3.68× 10−37 9.90× 10−13 1500 6.60× 10−37 2.94× 10−12 1800 1.10× 10−36 6.58× 10−12 2000 1.50× 10−36 1.02× 10−12 2500 2.94× 10−36 2.38× 10−11
(2) Platt, U; Perner, D.; Harris, G. W.; Winer, A. M.; Pitts, J. N., Jr. Nature 1980, 285, 312.
(3) Calvert, J. G.; Yarwood, G.; Dunker, A. M. Res. Chem. Intermed. 1994, 20, 463.
(4) Kotamarthi, V. R.; Gaffney, J. S.; Marley, N. B.; Doskey, P. V. Atmos. Environ. 2001, 35, 4489.
(5) Stutz, J.; Alicke, B.; Neftel, A. J. Geophys. Res. Atmos. 2002, 107, 8192.
(6) Perner, D.; Platt, U. Geophys. Res. Lett. 1979, 6, 917.
(7) Finlayson-Pitts, B. J.; Wingen, L. M.; Sumner, A. L.; Syomin, D.; Ramazan, K. A. Phys. Chem. Chem. Phys. 2003, 5, 223.
(8) Barney, W. S.; Finlayson-Pitts, B. J. J. Phys. Chem. A 2000, 104, 171.
(9) Carter, W. P. L; Atkinson, R.; Winer, A. M.; Pitts, J. N. Int. J. Chem. Kinet. 1981, 13, 735.
(10) Lammel, G.; Cape, J. N. Chem. Soc. Rev. 1996, 25, 361. (11) Ramazan, K. A.; Wingen, L. M.; Miller, Y.; Chaban, G. M.; Gerber, R. B.; Xantheas, S. S.; Finlayson-Pitts, B. J. J. Phys. Chem. A. 2006, 110, 6886.
(12) England, C.; Corcoran, W. H. Ind. Eng. Chem. Fundam. 1974, 13, 373.
(13) Chou, A.; Li, Z. R.; Tao, F. M. J.Phys. Chem. A 1999, 10, 7848. (14) Pimentel, A. S.; Lima, F. C. A.; da Silva, A. B. F. J. Phys. Chem. A 2007, 111, 2913.
(15) Miller, Y.; Finlayson-Pitts, B. J.; Gerber, R. B. J. Am. Chem. Soc. 2009, 131, 12180.
(16) MedeirosDiogo de Jesus, Diogo de Jesus; Pimentel, Andre Silva. J. Phys. Chem. 2011, 115, 6357.
(17) Becke, A. D. J. Chem. Phys. 1993, 98, 5648. (18) Becke, A. D. J. Chem. Phys. 1992, 96, 2155. (19) Becke, A. D. J. Chem. Phys. 1992, 97, 9173.
(20) Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. 1988, B37, 785. (21) Gonzalez, C.; Schlegel, H. B. J. Phys. Chem. 1989, 90, 2154. (22) Frisch, M. J.; Trucks, G. W.; Head-Gordon, M.; Gill, P. M. W.; Wong, M. W.; Foresman, J. B.; Johnson, B. G.; Schlegel, H. B.; Robb, M. A.; Replogle, E. S. et al. Gaussian 92/DFT, Revision B; Gaussian, Inc.: Pittsburgh, PA, 2003.
(23) Klippenstein, S. J.; Wagner, A. F.; Dunbar, R. C.; Wardlaw, D. M. Robertson, S. H. VARIFLEX, VERSION 1.00, 1999.
(24) Gilbert, R. G.; Smith, S. C. Theory of Unimolecular and Recombination Reactions; Blackwell Scientific: Carlton, Australia, 1990.
(25) Troe, J. J. Chem. Phys. 1977, 66, 6745.
(26) Hippler, H.; Troe, J.; Wendelken, H. J. J. Chem. Phys. 1983, 76, 6709.
(27) Cressault, Y.; Connord, V.; Hingana, H.; Teulet, Ph.; Gleizes, A. J. Phys. D: Appl. Phys. 2011, 44, 495202.
(28) Eckart, C. Phys. Rev 1930, 35, 1303.
(29) Hase, W. L. Baer Unimolecular Reaction Dynamics; International Series of Monographs on Chemistry; Oxford University Press: New York, 1996.
(30) Mokrushin, V.; Bedanov, V.; Tsang, W.; Zachariah, M. R.; Knyazev, V. D. ChemRate, Version 1.19; National Institute of Standards and Technology: Gaithersburg, MD, 2002.
(31) Givan, A.; Loewenschuss, A. J. Chem. Phys. 1989, 90, 6135. (32) Givan, A.; Loewenschuss, A. J. Chem. Phys. 1989, 91, 5126. (33) Givan, A.; Loewenschuss, A. J. Chem. Phys. 1991, 94, 7562. (34) Pinnick, D. A.; Agnew, S. F.; Swanson, B. I. J. Phys. Chem. 1992, 96, 7092.
(35) Stirling, A.; Papai, I.; Mink, J.; Salahub, D. R. J. Chem. Phys. 1994, 100, 2910.
(36) McKee, M. L. J. Am. Chem. Soc. 1995, 117, 1629.
(37) Zakharov, I. I.; Kolbasin, A. I.; Zakharova, O. I.; Kravchenko, I. V.; Dyshlovoi, V. I. Theor. Exp. Chem. 2008, 44, No.1, 26−31.
(38) Wang, X.; Qin, Q.-Z.; Fan, K. J. Mol. Struct. (THEOCHEM) 1998, 432, 55.
(39) Lai, K-Y; Zhu, R. S.; Lin, M. C. Chem. Phys. Lett. 2012, In press. (40) Lee, T.J. Lee; Taylor, P. R. Int. J. Quantum Chem. 1989, S23, 199.
(41) Chase, Jr. M. W. NIST-JANAF Thermochemical Tables, 4th ed.; Woodbury, NY, 1998.
(42) Kaiser, E. W.; Wu, C. H. J. Phys. Chem. 1977, 81, 187. (43) Wallington, T. J.; Japar, S. M. J. Atmos. Chem. 1989, 9, 399.