The protective effect of prostacyclin on adriamycin-induced apoptosis
in rat renal tubular cells
Cheng-Hsien Chen
a, Heng Lin
c, Yung-Ho Hsu
a, Yuh-Mou Sue
a, Tzu-Hurng Cheng
a,
Paul Chan
a,b, Tso-Hsiao Chen
a,b,⁎
a
Department of Internal Medicine, Taipei Medical University-Wan Fang Hospital, Taipei, Taiwan
b
Graduate Institute of Medical Sciences, Taipei Medical University, Taipei, Taiwan
c
Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan Received 18 October 2005; accepted 25 October 2005
Abstract
Adriamycin-induced nephrosis in rats is a commonly used experimental model for pharmacological studies of human chronic renal diseases. Adriamycin-induced apoptosis of renal tubular cells has been reported in adriamycin-treated rats. In addition, prostacyclin (PGI2) is known to have various protective effects on many kinds of cells. To investigate the protective effect of PGI2on cells undergoing adriamycin-induced apoptosis, this study selectively augmented PGI2production via adenovirus-mediated transfer of genes for cyclooxygenase-1 (COX-1) and prostacyclin synthase (PGIS) (two key enzymes of PGI2synthesis) to renal tubular cells. This PGI2overexpression protected rat renal tubular cells from adriamycin-induced apoptosis. Ad-COX-1/PGIS transfection was found to reduce the adriamycin-stimulated activities of caspase-3 and caspase-9, inhibit adriamycin-induced release of cytochrome c, elevate the expression of Bcl-xL, and suppress the activation and translocation of nuclear factor-kappaB (NF-κB) in adriamycin-treated renal tubular cells. Our results reveal that selective augmentation of PGI2production can protect rat renal tubular cells from adriamycin-induced apoptosis via the NF-κB signaling pathway. This implies the therapeutic potential of combined COX-1 and PGIS gene transfer in gene therapy for chronic renal diseases.
© 2005 Elsevier B.V. All rights reserved.
Keywords: Prostacyclin (PGI2); Adriamycin; Renal tubular cell; Apoptosis; Cyclooxygenase-1 (COX-1); Prostacyclin synthase (PGIS)
1. Introduction
Adriamycin is the anti-tumor anthracycline antibiotic of choice for the treatment of many solid malignancies and lymphomas. Rats treated with adriamycin develop heart failure as well as a self-perpetuating glomerular nephropathy. Even in the absence of continued adriamycin exposure, the glomerular damage progresses and late-onset tubular lesions are observed (Bertani et al., 1982; Scholey et al., 1989). Within a few weeks after adriamycin administration, the
glomerular filtration rate declines gradually and the animals develop a nephrotic and a tubular syndrome. A long-term study of this pathological change in rats demonstrated severe renal damage with characteristic features of chronic progres-sive renal diseases in humans (Bertani et al., 1982; Okuda et al., 1986). Adriamycin-induced nephrosis in rats, therefore, is a common experimental model used for pharmacological studies of human chronic renal diseases. However, the correlative molecular mechanism of this model is not very well understood.
Induction of apoptosis is an important cytotoxic mechanism of adriamycin (Muller et al., 1998). The apoptosis of renal tubular cells has been reported in adriamycin-treated rats (Zhang et al., 1996). Renal tubular cell apoptosis is a key feature of tubular atrophy, which is a hallmark of chronic renal diseases (Khan et al., 1999; Schelling et al., 1998). Reactive oxygen species derived from redox activation of adriamycin is
⁎ Corresponding author. Nephrology Division, Department of Internal Medicine, Taipei Medical University-Wan Fang Hospital, Taipei, Taiwan. No 111, Sing-Lung Road, Sec. 3, Wen-Shan District, Taipei City 116, Taiwan. Tel.: +886 2 29307930x8117; fax: +886 2 29335221.
E-mail address: [email protected] (T.-H. Chen).
0014-2999/$ - see front matter © 2005 Elsevier B.V. All rights reserved. doi:10.1016/j.ejphar.2005.10.057
a proposed cause of adriamycin cytotoxicity (Singal et al., 2000). Previous studies have indicated that, at submicromolar concentrations, adriamycin induces apoptosis with the activa-tion of caspases in endothelial cells and myocytes (Kotamraju et al., 2000; Sawyer et al., 1999). In mammalian cells, a major caspase activation pathway is the cytochrome c-initiated path-way. In this pathway, a variety of apoptotic stimuli cause cytochrome c release from mitochondria, which in turn in-duces a series of biochemical reactions that result in caspase activation and subsequent cell death (Jiang and Wang, 2004). Cytochrome c release is known to be regulated by Bcl-2 family proteins, including Bcl-2 and Bcl-xL, which bind to the
mitochondrial outer membrane and block cytochrome c efflux (Yang et al., 1997). Therefore, mitochondria-mediated apo-ptosis signaling plays a major role in adriamycin-induced cytotoxicity.
Prostacyclin (PGI2), largely produced in vascular
endo-thelial cells, acts on platelets and blood vessels through its specific cell surface receptor, thereby inhibiting platelet function, dilating blood vessels, and protecting the vascular endothelium (Moncada, 1982). PGI2is also known to inhibit
leukocyte functions such as migration and reactive oxygen species production (Boxer et al., 1980) and inhibit mesangial cell proliferation (Mene et al., 1990). PGI2 is produced by
the cyclooxygenase (COX) system in which COX converts arachidonic acid to PGH2, and PGH2 is subsequently
converted to PGI2 by the action of PGI2 synthase (PGIS)
(Vane and Botting, 1995). The production of PGI2 is
exe-cuted by either COX-1 or COX-2 coupled to PGIS (Smith et al., 2000). Because of its unstable property and valuable clinical implications in vascular physiology, several synthetic analogues of PGI2 with more stable chemical structures
have been developed (Sturzebecher et al., 1986). Among these analogues, beraprost has been reported to prevent radiocontrast nephropathy in LLC-PK1 cells (Yano et al., 2005).
The possibility of protecting the heart during the administration of adriamycin by prior administration of prostacyclin has been reported (Dowd et al., 2001). We suppose that PGI2 is able to protect kidney cells from
adriamycin-induced injury. To investigate the possible effect of PGI2on adriamycin-induced apoptosis and its potential in a
gene therapy treatment for renal tubular atrophy, an adenovirus-mediated gene transfer of PGI2 was chosen in
this study to provide a more physiologically relevant augmentation of PGI2 production. Previous experimental
work (Shyue et al., 2001) used a single gene transfer to augment PGI2 production. Overexpression of COX-1 alone
was accompanied by overproduction of PGE2 (a key
proin-flammatory mediator that may contribute to vascular inflam-mation), whereas overexpression of PGIS alone had a minimal effect on increasing PGI2 synthesis. In contrast, combined
COX-1/PGIS gene transfer selectively augmented PGI2
production and is thus better than the single gene transfer approach (Lin et al., 2002). Here, the adenovirus-mediated bicistronic COX-1/PGIS gene transfer was executed in renal tubular cells, and a selective augmentation of PGI2production
was found. The protective effect of endogenous PGI2 on
adriamycin-induced apoptosis was evaluated by analyzing the apoptosis profile of renal tubular cells. Our results reveal that the selective augmentation of endogenous PGI2production can
reduce adriamycin-induced apoptosis. This implies the poten-tial of combined COX-1 and PGIS gene transfer as a gene therapy for chronic renal diseases.
2. Materials and methods 2.1. Materials
Dulbecco's modified Eagle's medium (DMEM), fetal calf serum, and tissue culture reagents were from Invitrogen Corporation (PO, USA). All other chemicals of reagent grade were obtained from Sigma (MO, USA). Antibodies used in this research were purchased from BD Laboratories (CA, USA) and Santa Cruz Biotechnology (CA, USA).
2.2. Cell culture
Rat renal proximal tubular cells (NRK-52E) were purchased from Food Industry Research and Development Institute (Taiwan), and cultured in DMEM supplemented with antibi-otic/antifungal solution and 10% fetal bovine serum. They were grown until the monolayer became confluent. The medium for the cultured cells was then changed to the serum-free medium, and the cells were incubated overnight before the experiment.
2.3. Preparation of replication-defective recombinant adeno-viral vectors
We constructed in the replication-defective recombinant adenoviral (rAd) vector with two separate human phospho-glycerate kinase (HPGK) promoters (bicistronic) to drive COX-1 and PGIS (Ad-COX-1/PGIs), and a HPGK alone to serve as control (Ad-HPGK) as previously described (Lin et al., 2002). Replication-defective rAd vectors were generated by homologous recombination and amplified in 293 cells as described previously (Lin et al., 2002). rAd stocks were prepared by CsCl gradient centrifugation, aliquoted, and stored at −80 °C. Viral titers were de-termined by a plaque-assay method. Two hundred ninety-three cells were infected with serially diluted viral prepa-rations and then overlaid with low melting-point agarose after infection. Numbers of plaques formed were counted within 2 weeks.
2.4. Measurements of eicosanoids by enzyme immunoassay Cells were sonicated in 1 ml of ice-cold buffer (0.05 M Tris at pH 7.0, 0.1 M NaCl, and 0.02 M EDTA) and centrifuged at 55,000 ×g for 1 h. The supernatant was acidified and passed through a Sep-Pak C18 cartridge. Eicosanoids were eluted with 100% methanol, dried under nitrogen gas, redissolved in a small amount of buffer, and
analyzed using 6-keto-PGF1α, PGE2, and 15-d-PGJ2 ELISA
kits from R&D Systems Inc. 2.5. DAPI stain
The cells grown on slides were washed twice in phosphate buffered saline (PBS) for 1 min. The slides were overlaid with DAPI (4′-6-diamidino-2-phenyindole) (1 μg/ml) in PBS plus 0.5% 1,4-diazabicyclo[2,2,2]octane and analyzed immediately.
2.6. TUNEL Stain
Adriamycin-mediated apoptosis in NRK-52E cells was detected by enzymatic labeling of DNA strand breaks using terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end-labeling (TUNEL). The cells grown on slides were washed twice in PBS for 1 min. Following the incubation of slides with the permeabilisation solution (0.1% Triton X-100 in 0.1% sodium citrate) for 8 min at 4 °C and washing twice with PBS for 5 min, the labeling reaction was performed using 50 μl of TUNEL reagent for each sample, except the negative control, in which reagent without enzyme was added and incubated for 1 h at 37 °C. Following PBS washing, slides were incubated with converter reagent for 30 min at 37 °C. After washing, slides were incubated with Fast Red substrate solution for 10 min to stain cells containing labeled DNA strand breaks. TUNEL labeling was conducted using a Cell Death Detection kit (Roche; Mannheim, Germany) and performed according to the manufacturer's instructions.
2.7. Western blot analysis
For Western blot analysis, the protein concentration of each sample was measured using Bio-Rad Protein Assay Dye (Bio-Rad Laboratories, Inc., CA, USA) according to the manufacturer's directions. A total of 30 μg of NRK-52E lysate proteins were applied to each lane and analyzed by Western blotting. Cytosol cytochrome c was extracted by using a Cytochrome c Release Apoptosis Assay Kit (Calbiochem Inc., CA, USA). The antibodies of caspase-3, caspase-9, cytochrome c, Bcl-2 and nuclear factor-kappaB (NF-κB) were diluted to 1:1000 for assay. Peroxidase-conjugated anti-rabbit or anti-mouse IgG (1 : 5000 dilution) was used as the second antibody to detect caspase-3, caspase-9, cytochrome c, Bcl-xL, and NF-κB bands by
enhanced chemiluminescence (Amersham Biosciences Corp, NJ, USA).
2.8. Lactate dehydrogenase cytotoxicity assay
For lactate dehydrogenase (LDH) cytotoxicity assays, NRK-52E cells were plated at 10,000 cells/well in 96-well plates and grown overnight. The culture medium from cells treated with adriamycin was collected and assayed using the LDH Cytotoxicology Detection kit (Roche) according to the
manufacturer's directions. Each data point was determined in triplicate.
2.9. Electrophoretic mobility shift assay (EMSA)
To prepare nuclear protein extracts, cultured NRK-52E cells were washed with cold PBS and then immediately removed by scraping in PBS. After centrifugation of the cell suspension at 1000 ×g, the cell pellets were resuspended in cold buffer A (containing KCl 10 mM, EDTA 0.1 mM, DTT 1 mM, and PMSF 1 mM) for 15 min. The cells were lysed by adding 10% NP-40 and then centrifuged at 5000 ×g to obtain pellets of nuclei. The nuclear pellets were resuspended in cold buffer B (containing HEPES 20 mM, EDTA 1 mM, DTT 1 mM and PMSF 1 mM, and NaCl 0.4 mM), vigorously agitated, and then centrifuged. The supernatant containing the nuclear proteins was used for the Western blot assay or stored at −70 °C until used. A double-stranded containing a high affinity sequence for NF-κB from the mouse kappa-light chain enhancer (5′AGC TTC AGA GAC TTT CCG AGA GG3′) was prepared. The oligonucleotides were end-labeled with [32P]ATP. Extracted nuclear proteins (10μg) were incubated with 0.1 ng of 32P-labeled DNA for 15 min at room temperature in 25 μL of binding buffer containing 1 μg of poly (dI–dC). The mixtures were electrophoresed on 5% non-denaturing polyacrylamide gels. Gels were dried and imaged by autoradiography.
2.10. Statistical analysis
Analysis of variance (ANOVA) was used to compare the levels of eicosanoids. The level of differences among groups was analyzed by Student t tests. Pb0.05 was considered statistically significant.
3. Results
3.1. Increase of COX-1 and PGIS protein and prostacyclin caused by Adv-COX-1/PGIS transfection in NRK-52E cells
NRK-52E cells were transfected with Ad-COX-1/PGIS at different pfu/cell to determine the transfection quantity of Ad-COX-1/PGIS sufficient for COX-1 and PGIS overexpression. The transfected cells were lysed, and COX-1 and PGIS protein levels were determined by Western blot analysis. The transgenic COX-1 and PGIS protein levels were markedly elevated by Ad-COX-1/PGIS transfection with 20 moi (multiplicity of infec-tion; pfu per cell), and gradually increased along raising transfection dose (Fig. 1A). The efficiency of gene expression of adenoviral administration in rat renal tubular cells was also evaluated by determining COX-1 and PGIS protein levels 1 to 3 days after administration. Compared with Adv-HPGK control, Adv-COX-1/PGIS augmented COX-1 and PGIS protein levels in a time-dependent manner (Fig. 1B). Maximal augmentation was noted at 72 h after administration. Several prostanoids in NRK-52E were measured at different times or at different transfection doses of adenoviral administration. The production
of PGI2 was typically monitored by measurement of
6-keto-prostaglandin F1α (6-keto-PGF1α) because 6-keto-PGF1α is a
stable product of the non-enzymatic hydration of PGI2.
Adv-Fig. 2. Prostanoid levels in NRK-52E cells transfected with Ad-COX-1/PGIS. (A) Time course of prostanoid levels in NRK-52E cells infected with 20 moi of adenoviral particles. (B) Prostanoid levels in NRK-52E cells transfected with Ad-COX-1/PGIS at different moi for 2 days. Adv-HPGK transfection was included as a control. A bar is mean ± S.D. of 3 experiments. *Pb0.05 compared with the expression level of 6-keto-PGF1αin control cells.
Fig. 1. COX-1 and PGIS protein levels in NRK-52E cells transfected with Ad-COX-1/PGIS. (A) COX-1 and PGIS protein levels in NRK-52E cells transfected with Ad-COX-1/PGIS at different moi for 3 days. (B) Time course of COX-1 and PGI2protein levels in NRK-52E cells transfected with Ad-COX-1/PGIS at
20 moi. Protein levels in transfected cells were determined by Western blot analysis with specific antibodies.
Fig. 3. Adriamycin-induced injury in NRK-52E cells. (A) Cytotoxicity induced by adriamycin in NRK-52E cells. NRK-52E cells were treated with 1.5, 3 and 6 μM of adriamycin from 6 to 18 h. The lactate dehydrogenase (LDH) released from the cytosol of damaged cells was measured to determine the cytotoxicity of adriamycin. A bar is mean ± S.D. of 3 experiments.#Pb0.05 compared with
control cells at 6 h.&Pb0.05 compared with control cells at 12 h. *Pb0.05
compared with control cells at 18 h. (B) Adriamycin-induced apoptosis in NRK-52E as shown by the TUNEL assay. NRK-NRK-52E cells were treated for 6 h with 3 μM adriamycin as indicated, harvested, stained with DAPI and TUNEL, and examined by fluorescence microscopy as described under Materials and methods (original magnification, ×100). (C) The protective effect of Ad-COX-1/PGIS transfection against the cytotoxicity of adriamycin in NRK-52E cells. NRK-52E cells were transfected with 20 moi of COX-1/PGIS or Ad-HPGK for 2 days, and then treated with 3μM adriamycin from 6 to 18 h. A bar is mean ± S.D. of 3 experiments.&Pb0.05 compared with Ad-HPGK control at 12 h. *Pb0.05 compared with Ad-HPGK control at 18 h.
COX-1/PGIS increased PGI2 levels in a time- and
dose-dependent manner (Fig. 2), which was similar to the COX-1 and PGIS expression patterns. However, the PGE2and 15-D-PGJ2
levels were not significantly affected by Adv-COX-1/PGIS transfection. Apparently, Adv-COX-1/PGIS transfection can selectively augment the endogenous PGI2 production in rat
renal tubular cells.
3.2. Protective effect of Ad-COX-1/PGIS transfection against adriamycin-induced apoptotic injury in NRK-52E cells
To determine the cytotoxicity of adriamycin in rat renal tubular (NRK-52E) cells, the lactate dehydrogenase (LDH) released from the cytosol of damaged cells was measured. NRK-52E cells were treated with 1.5, 3, and 6 μM of adriamycin from 6 to 18 h. As shown in Fig. 3A, there was already a significant injury to cells treated with as little as 3 μM of adriamycin for 6 h, and the increase in adriamycin cytotoxicity paralleled the increase in its dose. TUNEL staining indicated that NRK-52E cell injury was caused by adriamycin-induced apoptosis (Fig. 3B). Subsequently, the protective effect of Ad-COX-1/PGIS was also examined using an LDH detection system. As shown in Fig. 3C, Ad-COX-1/PGIS transfection reduced the cytotoxicity induced by adriamycin treatment. This result reveals that the endogenous PGI2 increase caused by Ad-COX-1/PGIS
transfection protects rat renal tubular cells from adriamycin-induced apoptosis.
3.3. Effect of Ad-COX-1/PGIS transfection on adriamycin-induced activities of caspases in NRK-52E cells
Caspase-dependent apoptotic signaling plays a major role in adriamycin-induced apoptotic injury (Kotamraju et al., 2000; Sawyer et al., 1999). The activities of caspase-3 and caspase-9 stimulated by adriamycin were also evaluated by monitoring the quantity of cleaved subtypes of caspase-3 and caspase-9 in NRK-52E cells. As shown inFig. 4A, the cleaved subtypes of both caspase-3 and caspase-9 were significantly elevated in the cells treated with adriamycin, even for 4 h. Compared with Ad-HPGK transfection, Ad-COX-1/PGIS transfection significantly reduced the quantity of cleaved subtypes of both caspase-3 and caspase-9 in adriamycin-treated NRK-52E cells (Fig. 4B). That is, adriamycin stimulates the activities of caspase-3 and caspase-9 in rat renal tubular cells, and endogenous PGI2
increase caused by Ad-COX-1/PGIS transfection reduces these activities.
3.4. Effect of Ad-COX-1/PGIS transfection on mitochondria-mediated apoptosis signaling induced by adriamycin in NRK-52E Cells
Cytochrome c and Bcl-2 family proteins are supposed to be important in mitochondria-mediated apoptosis signaling. To evaluate the influence of transfection on the variation of cytochrome c and Bcl-xLcaused by adriamycin, NRK-52E cells
transfected with Ad-HPGK or Ad-COX-1/PGIS were treated with 1.5 μM adriamycin for different periods. In Ad-HPGK transfected NRK-52E cells, released cytochrome c was increased significantly within 4 h (Fig. 5). However, in Ad-COX-1/PGIS transfected cells, this increase was inhibited and expression of Bcl-xLwas even elevated.
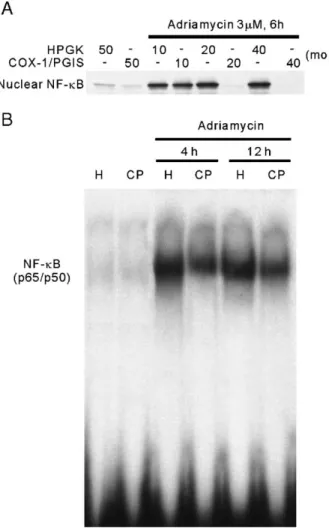
3.5. Inhibition effect of Ad-COX-1/PGIS transfection on NF-κB translocation caused by adriamycin in NRK-52E cells
NF-κB is known as an important transcriptional factor. Recent evidence has shown that NF-κB activation and translocation are pro-apoptotic in adriamycin-treated endothe-lial cells and cardiomyocytes (Wang et al., 2002). To evaluate
Fig. 4. Western blot of cleaved caspase-9 and caspase-3 in adriamycin-treated 52E cells. (A) Protein levels of cleaved caspase-9 and caspase-3 in NRK-52E cells treated with adriamycin under various conditions. NRK-NRK-52E cells were treated with 3μM adriamycin for 4, 6, or 12 h, or 6 h at 1.5, 3, or 6 μM as indicated. (B) Protein levels of cleaved caspase-9 and caspase-3 in adenoviral transfected NRK-52E cells treated with adriamycin. NRK-52E cells were transfected 2 days with 20 moi of Ad-COX-1/PGIS (CP) or Ad-HPGK (H) as a control, and treated with 3μM adriamycin for 4, 6, or 12 h as indicated. Western blotting was carried out with the specific antibody against cleaved caspase-9 and caspase-3.
Fig. 5. The levels of released cytochrome c and Bcl-xLin adenoviral transfected
NRK-52E cells. NRK-52E cells were transfected 2 days with 20 moi of Ad-COX-1/PGIS (CP) or Ad-HPGK (H) as a control, and treated with 3 μM adriamycin for 4, 6, or 12 h as indicated. Cytosol cytochrome c was extracted as described in Materials and methods section. Western blotting was carried out with the specific antibodies against cytochrome c and Bcl-xL.
the effect of Ad-COX-1/PGIS transfection on NF-κB translo-cation caused by adriamycin, NRK-52E cells transfected with different doses of viral vectors were treated with 3 μM adriamycin for 6 h, and NF-κB translocated in nuclei was monitored by Western blotting. As shown in Fig. 6A, adriamycin greatly elevated NF-κB in Ad-HPGK-transfected NRK-52E cell nuclei. This nuclear NF-κB elevation was totally blocked by 20 and 40 moi of Ad-COX-1/PGIS. The activation of NF-κB caused by adriamycin was also measured in terms of DNA-binding activity using the Electrophoretic Mobility Shift Assay (EMSA). NRK-52E cells were transfected with 20 moi of viral vectors for 2 days, and then incubated with 3 μM adriamycin for different time periods. The DNA-binding activity was monitored in nuclear extracts. As shown in Fig.
6B, the DNA binding activity of NF-κB increased in NRK-52E
cells treated with adriamycin for 4 and 12 h. This increase in DNA-binding activity of NF-κB was reduced apparently by Ad-COX-1/PGIS transfection. This result reveals that NF-κB translocation and activation present in adriamycin-treated NRK-52E cells are inhibited by PGI2endogenously augmented
by Ad-COX-1/PGIS transfection. 4. Discussion
Results from this study indicate that Adv-COX-1/PGIS transfection is effective in augmenting COX-1 and PGIS expression in rat renal tubular (NRK-52E) cells and in reducing apoptotic death caused by adriamycin. Adv-COX-1/PGIS transfection increases PGI2 without overproduction of other
prostanoids in rat renal tubular cells. PGI2level augmented by
Ad-COX-1/PGIS transfection presumably accounts for this protective action, as PGI2 is a potent protective effector for
many kinds of cell injury (Boxer et al., 1980; Mene et al., 1990; Moncada, 1982; Yano et al., 2004). Besides monitoring reduction in adriamycin-mediated death of NRK-52E cells, also monitored were the apoptotic signals associated with adriamycin, such as the activation of caspase-3 and caspase-9, increase in cytochrome c release, and decrease in Bcl-xL. These
apoptotic signals present in adriamycin-treated NRK-52E cells are reversed by Ad-COX-1/PGIS transfection. Even the activation and translocation of transcription factor NF-κB induced by adriamycin are also inhibited by Ad-COX-1/PGIS transfection. It is supposed that endogenous selective PGI2
augmentation caused by Ad-COX-1/PGIS transfection protects adriamycin-treated rat renal tubular cells through the inhibition of NF-κB.
Previous studies have shown that prostacyclin synthesis can be selectively augmented by cotransfecting endothelial and cerebral cells with COX-1 and PGIS (Lin et al., 2002; Shyue et al., 2001). Our data indicate that the cotransfection of these two enzymes also selectively augments prostacyclin synthesis in rat renal tubular cells, and protects cells from adriamycin injury. COX-1/PGIS cotransfection is potentially a therapeutic strategy for reducing adriamycin-induced nephropathy. However, the etiology of adriamycin-induced nephrotoxicity is multiple and includes glomerular and interstitial inflammation and fibrosis (Bertani et al., 1982; Okuda et al., 1986; Wang et al., 2000). In the present study, we focused only on the effect of endogenous PGI2augmentation on the direct toxic action of adriamycin on
renal tubular cells. Still unknown is whether COX-1/PGIS cotransfection is effective in adriamycin-induced nephropathy in vivo. Nevertheless, renal tubular cell apoptosis is a key feature of tubular atrophy, which is a hallmark of chronic renal diseases (Khan et al., 1999; Schelling et al., 1998). Our results suggest that PGI2could be useful in the therapy of a chronic
nephropathy in vivo. In addition, systemic administration of PGI2 and its analogues is associated with undesirable side
effects. Prostacyclin acts locally in an autocrine and paracrine manner. Delivery of PGI2to the targeted vascular region would
be preferable. Local administration of PGI2and its more stable
analogs remains a challenge because of the relatively short
half-Fig. 6. The translocation and activation of NF-κB in adenoviral transfected NRK-52E cells treated with adriamycin. (A) The levels of nuclear NF-κB in adenoviral transfected NRK-52E cells treated with adriamycin. NRK-52E cells were transfected with Ad-COX-1/PGIS or Ad-HPGK at varied moi as indicated for 2 days, and then treated with 3μM adriamycin for 6 h. Nuclear protein was extracted as described in Materials and methods section. Western blotting was carried out with the specific antibody against NF-κB p65. The quantity of nuclear NF-κB represents the level of NF-κB translocation. (B) The DNA-binding activity of NF-κB in adenoviral transfected NRK-52E cells treated with adriamycin. NRK-52E cells were transfected with 20 moi of Ad-COX-1/PGIS (CP) or Ad-HPGK (H) for 2 days, and then incubated with 3μM adriamycin for 4 and 12 h. The nuclear proteins were extracted and analyzed by EMSA with NF-κB binding nucleotides. The DNA-binding activity of NF-κB is responsible for the majority of NF-κB activity.
life of these drugs. On the other hand, gene transfer approaches can prolong expression of PGI2synthetic enzymes and thereby
maintain drug concentrations for a longer period than the drugs themselves. Therefore, COX-1/PGIS cotransfection has a potential as an in vivo therapeutic approach.
Similar to the findings in endothelial cells and myocytes (Kotamraju et al., 2000; Sawyer et al., 1999), the adriamycin-induced apoptosis in NRK-52E cells is dependent on the activation of caspase-3 and caspase-9, since the cleavage subforms of both caspase-3 and caspase-9 are markedly induced by adriamycin. Caspase-9 is considered to be a critical apoptosis regulator in a variety of cells. Activated caspase-9 cleaves procaspase-3 to its active form, which in turn, stimulates the caspase-dependent deoxyribonuclease by inactivating its inhib-itor and thereby allowing chromosomal degradation (Enari et al., 1998; Sakahira et al., 1998). Notably, Ad-COX-1/PGIS transfection inhibited the adriamycin-induced activation of caspase-9 and caspase-3. On the other hand, caspase-9 is activated from procaspase-9 by cytosolic cytochrome c (Jiang and Wang, 2004). The mitochondrial release of cytochrome c is regulated by Bcl-2 family proteins, including Bcl-2 and Bcl-xL,
which bind to the mitochondrial outer membrane and block cytochrome c efflux (Yang et al., 1997). In our results, adriamycin markedly reduced the Bcl-xL expression, which
was reversed by Ad-COX-1/PGIS transfection. Therefore, endogenous PGI2 augmentation likely attenuates
adriamycin-induced renal tubular cell injury by acting on a site upstream to the one for Bcl-xLexpression.
In the present study, we demonstrate that adriamycin induces NF-κB activation and translocation in rat renal tubular cells. NF-κB has been reported to be involved in regulating adriamycin-induced apoptosis in various cancer cells and carcinomas (Arlt et al., 2001; Manna and Aggarwal, 1999; Somerville and Cory, 2000). Adriamycin-induced NF-κB
activation in tumor cells is anti-apoptotic. Inhibition of NF-κB activation sensitizes cancer cells to adriamycin-induced apoptosis (Arlt et al., 2001; Manna and Aggarwal, 1999; Somerville and Cory, 2000). The present data provide evidence that NF-κB activation promotes adriamycin-induced apoptosis in rat renal tubular cells. The proapoptotic character of NF-κB might be due to its direct activation of apoptotic genes, including Fas ligand, Fas, c-Myc and p53 (Chan et al., 1999; Qin et al., 1999), or to downregulation of the activities of some anti-apoptotic factors, e.g. Bcl-xL(Hettmann et al., 1999). The
involvement of NF-κB in regulating the cell cycle can also be a mechanism for its proapoptotic effect (Qin et al., 1999). In addition, recent studies reveal that H2O2 is responsible for
adriamycin-induced NF-κB activation in adriamycin-treated endothelial cells and cardiomyocytes (Wang et al., 2002). The ability of PGI2 to inhibit reactive oxygen species production
may be the reason our results showed Ad-COX-1/PGIS transfection inhibited adriamycin-induced NF-κB activation. However, the signal-transduction pathways involved in this inhibition remain to be determined.
In summary, Adv-COX-1/PGIS transfection can selectively augment the endogenous PGI2production in rat renal tubular
cells. Through the inhibition of NF-κB, this endogenous PGI2
increase enhances Bcl-xLexpression and inhibits cytochrome c
release, reduces the activity of caspases, and eventually protects rat renal tubular cells from adriamycin-induced apoptosis. Combined COX-1/PGIS gene transfer is a potentially useful gene therapy for chronic renal diseases.
Acknowledgements
This study was sponsored by Taipei Medical University and National Science Council, Taiwan.
References
Arlt, A., Vorndamm, J., Breitenbroich, M., Folsch, U.R., Kalthoff, H., Schmidt, W.E., Schafer, H., 2001. Inhibition of NF-kappaB sensitizes human pancreatic carcinoma cells to apoptosis induced by etoposide (VP16) or doxorubicin. Oncogene 20, 859–868.
Bertani, T., Poggi, A., Pozzoni, R., Delaini, F., Sacchi, G., Thoua, Y., Mecca, G., Remuzzi, G., Donati, M.B., 1982. Adriamycin-induced nephrotic syndrome in rats: sequence of pathologic events. Lab. Invest. 46, 16–23.
Boxer, L.A., Allen, J.M., Schmidt, M., Yoder, M., Baehner, R.L., 1980. Inhibition of polymorphonuclear leukocyte adherence by prostacyclin. J. Lab. Clin. Med. 95, 672–678.
Chan, H., Bartos, D.P., Owen-Schaub, L.B., 1999. Activation-dependent transcriptional regulation of the human Fas promoter requires NF-kappaB p50–p65 recruitment. Mol. Cell. Biol. 19, 2098–2108.
Dowd, N.P., Scully, M., Adderley, S.R., Cunningham, A.J., Fitzgerald, D.J., 2001. Inhibition of cyclooxygenase-2 aggravates doxorubicin-mediated cardiac injury in vivo. J. Clin. Invest. 108, 585–590.
Enari, M., Sakahira, H., Yokoyama, H., Okawa, K., Iwamatsu, A., Nagata, S., 1998. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 391, 43–50.
Hettmann, T., DiDonato, J., Karin, M., Leiden, J.M., 1999. An essential role for nuclear factor kappaB in promoting double positive thymocyte apoptosis. J. Exp. Med. 189, 145–158.
Jiang, X., Wang, X., 2004. Cytochrome c-mediated apoptosis. Annu. Rev. Biochem. 73, 87–106.
Khan, S., Cleveland, R.P., Koch, C.J., Schelling, J.R., 1999. Hypoxia induces renal tubular epithelial cell apoptosis in chronic renal disease. Lab. Invest. 79, 1089–1099.
Kotamraju, S., Konorev, E.A., Joseph, J., Kalyanaraman, B., 2000. Doxorubi-cin-induced apoptosis in endothelial cells and cardiomyocytes is ameliorated by nitrone spin traps and ebselen. Role of reactive oxygen and nitrogen species. J. Biol. Chem. 275, 33585–33592.
Lin, H., Lin, T.N., Cheung, W.M., Nian, G.M., Tseng, P.H., Chen, S.F., Chen, J.J., Shyue, S.K., Liou, J.Y., Wu, C.W., Wu, K.K., 2002. Cyclooxygenase-1 and bicistronic cyclooxygenase-1/prostacyclin synthase gene transfer protect against ischemic cerebral infarction. Circulation 105, 1962–1969.
Manna, S.K., Aggarwal, B.B., 1999. Lipopolysaccharide inhibits TNF-induced apoptosis: role of nuclear factor-kappaB activation and reactive oxygen intermediates. J. Immunol. 162, 1510–1518.
Mene, P., Abboud, H.E., Dunn, M.J., 1990. Regulation of human mesangial cell growth in culture by thromboxane A2 and prostacyclin. Kidney Int. 38, 232–239.
Moncada, S., 1982. Eighth Gaddum memorial lecture. University of London Institute of Education, December 1980. Biological importance of prostacy-clin. Br. J. Pharmacol. 76, 3–31.
Muller, I., Niethammer, D., Bruchelt, G., 1998. Anthracycline-derived chemotherapeutics in apoptosis and free radical cytotoxicity (Review). Int. J. Mol. Med. 1, 491–494.
Okuda, S., Oh, Y., Tsuruda, H., Onoyama, K., Fujimi, S., Fujishima, M., 1986. Adriamycin-induced nephropathy as a model of chronic progressive glomerular disease. Kidney Int. 29, 502–510.
Qin, Z.H., Chen, R.W., Wang, Y., Nakai, M., Chuang, D.M., Chase, T.N., 1999. Nuclear factor kappaB nuclear translocation upregulates c-Myc and p53
expression during NMDA receptor-mediated apoptosis in rat striatum. J. Neurosci. 19, 4023–4033.
Sakahira, H., Enari, M., Nagata, S., 1998. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature 391, 96–99. Sawyer, D.B., Fukazawa, R., Arstall, M.A., Kelly, R.A., 1999.
Daunorubicin-induced apoptosis in rat cardiac myocytes is inhibited by dexrazoxane. Circ. Res. 84, 257–265.
Schelling, J.R., Nkemere, N., Kopp, J.B., Cleveland, R.P., 1998. Fas-dependent fratricidal apoptosis is a mechanism of tubular epithelial cell deletion in chronic renal failure. Lab. Invest. 78, 813–824.
Scholey, J.W., Miller, P.L., Rennke, H.G., Meyer, T.W., 1989. Effect of converting enzyme inhibition on the course of adriamycin-induced nephropathy. Kidney Int. 36, 816–822.
Shyue, S.K., Tsai, M.J., Liou, J.Y., Willerson, J.T., Wu, K.K., 2001. Selective augmentation of prostacyclin production by combined prostacyclin synthase and cyclooxygenase-1 gene transfer. Circulation 103, 2090–2095. Singal, P.K., Li, T., Kumar, D., Danelisen, I., Iliskovic, N., 2000.
Adriamycin-induced heart failure: mechanism and modulation. Mol. Cell. Biochem. 207, 77–86.
Smith, W.L., DeWitt, D.L., Garavito, R.M., 2000. Cyclooxygenases: structural, cellular, and molecular biology. Annu. Rev. Biochem. 69, 145–182. Somerville, L., Cory, J.G., 2000. Enhanced roscovitine-induced apoptosis is
mediated by a caspase-3-like activity in deoxyadenosine-resistant mouse leukemia L1210 cells. Anticancer Res. 20, 3347–3355.
Sturzebecher, S., Haberey, M., Muller, B., Schillinger, E., Schroder, G., Skuballa, W., Stock, G., Vorbruggen, H., Witt, W., 1986. Pharmacological
profile of a novel carbacyclin derivative with high metabolic stability and oral activity in the rat. Prostaglandins 31, 95–109.
Vane, J.R., Botting, R.M., 1995. Pharmacodynamic profile of prostacyclin. Am. J. Cardiol. 75, 3A–10A.
Wang, Y., Wang, Y.P., Tay, Y.C., Harris, D.C., 2000. Progressive adriamycin nephropathy in mice: sequence of histologic and immunohistochemical events. Kidney Int. 58, 1797–1804.
Wang, S., Kotamraju, S., Konorev, E., Kalivendi, S., Joseph, J., Kalyanaraman, B., 2002. Activation of nuclear factor-kappaB during doxorubicin-induced apoptosis in endothelial cells and myocytes is pro-apoptotic: the role of hydrogen peroxide. Biochem. J. 367, 729–740.
Yang, J., Liu, X., Bhalla, K., Kim, C.N., Ibrado, A.M., Cai, J., Peng, T.I., Jones, D.P., Wang, X., 1997. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science 275, 1129–1132. Yano, T., Itoh, Y., Kubota, T., Sendo, T., Oishi, R., 2004. A prostacyclin analog
beraprost sodium attenuates radiocontrast media-induced LLC-PK1 cells injury. Kidney Int. 65, 1654–1663.
Yano, T., Itoh, Y., Kubota, T., Sendo, T., Koyama, T., Fujita, T., Saeki, K., Yuo, A., Oishi, R., 2005. A prostacyclin analog prevents radiocontrast nephropathy via phosphorylation of cyclic AMP response element binding protein. Am. J. Pathol. 166, 1333–1342.
Zhang, J., Clark Jr., J.R., Herman, E.H., Ferrans, V.J., 1996. Doxorubicin-induced apoptosis in spontaneously hypertensive rats: differential effects in heart, kidney and intestine, and inhibition by ICRF-187. J. Mol. Cell Cardiol. 28, 1931–1943.