Correspondence: Hsuan-Liang Liu, Department of Chemical Engineering and Biotechnology, National Taipei University of Technology, 1 Sec. 3 ZhongXiao E. Rd., Taipei 10608.Tel: +886-2-27712171 ext. 2542;
Fax: +886-2-27317117; Email: [email protected]
Copyright in this article, its metadata, and any supplementary data is held by its author or authors. It is published under the
Chemical Chaperone and Inhibitor Discovery: Potential
Treatments for Protein Conformational Diseases
Jian-Hua Zhao1, Hsuan-Liang Liu1,2, Hsin-Yi Lin1,2, Chih-Hung Huang1,2, Hsu-Wei
Fang1,2, Shiao-Shing Chen3, Yih Ho4, Wei-Bor Tsai5 and Wen-Yih Chen6
1Department of Chemical Engineering and Biotechnology, National Taipei University of Technology,
1 Sec. 3 ZhongXiao E. Rd., Taipei 10608. 2Graduate Institute of Biotechnology, National Taipei
Uni-versity of Technology, 1 Sec. 3 ZhongXiao E. Rd., Taipei, Taiwan 10608. 3Institute of Environmental
Engineering and Management, National Taipei University of Technology, 1 Sec. 3 ZhongXiao E. Rd., Taipei, Taiwan 10608. 4School of Pharmacy, Taipei Medical University, 250 Wu-Hsing St., Taipei,
Taiwan 110. 5Department of Chemical Engineering, National Taiwan University, 1 Sec. 4 Roosevelt
Rd., Taipei, Taiwan 106. 6Department of Chemical and Materials Engineering, National Central
University, 300 Jhongda Rd., Jhongli City, Taoyuan County, Taiwan 32001.
Abstract: Protein misfolding and aggregation cause a large number of neurodegenerative diseases in humans due to (i) gain
of function as observed in Alzheimer’s disease, Huntington’s disease, Parkinson’s disease, and Prion’s disease or (ii) loss of function as observed in cystic fi brosis and α1-antitrypsin defi ciency. These misfolded proteins could either lead to the formation of harmful amyloids that become toxic for the cells or to be recognized and prematurely degraded by the protein quality control system. An increasing number of studies has indicated that some low-molecular-weight compounds named as chemical chaperones can reverse the mislocalization and/or aggregation of proteins associated with human conformational diseases. These small molecules are thought to non-selectively stabilize proteins and facilitate their folding. In this review, we summarize the probable mechanisms of protein conformational diseases in humans and the use of chemical chaperones and inhibitors as potential therapeutic agents against these diseases. Furthermore, recent advanced experimental and theo-retical approaches underlying the detailed mechanisms of protein conformational changes and current structure-based drug designs towards protein conformational diseases are also discussed. It is believed that a better understanding of the mecha-nisms of conformational changes as well as the biological functions of these proteins will lead to the development and design of potential interfering compounds against amyloid formation associated with protein conformational diseases.
Keywords: misfolding, Alzheimer’s disease, Prion’s disease, Parkinson’s disease, Huntington’s disease, amyloid, chemical
chaperone, molecular dynamics simulation, structure-based drug design, protein conformational disease
Protein Conformational Diseases
Protein misfolding is believed to be the primary cause of several neurodegenerative diseases in humans, such as Alzheimer’s disease (AD), Creutzfeldt-Jakob disease, Gaucher’s disease, Huntington’s disease, Parkinson’s disease, Prion’s disease, cystic fi brosis (CF), α1-antitrypsin defi ciency, and many others. In most of the cases, protein misfolding takes place due to an undesirable mutation in the polypeptide chain, an unfavorable physiological environment or, in a few cases, some less known reasons.
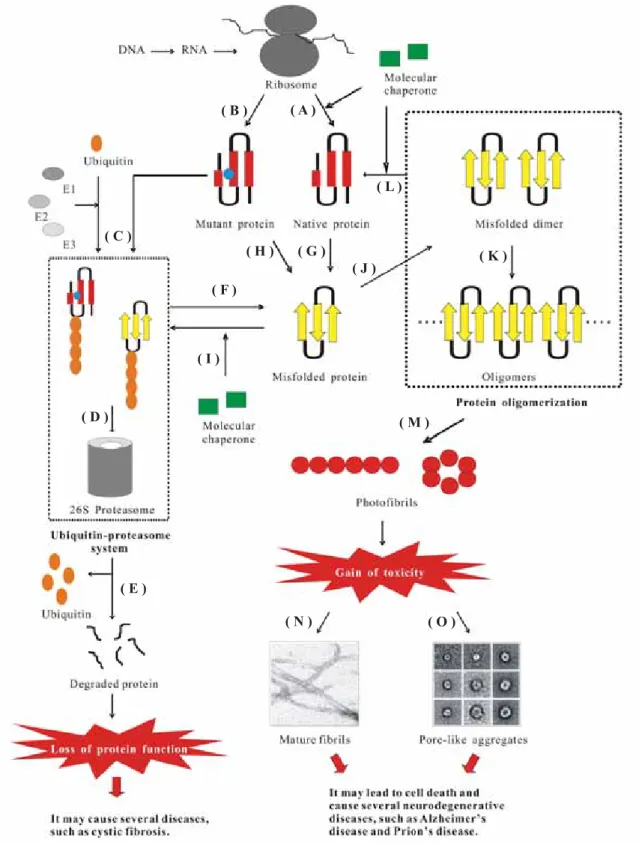
The misfolded protein may lead to harmful effects, which can be divided into two categories (see Fig. 1 for details): (i) gain of function as observed in AD, Huntington’s disease, Parkinson’s disease, and Prion’s disease and (ii) loss of function as in the cases of CF and α1-antitrypsin defi ciency.1 In the former
category, the misfolded proteins may further aggregate to form amyloids, which fi nally cause cell toxic-ity and eventually death. More than twenty proteins in humans are involved in aberrant aggregation, include Aβ, prion, β2-microglobulin, tau, α-synuclein, etc. Aggregation can also occur in proteins with native conformations, although mutations usually accelerate this process. In other words, mutations are not an absolute requirement for protein misfolding and diseases. In the latter category, mutations are associated with defect in folding, leading to the accumulation of intermediate structure in endoplasmic reticulum (ER), as a consequence of increasing ER stress.2 Moreover, such mutations lead to the absence
Protein Quality Control System:
Molecular Chaperones and Ubiquitin
Proteasome System
Proteins that are not able to reach their native states are recognized as misfolded and subsequently targeted to a degradation pathway (Fig. 1C). This is referred to the “protein quality control” system, which plays a critical role in cell function and survival and consists of two components: molecu-lar chaperones and ubiquitin-proteasome pathway (UPP).3
Many molecular chaperones aid in normal fold-ing of proteins and also in refoldfold-ing of the abnor-mal conformations back to their native states, thus prevent the formation of misfolded or aggregated structures (Fig. 1A and L) and slow, arrest, or revert disease progression.1 They are capable of
distin-guishing between the native and non-native states of the targeted proteins. However, until now, it is still unclear how they distinguish between the cor-rectly and incorcor-rectly folded proteins and how they selectively target the latter for degradation. Molec-ular chaperones further facilitate to direct the misfolded proteins to the proteasomal pathway (Fig. 1I). Abnormal proteins can also be targeted to proteasome for degradation by covalent attach-ment to polyubiquitin, which is an energy-requiring process (Fig. 1C).4
However, in some cases, the protein quality control system is imperfect (Fig. 1F). There are two types of protein quality control defect, which have been linked to the etiology of an increasing list of congenital and acquired conformational diseases. In the fi rst type, it is possible that the misfolded proteins are stable folding intermediates, thus they can escape from the degradation pathway and accumulate as aggregates, resulting in the formation of fi brilar deposits known as amyloids. In the second type, proteins harboring modest mutations not compromising their functional integ-rity may be recognized as misfolded, resulting in unnecessary degradation and consequently leading to the loss of functional phenotypes.
From Protein Misfolding and
Aggregation to Amyloid Fibrils
Protein misfolding and aggregation
Protein misfolding and its pathogenic conse-quences have become an important research topic
over the past decades. It is now well known that the molecular basis of protein aggregation into amyloid structures involves the existence of the misfolded forms of proteins (Fig. 1G and H). However, the critical problem is what factors are responsible for protein conformational changes leading to the misfolded forms. Over the past few years, a number of factors, such as mutations that destabilize the folded structure (Fig. 1H), changes in the environmental conditions (pH, oxidative stress, and metal ions), and the activity of certain proteins collectively named pathological chaper-ones (apolipoprotein E, amyloid P component, and protein X), have been identifi ed to play such a critical role.5 Once a certain concentration of the
misfolded protein is reached, the formation of their aggregates can occur in the cells (Fig. 1J and K), leading to the formation of an amyloid-like struc-ture, which eventually causes different types of neurodegenerative disorders and ultimately leads to cell death.3
The formation of an aggregate is a common feature of all protein conformational diseases (PCDs). It is caused by the destabilization of the α-helical structures and the simultaneous forma-tion of the β-sheet structures (Fig. 1G and H).6
These β-sheet structures are formed between alter-nating peptide strands. Linkages between these strands result from hydrogen bonding between their aligned pleated structures.7 Such β-linkages
with a pleated strand from one molecule inserting into a pleated sheet of the next lead to the formation of intermolecular hydrogen bonding networks.
Before oligomerization, the fi brillogenesis often starts with dimers, which have been recognized as the initial building blocks of amyloid fibrils (Fig. 1J).8 They further polymerize into oligomers
(Fig. 1K). During this nucleation process, the content of the secondary β-structure of the oligo-meric assemblies is generally increased.9 After the
nucleation or seeding step, the growing assemblies, ordered prefibrillar aggregates or protofibrils (Fig. 1M), are formed via an elongation process and eventually give rise to mature amyloid fi brils.10
The relationship between protein
misfolding and aggregation
Until now, it is still not clear whether conforma-tional changes induce protein oligomerization and whether misfolding triggers protein aggregation.11
( A ) ( B ) ( C ) ( D ) ( E ) ( N ) ( O ) ( M ) ( I ) ( F ) ( H ) ( G ) ( J ) ( L ) ( K )
Figure 1. The pathway of protein synthesis and degradation in the cell. (A) Nascent polypeptide chain is converted into its native folded
protein with the help of molecular chaperones. (B) Nascent polypeptide chain with a mutation (blue ball) folds into its native-like protein (or partial unfolded protein). (C) Mutant (or partial unfolded) protein may be re-recognized as imperfect proteins, leading to ubiquitination by E1 (ubiquitin activating enzyme), E2 (ubiquitin conjugating enzyme), and E3 (ubiquitin ligase). (D) Misfolded protein enters into the protea-some system with the help of the ubiquitin complex. (E) Misfolded protein is degraded into small peptide by proteaprotea-some and ubiquitin is regenerated. (F) Impaired proteasome system could not degrade the misfolded protein. (G) Native protein molecule is converted into mis-folded structure, which is caused by destabilization of the α-helical structure and the simultaneous formation of the β-sheet structure. (H) Mutations accelerate protein misfolding. (I) Molecular chaperones facilitate to direct the misfolded proteins to the proteasomal pathway. (J) Misfolded monomers aggregate into dimer as initial building blocks for the formation of amyloid fi brils. (K) These building blocks further polymerize to form oligomers. (L) Molecular chaperones disaggregate the compact aggregates and develop native folded monomer. (M) Oligomers further form photofi brils. (N) Amyloid hypothesis. (O) Channel hypothesis.
it has been proposed that the critical event in PCDs is the formation of protein oligomers that can act as the seeds to induce protein misfolding. In this model, misfolding occurs as a consequence of aggregation (polymerization hypothesis),11 which
follows a crystallization-like process dependent on nucleus formation.
An alternative model indicates that the underlying protein is stable in both the folded and misfolded forms in solution (conformation hypothesis).5 It
sug-gests that spontaneous or induced conformational changes result in the formation of the misfolded protein, which may or may not form an aggregate. Several factors, such as those mentioned above, may cause the protein conformational changes and further lead to the misfolded forms of proteins.
In the third hypothesis, the native proteins undergo slightly conformational changes, resulting in the formation of an unstable amyloidogenic intermediate in the cellular environment. This unstable intermediate exposes many of its hydro-phobic regions, leading to the growth of small oligomers, which mainly consist of β-sheets via intermolecular interactions. These small oligomers further form an ordered fi bril-like structure named amyloid also via intermolecular interactions. In this model, the conversion of the folded protein into the pathological form is triggered by confor-mational changes, but the complete misfolding is dependent on oligomerization.11 So far, most of
the experimental fi ndings can be explained by the above three models. However, the conformation/ oligomerization hypothesis is still the most comprehensive and accepted model for protein misfolding and aggregation.
Amyloid aggregates lead to cell toxicity
Two hypotheses have been proposed to explain how the aggregates cause toxic effects to the cells (Fig. 1N and O). The fi rst one, known as the “amy-loid hypothesis” (Fig. 1N),9 directly believes that
the huge amount of aggregates may damage organs simply by hindering a proper fl ow of nutrient to the cells, thus impairing tissue functions in the case of the peripheral amyloidosis. Recent studies have reported that an important role of modifi cation to the intracellular free calcium and reactive oxygen species (ROS) occurs in cells exposed to toxic aggregate.12 It suggests that the oxidative stress
following exposure to the early species involved in amyolid formation could damage cells and
eventually cause cell death. However, it is still not clear why protein aggregation is followed by the production of ROS.
The second one, the so-called “channel hypoth-esis” (Fig. 1O), has been proposed to explain the biochemical mechanism of amyloid toxicity.13 This
hypothesis also supports the changes of intracel-lular ion content and ROS state mentioned above. Protofi brils, the precursors of longer protofi laments and mature fi brils, typically appear as globular assemblies (2.5–5 nm in diameter) with a high β-sheet content and spontaneously organize into beaded chains and variously sized annular rings comprising small “doughnut” shaped species form-ing a central pore in membranes.14 It is still not
possible to detect these channels in the cells involved in amyloid diseases due to the technical difficulties, but channels have been observed
in vitro from a number of amyolid peptides and
proteins, including islet amyloid polypeptide (IAPP or Amylin), a neurotoxic fragment of the prion protein (PrP106–126), serum amyloid A, polygluta-mine, transthyretin (TTR), α-synuclein, and lyso-zyme. The observation of channels amongst such a diverse variety of peptide sequences suggests a deep underlying similarity in their physical chem-ical structures. This special amyloid pore may account for the toxicity of the amyloid aggregates. Thus, protein oligomers may act as a biological signal killing the target cells by forming non-specifi c membrane pores, which further result in the unbalance of the ion concentration.
Toxic Amyloid Formation Causes
Many Human Neurodegenerative
Disorders: The Cases of AD
and Prion’s Disease
AD and prion disease are the primary and frequent discovered diseases involving selective neuronal vulnerability with degeneration in specifi c brain regions and deposits of the corresponding misfolded protein in neurons and other cells. In this section, we aim to elucidate the causes of human neurode-generative disorders through the formation of toxic amyloids by the cases of AD and Prion’s disease.
Alzheimer’s disease
AD is a progressive neurodegenerative disease characterized by extracellular amyloid plaques and intraneuronal fi brillary tangles in the brain. It has
been shown that Aβ1–40 and Aβ1–42 are the main
alloforms of amyloid β (Aβ) peptides found in amyloid plaques. Aβ peptides are derived from proteolytic processing of the amyloid precursor protein (APP). APP can be cleaved by three differ-ent proteases, named α-, β-, and γ-secretases. In general, β-secretase cleaves the extracellular domain of APP to generate the N terminus of Aβ, while γ-secretase performs proteolysis in the middle of the transmembrane domain of APP to produce the C terminus of Aβ.15 Finally, the two
main products, Aβ1–40 and Aβ1–42, migrate outside
the cell and give rise to fi brils.
Several mechanisms have been proposed to explain the neuron toxicity of Aβ peptides, such as the formation of ion channels on the cell mem-brane,16 the generation of free radicals,17 and the
interaction of Aβ peptides with various receptors, such as apolipoprotein E18 and mitochondrial
hydroxyacyl-CoA dehydrogenase.19 However, the
cytotoxic mechanism of Aβ has not yet been fully understood at present. Compelling evidences have demonstrated that the soluble oligomers (amyloid-derived diffusible ligands; ADDLs) and fi brils of Aβ are the toxic identities that cause neuronal injury and death in patients suffering from AD.20
Prion’s disease
The prion protein is thought to cause a disease in cattle called bovine spongiform encephalopathy (BSE) or “mad cow disease” and a disease in human named variant Creutzfeldt-Jakob disease (vCJD).5 It is known that normal prion protein is
protease sensitive, soluble, innocuous, and has a high α-helical content. This protein is thought to undergo a conformational change in which α-helices of the wild-type protein PrPC (normal
cellular form) are converted into β-sheet-dominant PrPSC (pathogenic isoform), resulting in misfolding
and aggregation. PrPC and PrPSC share the same
covalent structure but possess different folds. PrPSC
has been identifi ed as the causative agent in trans-missible spongiform encephalopathies because it is capable of aggregating into a variety of forms from amorphous to highly structured aggregates.
A central theme in prion disease research is the detection of the process underlying the conforma-tional transition from PrPC to PrPSC. PrPC has been
found to undergo a pH-dependent conformational change in the range of pH 4.4–6.0, with a loss of α-helical content and a gain of β-structure.21 PrPSC
also acts as a template for the structural conversion of PrPC as well as huntingtin proteins, subsequently
forming aggregates.22 Moreover, unlike AD, it is
believed that in Prion’s disease, the conformational infection and aggregation can take place both extracellularly and intracellularly.23
Protein Misfolding with the Loss
of Function Lead to Several Lethal
Diseases: The Case of CF
The human cystic fi brosis transmembrane conduc-tance regulator (CFTR) gene encodes an integral membrane glycoprotein of 1,480 amino acid resi-dues with two N-linked glycosylation sites.24 The
CFTR is a cAMP-regulated chloride (Cl−) channel
localized at the apical membrane of secretory epithelia. Mutations in this channel cause CF, a disease characterized by the inability of epithelial cells to secrete chloride, result in the production of thick and viscous mucus that causes severe functional obstruction of lungs and pancreas. A majority of CF patients has a deletion of a phe-nylalanine residue at position 508, resulting in an F508del-CFTR protein.25 The clinical importance
of this mutation becomes evident because it accounts for 90% of patients diagnosed with CF.26
This mutation results in a misfolded channel retain-ing in the ER in an immature state and are rapidly degraded by a process involving the ubiquitin-dependent proteasomal system.24 Thus very little
of this protein can reach the cellular membrane, resulting in the loss of function phenotype.
Potential Treatments for PCDs
Drugs against PCDs in humans aim to inhibit aggregation and/or to enable the mutant proteins to escape from the protein quality control systems so that their function can be rescued. Recently, several low-molecular-weight compounds, named chemical chaperones,27 have been shown to act as
the potential therapeutic agents for the control of many PCDs. In this section, we provide some evidences showing that chemical chaperones or inhibitors can be used as potential therapeutic agents for the control of several PCDs in humans, such as AD, Prion’s disease, and CF.
Potential treatment for AD
Aβ has emerged as the most promising target in the treatment or prevention of AD. Inhibition of
Aβ-fi bril formation might be a reasonable therapeutic strategy because familial mutations that lead to an increase in Aβ concentration or to its aggregation increase neuropathology.28 Unfortunately, no
effec-tive therapy using a chemical chaperone system has been successfully conducted so far. A previous study has shown that osmolytes such as glycerol and tri-methylamine N-oxide (TMAO), acting as chemical chaperones, correct folding defects by preferentially hydrating partially denatured proteins and entropi-cally stabilize native conformations.29 Such
infor-mation could potentially be used to develop cellular models of Aβ aggregation and the assessment of agents that modulate fi bril formation. In this case, chemical chaperones could exacerbate the patho-physiologic state. However, a variety of chemical compounds have been found to inhibit the fi brilla-tion of Aβ, including antibiotics, benzofuran derivatives, sulfonated dyes, styryl benzene, fl avone, and peptidic β-sheet breakers. Cell culture experi-ments have indicated that Aβ inhibitors could reduce the cytotoxicity of Aβ peptides and the amount of amyloid deposition in mice with acute amyloidosis.30
However, very few of them have entered into clinic trial phases because of the problems inherent in these inhibitors, including low bioavailability, poor biostability, toxicity, and inability across the blood-brain barrier (BBB), limiting their therapeutic applications. Thus, structural modification of inhibitors as well as design of new inhibitors with alternative structural scaffolds is necessary to improve the physiochemical properties of these compounds in the future.
Potential treatment for Prion’s disease
Therapeutic agents are often designed in an attempt to destroy the PrPSC structure and hopefully
recov-ery the PrPC structure or any other innocuous
isoform. For example, several β-sheet breaker peptides, which could slow or reverse disease progression, have been designed to disrupt the β-rich amyloidgenic PrPSC species. However, it has
been shown that disrupting PrPSC is not suffi cient
to inhibit Prion’s disease and that this strategy may increase propagation of PrPSC and infectivity.31
Thus, strategies for stabilizating the PrPC
conforma-tion also appear as alternative approaches.
Some chemical chaperons, such as TMAO, dimethyl sulfoxide (DMSO), and glycerol, are thought to stabilize the PrPC conformation and
have been suggested to be effective in the
destruction/protection strategy. In a previous study, Tatzelt and co-workers have shown that TMAO and other protective osmolytes success-fully prevent scrapie formation in vitro.32
More-over, a number of other compounds, such as anthracyclines, porphyrins, and diazo dyes, have also been shown to inhibit prion replication when administered with PrPSC in animal models.33 It is
regretful that this is not a clinically relevant model for therapeutic intervention because subclinical disease exists for months in mice and years in humans.7 However, a recent study by Vogtherr and
co-workers has suggested that quinacrine binds specifi cally to PrPC and inhibit the conversion of
PrPC to PrPSc, which in turn suppresses the
pro-gression of Creutzfeldt-Jacob disease.34 Currently,
quinacrine has been clinically approved and clinical trials are being carried out to test the use-fulness of this molecule in patients suffering from Creutzfeldt-Jacob disease.
Potential treatment for CF
There are numerous ongoing efforts towards fi nding agents that promote the folding or block the degradation of nascent F508del-CFTR with the potential to provide a therapeutic basis for the treatment of CF. For example, Sato et al. have examined the effect of glycerol on the fate of the mutant F508del-CFTR and concluded that 10% glycerol (vol/vol) could mediate an increase in the transport of the mutant protein from the ER to the plasma membrane in a cell culture model, which is associated with an increase in the functional activity of CFTR.35 In addition, two other
low-molecular-weight organic compounds, deuterated water and TMAO, have also been shown to increase the post-translational maturation of F508del-CFTR in a cell culture model, leading to the increase of the chloride transport activity.36
Moreover, other study has also reported that chlo-ride transport is increased signifi cantly by the use of DMSO on polarized epithelial cell lines express-ing F508del-CFTR.37 These results all suggest that
these low-molecular-weight compounds could stabilize this mutant, promoting the proper folding and transport to its site of function.
The role of chemical chaperones
in the treatment for PCDs
Until now, the precise mechanisms of the action of chemical chaperones are still not fully
understood. However, they are thought to provide positive effect on protein folding, to reduce protein aggregation, and to stabilize a conformation capable of escaping from the degradation pathway. For example, osmolytes, such as glycerol, are believed to stabilize the native structures of proteins, which can be explained by their prefer-ential exclusion from the surfaces of proteins. This effect increases the chemical potential of the pro-tein and is proportional to the solvent-exposed surface area of the protein, thus more expanded conformations are disfavored in the presence of osmolytes and the protein tends to adopt the more compact native structure.38 Recently, Carprnter
et al. have investigated the effects of sucrose, a model osmolyte, on the conformational equilibria and fl uctuations within the native-state ensembles of bovine pancreatic ribonuclease A and S and horse heart cytochrome c and concluded that the presence of sucrose shifts the conformational equilibria toward the most compact protein species within the native-state ensembles due to the pref-erential exclusion of sucrose from the protein surface.39
Besides the human diseases and their corre-sponding therapies mentioned above, lysosomal storage diseases have been tested for possible chemical chaperone therapy in humans. For example, Matsuda et al. have synthesized a compound, N-octyl-4-epi-β-valienamine (NOEV), for molecular therapy of brain pathology in β-galactosidosis and confirmed its restorative effect on the model mouse brain after short-term oral administration.40 Moreover, Tropak and
co-workers have shown that increasing the amount of hexosaminidase A is capable of exiting the endo-plasmic reticulum for transport to the lysosome and propose that such hexosaminidase inhibitors can function as pharmacological chaperones by enhancing the stability of the native conformation of the enzyme.41
However, these chemical chaperones are a far from practical therapeutic approach in humans because the necessary effi cacious dose is toxic. Nevertheless, exploiting the mechanism by which they are effective may yield clues for the design of new compounds, which are less toxic. For example, Zhang et al. have reported that some organic sol-utes, such as myo-inositol, betaine, and taurine, exhibit the ability to restore the folding defect of F508del-CFTR.42 Despite their reduced toxicity in
comparison to glycerol, the robustness and
corrective effi cacy of such compounds remain to be demonstrated in the whole organisms. Encourag-ingly, some in vivo data has already shown that such strategy may be valid.43 Moreover, recent study has
reported that various disaccharides can inhibit the polyglutamine-mediated protein aggregation.44
Tanaka et al. have found that trehalose, the most effective disaccharides, can decrease polyglutamine aggregates in cerebrum and liver due to the improved motor dysfunction and extended lifespan in a transgenic mouse model of Huntington disease.44 Most importantly, trehalose is nontoxic
with high solubility and can be coupled with effi cacy upon oral administration, which makes trehalose a promising therapeutic drug or a leading compound for the treatment of polyglutamine diseases.44 These evidences together suggest that
chemical chaperones are potential pharmaceuticals for the treatment of protein misfolding diseases.
Insights into the Process of Protein
Misfolding: Computational Studies
Understanding the misfolded structure of the amyloid-associated proteins and how they change their conformations to the misfolded and/or toxic forms can help to elucidate their aggregation mechanisms and may contribute to the develop-ment of some effective therapies for treating PCDs in humans. Drug compounds are generally designed to inhibit, restore, or otherwise modify the structure and behavior of the disease-associated proteins. Target proteins are typically the key molecules involved in a specifi c metabolic or cell signaling pathway that is known or is believed to be related to a particular disease state.
The early stage in drug discovery involves tar-get discovery, validation, and identifi cation by high-throughput physical and/or virtual screening. So far, structure-based drug design is considered as one of the most powerful approaches in drug discovery platform and ca be applied computation-ally when the structure of the target protein is (i) known based on the crystallographic, nuclear magnetic resonance (NMR) techniques, or other experimental methods, or is (ii) unknown yet to be built using homology modeling. The process often involves the generation of a very large in silico library of potential derivatives and the use of molecular docking to select derivatives that may interact with the target protein on the basis of shape complementarities and charge placement.
However, the critical problem is the structural information about the target protein must be avail-able. Unfortunately, due to the diffi culties in crys-tallizing amyloid fibrils, the detailed intrinsic structure has yet to be determined by x-ray diffrac-tion. Thus, some biophysical techniques, such as transmission and cryo-electron microscopy, atomic force microscopy (AFM), and solid-state NMR, have been used to disclose the structural features of amyloid fi brils. Nevertheless, these techniques are still diffi cult to provide structural information in atomic detail. For the case of Aβ, some evi-dences have suggested that Aβ exists as a mixture of α-helix, β-sheet, and random coil in aqueous solution.45 However, in fl uorinated alcohols, such
as trifl uoroethanol (TFE) or hexfl uoro 2-propanol (HFIP), it adopts a stable α-helical conformation.46
It is known that the α-helical conformation of Aβ is temperature sensitive but its β-sheet conforma-tion is not. Moreover, the α-helical conformation of Aβ is favored at pHs 1–4 and 7–10; whereas the β-sheet conformation of Aβ is favored at pH 4–7.47
X-ray diffraction analyses and NMR determina-tions have reported that Aβ stacks in the form of pleated β-sheet structures oriented perpendicular to the fi bril axis.48 Furthermore, several
conforma-tional studies on Aβ fi brillogenesis have suggested that the conformational transition of Aβ into β-sheet structure in fi brils goes through an α-helix-containing intermediate conformation.49 Thus, it
is extraordinarily diffi cult to design an inhibitor due to the structural complexity of Aβ. Therefore, computational studies provide an alternative tool to elucidate the transition between the unaggre-gated and aggreunaggre-gated proteins, where experimental techniques cannot yet probe.
Molecular dynamics (MD) simulation has been employed as a powerful tool to provide structural information in atomic detail under various condi-tions. For the case of Aβ, numbers of MD simula-tions have been performed by several groups with a view to elucidating the conformational transi-tion and assembly mechanism based on the mod-eled structures of Aβ, i.e. fragments or synthesized analogs of Aβ. For example, Klimov and Thirumalai have performed a series of MD simu-lations towards Aβ peptide and their results showed that Aβ16–22 peptides form antiparallel
β-sheet structures and the α-helical intermediates are transiently populated.50 They further proposed
that fi bril formation by Aβ peptides, which occurs by maximizing the number of slat bridges and
hydrophobic interactions, must involve oligomers with high α-helical content. In a more recent study, atomic detail MD simulations with explicit solvent have been conducted to show that only large Aβ16–22 oligomers with at least 8–16
mono-mers form a stable β-sheet aggregate through better hydrophobic contacts and a better shielding of backbone-backbone hydrogen bonds from the solvent.51 In the case of prion, two 10-ns
trajec-tories generated by MD simulations have been used to probe the initial events in the conforma-tional transition of PrPC to the aberrant
aggregation-prone form.52 More recently, DeMarco and
Daggeat have simulated the fragment of human prion protein (residues 90–230) at low pH, which triggers misfolding of this protein, and observed a conformational transition to a PrPSC-like
iso-form.53 Their results further demonstrated that the
N-terminal portion of this protein, which has been identifi ed as a probable aggregation site, under-goes extensive conformational rearrangement, leading to the formation of a large solvent-accessible hydrophobic cluster.
In addition to prion protein and Aβ peptide, other amyloid-associated proteins have also been investigated intensively by MD simulations. For example, TTR has been shown to be the cause of or involved in senile systemic amyloidosis.54 From
the MD simulation results, Armen et al. have proposed that the formation of the “α-sheet” structure by alternating αL and αR residues may
represent a key pathological conformation during amyloidogenesis.55 Another case is human cystatin
C (HCC), which is thought to co-localize with Aβ peptide in the dimeric or oligomeric form in brain amyloid deposits of patients, particularly in elderly individuals and patients suffering from AD or Down’s syndrome.56 The structure of HCC
con-sists of a core with a fi ve-stranded anti-parallel β-sheet (β-region) wrapped around a central helix.57 The monomeric HCC is thought to form
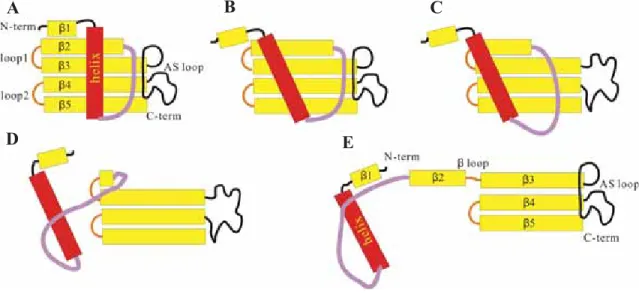
a dimeric structure through the so-called “3D domain swapping” process.58 The results from our
previous MD study has allowed us to propose a possible mechanism for HCC domain swapping as follows: (i) fi rst, the central helix departs from the β-region via the disruption of the interior hydrophobic core (Fig. 2A,B); (ii) subsequently, the native contacts within β2 and AS loop disap-pear (Fig. 2B,C); (iii) then, the β-hairpin between β2 and β3 unfolds through the destruction of three important salt bridges following the so-called
“zip-up” mechanism (Fig. 2C,D); and (iv) fi nally, the open form of the monomeric HCC is generated (Fig. 2E).59
Most importantly, the results of the above men-tioned MD simulations are all in good agreement with the available experimental observations, pro-viding atomic insights into the conformational changes associated various PCDs in humans and may contribute to the development of structure-based drug design.
Possible Therapeutic Strategies
for PCDs: Combining MD
Simulations and Structure-Based
Drug Design
Recently, a combined approach of MD simulation and structure-based drug design was conducted by Liu and co-workers.60 Long time MD simulations
were conducted to investigate the conformational transition of Aβ1–40 and their results showed that Aβ
adopts a α-helix/β-sheet intermediate structure, which exhibits a core domain constituted by the seg-ment of residues 24–37. In their later study, they performed virtual screening based on molecular docking towards this Aβ intermediate structure and aimed to design inhibitors, which can bind to the β-sheet region of the Aβ intermediate to interrupt the formation of the pleated β-sheet structure found in amyloid fibrils.55 From the results of thioflavin
T fl uorescence assay and AFM determination, they
have successfully identifi ed a new inhibitor, named as DC-AB1, to abolish Aβ fi brillation. This study not only reveals some clues to understanding the molec-ular events involved in Aβ aggregation, but also provides a strategy for inhibitor design based on the fl exible intermediate structures of Aβ peptides.
From their promising results, we can expect that the combined experimental and computational approaches to design potential interfering com-pounds for inhibiting protein aggregation associ-ated with PCDs in humans will increase dramatically in the near future of drug discovery and develop-ment history.
Conclusions
In this review, we discussed the mechanisms of various PCDs in humans, including AD, Prion’s disease, and CF, and the strategies of using chem-ical chaperones and inhibitors as therapeutic agents against these diseases. To further understand the biological meanings and aggregation mechanisms of either amyloid-associated proteins or normal proteins with the ability to aggregate into amyloid fi brils, their structures in atomic detail must be available. However, the lack of the detailed struc-tural information of amyloid fi bril makes it diffi cult to elucidate their aggregation mechanisms. At this stage, MD simulation combined with some exper-imental approach may provide a powerful tool to gain the atomic insights into the possible
A B C
D E
Figure 2. The proposed mechanism of the 3D domain swapping process of HCC. (A) The closed form of the monomeric HCC;52 (B) partially
unfolded monomeric HCC with the central α-helix moving away from the β-region via the disruption of the interior hydrophobic core; (C) partially unfolded monomeric HCC with the disappearance of the native contacts between β2 and β3-AS; (D) partially unfolded monomeric HCC with the β2-L1-β3 hairpin unfolded following the “zip-up” mechanism; and (E) the open form of the monomeric HCC.54
mechanisms of protein misfolding and aggregation associated with a number of PCDs in humans. Thus, it may further assist the structure-based drug design in the near future of drug discovery and development history.
Acknowledgment
The authors gratefully acknowledge the fi nancial support (Project number: NSC 96-2221-E-027-045-MY3) from the National Science Council of Taiwan.
References
[1] Cohen, F.E. and Kelly, J.W. 2003. Nature, 426:905. [2] Kaufman, R.J. 1999. Genes Dev., 13:1211.
[3] Berke, S.J.S. and Paulson, H.L. 2003. Curr. Opin. Genet. Dev., 13:253.
[4] Goldberg, A.L. 2003. Nature, 426:895.
[5] Prusiner, S.B. 1998. Proc. Natl. Acad. Sci. U.S.A., 95:13363. [6] Dobson, C.M. 1999. Trends. Biochem. Sci., 24:329. [7] Chaudhuri, T.K. and Paul, S. 2006. FEBS. J., 273:1331.
[8] Roher, A.E., Baudry, J., Chaney, M.O., Huo, Y.M. and Stine, W.B. 2000. Biochim. Biophys. Acta., 502:31.
[9] Kelly, J.W. 1998. Curr. Opin. Struct. Biol., 8:101. [10] Stefani, M. and Dobson, C.M. 2003. J. Mol. Med., 81:678. [11] Soto, C. 2001. FEBS. Lett, 498:204.
[12] Hyun, D.H., Lee, M.H., Hattori, N., Kubo, S.I., Mizuno, Y., Halliwell, B. and Jenner, P. 2002. J. Biol. Chem., 277:28572. [13] Kourie, J.I. 2001. Cell Mol. Neurobiol., 21:173.
[14] Lashuel, H.A., Petre, B.M., Wall, J., Simon, M., Nowak, R.J., Walz, T. and Lansbury, P.T. 2002. J. Mol. Biol., 322:1089.
[15] Selkoe, D.J. 1999. Nature, 399:A23.
[16] Kagan, B.L., Hirakura, Y., Azimov, R., Azimova, R. and Lin, M.C. 2002. Peptides, 23:1311.
[17] Monji, A., Utsumi, H., Ueda, T., Imoto, T., Yoshida, I., Hashioka, S., Tashiro, K. and Tashiro, N. 2001. J. Neurochem, 77:1425. [18] Lauderback, C.M., Kanski, J., Hackett, J.M., Maeda, N., Kindy, M.S.
and Butterfi eld, D.A. 2002. Brain Res., 924:90.
[19] Lustbader, J.W., Cirilli, M., Lin, C., Xu, H.W., Takuma, K., Wang, N., Caspersen, C. et al. 2004. Science, 304:448.
[20] Klein, W.L., Krafft, G.A. and Finch, C.E. 2001. Trends Neurosci., 24:219.
[21] Swietnicki, W., Petersen, R., Gambetti, P. and Surewicz, W.K. 1997. J. Biol. Chem., 272:27517.
[22] Zanata, S.M., Lopes, M.H., Mercadante, A.F., Hajj, G.N.M., Chiarini, L.B., Nomizo, R., Freitas, A.R.O. et al. 2002. EMBO J., 21:3307.
[23] Ma, J. and Lindquist, S. 2002. Science, 298:1785. [24] Kopito, R.R. 1999. Physiol. Rev., 79:S167.
[25] Kerem, B-S., Rommens, J.M., Buchanan, J.A., Markiewicz, D., Cox, T.K., Chakravarti, A., Buchwald, M. et al. 1989. Science, 245:1073.
[26] Amaral, M.D.J. 2006. Inherit. Metab. Dis., 29:477.
[27] Welch, W.J. and Brown, C.R. 1996. Cell Stress Chaperones, 1:109. [28] Findies, M.A. 2002. Curr. Top Med. Chem., 2:417.
[29] Yang, S.H., Upis, C.M., Huang, T.H.J., Chakrabartty, A. and Fraser, P.E. 1999. J. Biol. Chem., 274:32970.
[30] Kisilevsky, R., Szarek, W. and Weaver, D. 1999. US Patent, 5:972,328.
[31] Piening, N., Weber, P., Giese, A. and Kretzschmar, H. 2005. Biochem. Biophys. Res. Commun., 326:339.
[32] Tatzelt, J., Prusiner, S.B. and Welch, W.J. 1996. EMBO J., 15:6363.
[33] Korth, C., May, B.C.H., Cohen, F.E. and Pruisner, S.B. 2001. Proc. Natl. Acad. Sci. U.S.A., 98:9836.
[34] Vogtherr, M., Grimme, S., Elshorst, B., Jacobs, D.M., Fiebig, K., Griesinger, C. and Zahn, R.J. 2003. Med. Chem., 46:3563. [35] Sato, S., Ward, C.L., Krouse, M.E., Wine, J.J. and Kopito, R.R. 1996.
J. Biol. Chem., 271:635.
[36] Brown, C.R., Hong-Brown, L.Q., Biwersi, J., Verkman, A.S. and Welch, W.J. 1996. Cell Stress Chaperones, 2:117.
[37] Bebök, Z., Venglarik, C.J., Panczel, Z., Jilling, T., Kirk, K.L. and Sorscher, E.J. 1998. Am. J. Physiol., 275:C599.
[38] Bolen, D.W. and Baskakov, I.V. 2001. J. Mol. Biol., 310:955. [39] Kim, Y-S., Jones, L.S., Dong, A., Kendrick, B.S., Chang, B.S.,
Manning, M.C., Randolph, T.W. and Carpenter, J.F. 2003. Protein Sci., 12:1252.
[40] Matsuda, J., Suzuki, O., Oshima, A., Yamamoto, Y., Noguchi, A., Takimoto, K., Itoh, M., Matsuzaki, Y., Yasuda, Y., Ogawa, S., Sakata, Y., Nanba, E., Higaki, K., Ogawa, Y., Tominaga, L., Ohno, K., Iwasaki, H., Watanabe, H., Brady, R.O. and Suzuki, Y. 2003. Proc. Natl. Acad. Sci. U.S.A., 100:15912.
[41] Tropak, M.B., Reid, S.P., Guiral, M., Withers, S.G. and Mahuran, D. 2004. J. Biol. Chem., 279:13478.
[42] Zhang, X-M., Wang, X-T., Yue, H., Leung, S., Leung, W., Thibodeau, P.H., Thomas, P.J. and Guggino, S.E. 2003. J. Biol. Chem., 278:51232–51242.
[43] Zhang, Z., Ferraris, J.D., Brooks, H.L., Brisc, I. and Burg, M.B. 2003. Am. J. Physiol. Renal. Physiol., 285:F688.
[44] Tanaka, M., Machida, Y., Niu, S., Ikeda, T., Jana, N.R., Doi, H., Kurosawa, M., Nekooki, M. and Nukina, N. 2004. Nature Med., 10:148.
[45] Zhang, S., Iwata, K., Lachenmann, M.J., Peng, J.W., Li, S., Stimson, E.R., Lu, Y. et al. 2000. Struct. Biol., 130:130.
[46] Fezoui, Y. and Teplow, D.B. 2002. J. Biol. Chem., 277:36948. [47] Stine, W.B.J., Dahlgren, K.N., Krafft, G.A. and LaDu, M.J. 2003.
J. Biol. Chem., 278:11612.
[48] Luhrs, T., Ritter, C., Adrian, M., Riek-Loher, D., Bohrmann, B., Dobeli, H., Schubert, D. and Riek, R. 2005. Proc. Natl. Acad. Sci. U.S.A., 102:17342.
[49] Kirkitadze, M.D., Condron, M.M. and Teplow, D.B. 2001. J. Mol. Biol., 312:1103.
[50] Klimov, D.K. and Thirumalai, D. 2003. Structure, 11:295. [51] Röhrig, U.F., Laio, A., Tantalo, N., Parrinello, M. and Petronzio, R.
2006. Biophys. J., 91:3217.
[52] Alonso, D.O.V., DeArmond, S.J., Cohen, F.E. and Daggett, V. 2001. Proc. Natl. Acad. Sci. U.S.A., 98:2985.
[53] DeMarco, M.L. and Daggett, V. 2007. Biochemistry, 46:3045. [54] Sacchettini, J.C. and Kelly, J.W. 2002. Nat. Rev. Drug Disc, 1:267. [55] Armen, R.S., Alonso, D.O. and Daggett, V. 2004. Structure,
12:1847.
[56] Olafsson, I. and Grubb, A. 2000. Amyloid, 7:70.
[57] Janowski, R., Abrahamson, M., Grubb, A. and Jaskólski, M. 2004. J. Mol. Biol., 341:151.
[58] Bennett, M.J., Schlunegger, M.P. and Eisenberg, D. 1995. Protein Sci., 4:2455.
[59] Lin, Y-M., Liu, H-L., Zhao, J-H., Huang, C-H., Fang, H-W., Ho, Y. and Chen, W-Y. 2007. Biotechnol. Prog, in press, doi:10.1021/ bp060380d.
[60] Liu, D., Xu, Y., Feng, Y., Liu, H., Shen, X., Chen, K., Ma, J. and Jiang, H. 2006. Biochemistry, 45:10963.