行政院國家科學委員會專題研究計畫成果報告
一氧化氮與內皮素之間於新生鼠心臟細胞的交互作用
Inter action between nitr ic oxide and endothelin-1 in neonatal r at
car diomyocytes
計畫編號:NSC 89-2316-B-002-028
執行期限:88 年 8 月 1 日至 89 年 7 月 31 日
主持人:陳錦澤 國立台灣大學醫學院內科
一、中文摘要 內皮素會造成心肌細胞的肥大,而於 此過程中,心血管系統所釋放的一氧化氮 扮演著某種程度的對抗作用。本研究,在 於探討內生性一氧化氮對抗內皮素所誘發 c-fos 基因表現的作用機轉。實驗結果顯 示:內皮素經由活化其乙型接受器,會刺 激一氧化氮合成酵素,造成一氧化氮生成 增加。經由一氧化氮貢限劑如 SNAP、SIN-1 及一氧化氮清除劑 PTIO 的運用,以北方式 點墨法搭配報告者基因轉染的方法,顯示 一氧化氮可對抗內皮素所誘發 c-fos 的基因 表現;另外,以不同的負向突變基因 Ras (RasN17), Raf-1 (Raf301), 或 mERK,轉染 入心肌細胞皆可對抗內皮素所誘發 c-fos 的 基因表現,顯示內皮素誘發 c-fos 的基因表 現是經由 Ras/Raf/ERK 的訊息路徑;直接 觀查一氧化氮對內皮素所激活 ERK 的作 用,一氧化氮亦可對抗內皮素的激活 ERK 的活性;猶有甚者,一氧化氮尚且可對抗 內皮素所增加乙型重鍊肌蛋白基因啟動子 活性的作用。縱合觀之,內皮素會造成心 肌細胞一氧化氮生成增加,而一氧化氮可 進一步對抗內皮素的激活 ERK 的活性,造 成對抗內皮素所誘發 c-fos 的基因表現。 關鍵詞:內皮素、心肌細胞、一氧化氮、 Ras/Raf/ERK 訊息路徑、 c-fos AbstractEndothelin-1 (Et-1) treatment of cardiac myocytes (CM) induces cardiac hypertrophy. Cardiovascular release of nitric oxide (NO) may play a role during cardiac hypertrophy. The present study examined the protection mechanism of endogenously released NO on
c-fos induction by Et-1 in neonatal rat CM.
CM exposed to 1 induced NO release. Et-1 stimulated nitric oxide synthase (NOS) activity led to NO production that was attenuated by treating CM with an antagonist to endothelin B receptor. CM treated with a NO donor, S-nitroso-N-acetylpenicillamine (SNAP) or 3-morpholinosydnonimine (SIN-1), inhibited Et-1–induced c-fos expression.
Conversely, CM treated with a NO scavenger, 2-phenyl-4,4,5,5,-tetramethyl-imidazoline-l-oxyl-3-oxide (PTIO), augmented Et-1-induced c-fos expression. The attenuation of
NO on c-fos expression was shown by reducing either c-fos mRNA levels or c-fos
promoter activities using a chimera containing the c-fos promoter region (-2.25
kb) ligated to a reporter gene CAT. In contrast to the enhanced promoter activity in CM after PTIO treatment, attenuated Et-1-induced c-fos promoter activity was shown in CM treated with a NO donor. CM cotransfected with a dominant negative mutant of Ras (RasN17), Raf-1 (Raf301), or a catalytically inactive mutant of extracellular signal–regulated kinase (ERK)– 2 (mERK) inhibited Et-1–induced c-fos
promoter activity, indicating Ras/Raf/ERK pathway was involved. NO modulation of this signaling pathway was shown by its inhibitory effect on Et-1–induced ERK activity. CM treated with NO resulted in a decrease of Et-1-induced binding of nuclear proteins to the AP-1 binding sequences. Furthermore, CM treated with a NO donor significantly suppressed Et-1-induced β -myosin heavy chain promoter activities. These results indicate that CM under Et-1 treatment increases NO levels and the increased NO attenuated Et-1-induced c-fos
expression via the ERK signaling pathway.
These findings support the importance of NO as a negative regulator in Et-1–induced gene expression and cardiac hypertrophy.
Keywords: Endothelin-1, cardiomyocyte,
signaling pathway, c-fos
二、緣由與目的
Endothelin-1 (Et-1), a 21-amino acid peptide, is the most potent long-lasting vasoconstrictor (Yanagisawa et al., 1988). It is also a hypertrophy-promoting factor for various cells including cardiomyocytes (Wang et al., 1992 and 1993; Yamazaki et al., 1996). Cardiomyocytes contain EtA and EtB receptors (Kanno et al., 1993). We reported previously that Et-1 stimulated hypertrophic response by stimulating α– and β–myosin heavy chain gene expression in cardiomyocytes (Wang et al., 1992). We further showed that Et-1 induced c-fos expression and this induction was mediated via EtA receptor in cardiomyocytes (Cheng et al., 1999). In contrast, Et-1 triggering the release of NO from endothelial cells is mediated via EtB receptor (Tsukahara et al., 1994). The activation of EtA receptor in cardiomyocytes leads to the activation of phospholipase C and protein kinase C that result in an activation of mitogen-activated protein kinases (MAPK) (Clerk et al., 1994). The MAP kinases are a family of serine-threonine kinases, which include the extracellular signal-regulated kinases (ERKs), the c-Jun N-terminal kinases (JNKs/stress-activated protein kinases), and p38 MAPK. The ERKs that have been well studied are generally activated by mitogenic stimuli via a Ras/Raf1/MEK1 signaling
cascade. This leads to activation of transcription factors such as Elk1 and c-fos.
The signaling system leading to the activation of MAPK is subjected to diverse and complex regulation and the upstream of this kinase cascade has not been fully defined (Force and Bonventre, 1998; Schorb
et al., 1995; Duff et al., 1995). Recent studies suggested that the imbalance
of the intracellular oxidative and reductive status affected MAPK signaling pathways (Powis et al., 1997; Aikawa et al., 1997). When cells exposed to mitogens or cytokines, the intracellular redox status was altered by changing the oxidation-reduction state of essential thiol group in protein(s) (Lander et al., 1995; Sundaresan et al., 1995; Kamata et al., 1996). This redox-sensing mechanism underlying cellular responses is at least partly controlled at the level of transcription. Little is known of the molecular basis of such redox regulation in cells.
It is well documented that nitric oxide
(NO) or related molecules modulate cellular response. NO exerts its action by stimulating soluble guanylate cyclase, leading to an increase of cyclic GMP levels that activate cyclic GMP-dependent protein kinases (Schmidt and Walter, 1994). In addition, NO is capable of reacting with oxygen radicals such as superoxide anion to form peroxynitrate and the consequence of diminished No may contribute to the pathogenesis of atherosclerosis, hypertension-related complication (Lander et al., 1997; Akaike et al.,1998). Furthermore, recent studies have shown that NO or related molecules covently modify cysteine residues in proteins and such modification alters protein function and signaling amplitude (). Constitutive NO synthase (NOS3) and increased NO production were noticed during pathological states such as myocardial failure (Balligand et al., 1993; Haywood et al., 1996). It has been shown that NO antagonizes the actions of Et-1 in many cell types (Rizvi and Myers, 1997). Calderone et al (1998) reported that NO inhibited the growth-promoting effects of norepinephrine in cardiomyocytes. NO appears to attenuate the mitogenic effects despite the detailed mechanisms are not clear. The present study examined whether NO production was regulated by Et-1 in cardiomyocytes and the role of NO on Et-1-induced c-fos gene expression. We report
that cardiomyocytes exposed to Et-1 increased NO production that was mediated via EtB receptor. The released NO consequently attenuated Et-1–induced c-fos
expression via the ERK signaling pathway.
The inhibition of NO on Et-1-induced cardiac effects results in a decrease of β– myosin heavy chain transcriptional activity. Present results support the importance of NO derived from cardiomyocytes as a negative regulator for Et-1–induced cardiac hypertrophy.
三、結果
Effects of Et-1 on NO synthesis and NOS activity
The effect of Et-1 on NO generation and NOS activity in cardiomyocytes were analyzed. Exposure of cardiomyocytes to Et-1 (Et-10 nM) induced NO generation, with a maximal increase of 2-fold at 30 min after stimulation (Figure 1A). To specify whether the Et-1 receptor subtypes that were responsible for the generation of NO in cardiomyocytes, we pretreated
cardiomyocytes with Et-1 receptor type A antagonist (BQ485) or receptor type B antagonist (BQ788) before the Et-1 exposure. As shown in Figure 1B, BQ788 (100 nM) significantly inhibited the Et-1-induced generation of NO. In contrast, BQ485 (100 nM) had no significant effect on Et-1-induced generation of NO. Treatment cardiomyocytes with Et-1 receptor type B agonist IRL1620 (100 nM) alone also induced NO generation. These results suggested that an increase of NO generation upon Et-1 treatment was mediated mainly through the binding of Et-1 to Et-1 receptor type B. All three NOS isoforms characterized to date depend upon calmodulin activation, but constitutive NOS are distinguished from inducible NOS by their calcium sensitivity for the binding of activator calmodulin and enzymatic activity within the physiological range for intracellular calcium (Nathan and Xie, 1994). A constitutive NO synthase activity was detected and quantified in whole extracts of cardiomyocytes, and this activity was dependent upon the presence of calcium (Fig 1C). Exposure of cardiomyocytes to Et-1 (Et-10 nM) increased NOS activity, with a maximal increase of 2-fold at 30 min after stimulation (Figure 1C). To specify whether the Et-1 receptor subtypes that were responsible for the increase of NOS activity in cardiomyocytes, we pretreated cardiomyocytes with Et-1 receptor type A antagonist (BQ485) or receptor type B antagonist (BQ788) before the Et-1 exposure. As shown in Figure 1D, BQ788 (100 nM) significantly inhibited the Et-1-increased NOS activity. In contrast, BQ485 (100 nM) had no significant effect on Et-1-increased NOS activity. Treatment cardiomyocytes with Et-1 receptor type B agonist IRL1620 (100 nM) alone also increased NOS activity.
Figure 1: Effects of Et-1 on NO synthesis and NOS activity.
NO Modulates Et-1–Induced c-fos Gene
Expr ession in car diomyocytes
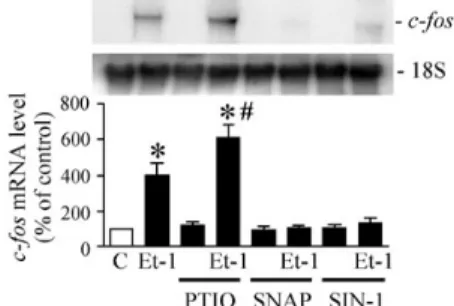
To explore whether NO modulates the induction of c-fos gene in Et-1-treated
cardiomyocytes. Cardiomyocytes were preincubated with an NO donor, ie, SNAP or
SIN-1, for 30 minutes before being treating with Et-1 in the presence of the NO donor. As shown in Figure 2, SNAP treatment of cardiomyocytes at a concentration of 100 µmol/L significantly suppressed Et-1– induced c-fos mRNA expression. Similarly,
treatment of cardiomyocytes with another NO donor, SIN-1 (100 µmol/L), also significantly attenuated Et-1–induced c-fos
expression. When cardiomyocytes were pretreated with PTIO, an NO scavenger, for 1 hour and then treatment with Et-1 in the presence of the agent. In contrast to the inhibitory effect by the NO donor, PTIO treatment of cardiomyocytes at a concentration of 100 µmol/L significantly augmented Et-1–induced c-fos mRNA levels
(Figure 2). These results suggest that Et-1– induced c-fos expression is modulated by
NO.
Figure 2: NO Modulates Et-1–Induced c-fos
mRNA
To further determine whether the modulation of NO in Et-1–induced c-fos
expression is a transcriptional event, an c-fos
promoter construct containing the c-fos
promoter region (–2.25 kb) and the reporter gene chloramphenicol acetyltransferase were transiently transfected into cardiomyocytes. As shown in Figure 3, cardiomyocytes exposed to 24 hours of Et-1 (10 nM) significantly increased c-fos promoter activity by 2.4-fold compared with untreated cells. The addition of both SNAP and SIN-1 to cardiomyocytes completely abolished this increased c-fos promoter activity. Conversely,
treatment of cardiomyocytes with PTIO enhanced this promoter activity. These results together suggest that NO modulation of c-fos induction by Et-1 involves
Figure 3: NO Modulates Et-1–Induced c-fos
promoter activity
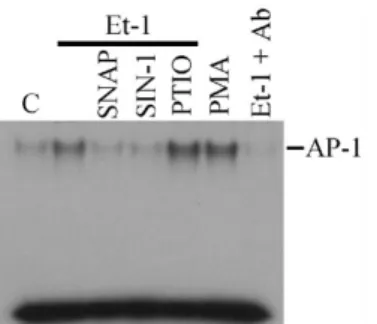
NO Regulates Et-1-Induced AP-1 Binding
The Et-1-induced c-fos gene expression was concordant with an increase in activator protein-1 (AP-I) activity. Using the electrophoretic mobility shift assay, the transcription factor AP-1 DNA binding activity was stimulated by Et-1 treatment for six hours (Figure 4). The Et-1-stimulated AP-1 binding was also blocked by the NO donor, SNAP or SIN-1, and enhanced by NO scavenger, PTIO, indicating that intracellular NO level is involved in the modulation of Et-1-stimulated AP-I activity. This binding was obviously specific to AP-1, because it was abolished by coincubation of nuclear proteins with c-Fos and c-Jun antibodies. These results indicate that NO inhibits the AP-1 protein induced by Et-1.
Figure 4: NO Regulates Et-1-Induced AP-1 Binding
Et-1–Induced c-fos Gene Expression Is
Mediated via the Ras/Raf/ERK Pathway
To dissect the signaling pathway in regulating c-fos induction by Et-1, we used
the dominant negative mutants of Ras (RasN17) and Raf-1 (Raf301) and a catalytically inactive mutant of ERK2 (mERK), all of which are associated with the Ras/Raf/ERK pathway, to examine the effect of these mutants on the induction of c-fos by
Et-1. As shown in Figure 5, cardiomyocytes that were cotransfected with the empty vector control PSR revealed no effect on Et-1– induced c-fos promoter activity. However,
cotransfection of the cells with RasN17, Raf301, or mERK resulted in a significant inhibition in Et-1–induced c-fos promoter
activity. Consistently, cardiomyocytes treated with a specific inhibitor to mitogen-activated
protein kinase kinase (MEK), ie, PD98059, attenuated Et-1–induced c-fos promoter
activity. In contrast, cardiomyocytes cotransfected with a dominant positive mutant of Ras (RasL61) or MEK1 greatly increased their c-fos promoter activity. These
results suggest that the Ras/Raf/ERK signaling pathway is involved in Et-1– induced c-fos gene expression in cardiomyocytes. ERKs are the upstream regulators of Et-1-induced c-fos gene expression.
Figure 5: Et-1–Induced c-fos Gene Expression Is Mediated via the Ras/Raf/ERK Pathway
NO Regulates Et-1–Induced ERK Phosphor ylation and Activity in car diomyocytes
Given that we showed that the ERK signaling pathway is involved in Et-1-induced c-fos expression and that NO
modulates Et-1-induced c-fos expression, we
further investigated whether NO modulates the activation of ERK in Et-1-treated cardiomyocytes. We first examined the ERK1/ERK2 phosphorylation in cardiomyocytes exposed to Et-1 (10 nM) in the presence of the NO donor or NO scavenger. As shown in Figure 6, treatment of Et-1 to cardiomyocytes for 15 minutes induced rapid phosphorylation of ERK1/ERK2 by 4.7-fold. Cardiomyocytes preincubated with an NO donor, SNAP, significantly inhibited Et-1–induced ERK1/ERK2 phosphorylation by 80%. In the same experiment, PTIO treatment of cardiomyocytes augmented Et-1–induced ERK1/ERK2 phosphorylation by 40% above that by Et-1-treatment only (Figure 6).
Figure 6: NO Regulates Et-1–Induced ERK Phosphorylation
NO modulation of the ERK signaling pathway was further elucidated by its inhibitory effect on Et-1–induced ERK activity. Cardiomyocytes exposed to Et-1 rapidly induced ERK activity by 3.4-fold, as indicated by an increase of 32
P-labeled phosphorylation of MBP by ERK (Figure 7). Treatment of cardiomyocytes with SNAP significantly attenuated this ERK activation by 90%. In contrast, PTIO treatment of cardiomyocytes enhanced this ERK activation by 50% (Figure 7). Taken together, these findings imply that NO modulates Et-1-induced c-fos gene expression via its
inhibitory effect on the ERK signaling pathway in cardiomyocytes.
Figure 7: NO Regulates Et-1–Induced ERK Activity
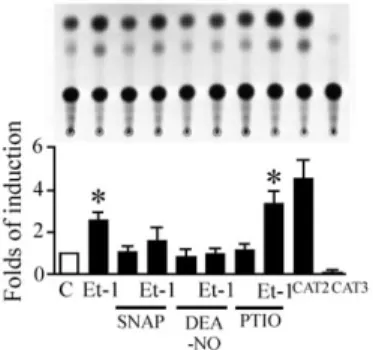
The activation of ß myosin heavy chain promoter (β -MHC) by Et-1 is modulated by NO
The hypertrophic responses in cardiac myocytes are characterized by the induction of a fetal gene program (Komuro and Yazaki, 1993). To get insight into the relation between Et-1 and NO, we examined whether NO modulates Et-1-induced expression of cardiac fetal genes. Incubation with Et-1 (10 nM) for 48 h increased β -MHC promoter activities by 2.3–fold as compaired with control level (Figure 8). When cardiac myocytes were pre-incubated with a NO donor, DEA-NO (100 µM) for 60 min, Et-1-induced activation of β -MHC promoter activities was significantly suppressed.
Figure 8: The activation of ß myosin heavy chain promoter (β -MHC) by Et-1 is modulated by NO
四、討論
Cardiac myocyte hypertrophy is a central feature of all types of heart muscle failure. Hypertrophic stimuli reach the nucleus via multiple signaling pathways within cardiac myocytes and elicit changes in cardiac gene expression. Et-1 is one of the local factors that play important roles in the development of heart failure. Further elucidation of the precise mechanisms by which this central pathway modulates the hypertrophic response may provide novel therapeutic approaches to human heart failure. Many hypertrophic stimuli, including Et-1 stimulation, have been shown to activate the Ras-mitogen-activated protein kinase pathway, also known as the ERK pathway (Post et al., 1996; Fuller et al., 1998). ERK1/2 are one element in a series of kinases that serve to connect the nucleus with cytosolic events. The present study demonstrated that inhibition of the ERK cascade with PD98059 or mERK blocked the NO modulation of Et-1-induced c-fos gene expression. These findings demonstrate that ERK1/2 plays a upstream role of NO modulation of Et-1-induced c-fos gene expression signaling of cardiac myocytes. The cytoplasmic mitogen-activated protein kinase (MAPK) cascade is a process through which extracellular stimuli can be transmitted from the cell surface to the nucleus (Cook et al., 19932). C-fos is an immediate-early gene encoding a DNA-binding protein of the AP-1 family, which can function in the transcription activation of many other gene (). MAPK, including ERK1/ERK2 and c-fos are all rapidly and transiently activated by Et-1 (Figure 5 and 6; Cheng et al., 1999). We reported here that the Et-1-induced activation of ERK1/ERK2 and the expression of c-fos are highly sensitively to the NO pretreatment. By using a MAPK kinase inhibitor, the involvement of MAPK in the NO modulation of Et-1-induced c-fos gene expression is also demonstrated.
The immediate proto-oncogene c-fos is
normally expressed at a minimal level in the mammalian heart. Increased proto-oncogene expression has been implicated in the development of cardiac hypertrophy (Barka et al., 1987). Induction of c-fos, a
transcriptional factor that interacts with
cis-regulatory element AP-1 in many genes, has important consequences for downstream gene expression that may determine the phenotypic response to Et-1. The activation of c-fos transcription is thought to be
mediated by the activation of MAPK (Saklatvala et al., 1993). In cardiomyocytes, Et-1 has been shown to activate two closely related MAPK isoforms, p42MAPK and p44MAPK (Post et al, 1996) via a
Ras-dependent pathway.
Exogenous NO has been reported to inhibit protein kinase C gene expression and its activity in a variety of cells. Jun et al. (1994) reported that lipopolysaccharide- or phorbol 12-myristate 13-acetate-induced NO or the NO donor sodium nitroprusside inhibited the expression of the protein kinase C delta gene in murine peritoneal macrophages. Studer et al. (1996) reported that the NO donor S-nitroso-N-acetylpenicillamine inhibited basal protein kinase C activity in mesangial cells. The modulation of protein kinase C activity is important for cellular proliferation and protein synthesis. We investigated whether the Et-1 increased expression of the intermediary signaling element mitogen-activated protein kinase, which is downstream of protein kinase C, and the proto-oncogen c-fos were modulated by NO treatment. Various growth factors, including Et-1, are known to activate mitogen-activated protein kinase and c-fos (Jones et al., 1995;
Cheng et al., 1999). We observed that mitogen-activated protein kinase and c-fos gene expression were increased in cardiomyocytes with Et-1 stimulation.
The present study implies that NO regulates Et-1-induced c-fos expression in cardiomyocytes via the inhibition of the ERK signaling pathway. However, the exact target molecule(s) of this NO modulation has not been defined. NO activates guagnlate cyclase to produce cGMP, which affects various targets, including cGMP-dependent protein kinase (Schmidt et al., 1993). The Ras/Raf/ERK signaling pathway is inhibited by the cGMP-dependent protein kinase via the phosphorylation of c-Raf kinase on Ser43 (Suhasini et al., 1998). In the present study, cardiomyocytes treated with KT5823, a cGMP-dependent protein kinase inhibitor,
did attenuate the inhibitory effect of NO on Et-1-induced promoter activity (data not shown). This implies that the inhibitory effect of NO on Et-1-induced c-fos induction is mediated via the cGMP-dependent protein kinase.
五、參考文獻
Akaike T, Suga M and Maeda H (1998) Proc Soc Exp Biol Med 217: 64-73.
Aikawa, R., Komuro, I., Yamazaki, T., Zou, Y., Kudoh, S., Tanaka, M., Shiojima, I., Hiroi, Y., and Yazaki, Y. (1997) J. Clin. Invest. 100, 1813-1821
Barka TH, van der Neon, Shaw PA.(1987) Oncogene 1:439-443.
Balligand JL, Kelly RA, Marsden PA, Smith TW and Michel T (1993) Proc Natl Acad Sci USA 90: 347-351.
Calderone A, Thaik CM, Takahashi N, Chang DLF and Colucci WS (1998) J Clin Invest 101: 812-818.
Cheng TH, Shih NL, Chen SY, Wang DL. and Chen JJ (1999) Cardiovas. Res. 41: 654-662.
Choukroun G, Hajjar R, Kyriakis JM, Bonventre JV, Rosenzweig A and Force T (1998) J Clin Invest 102: 1311-1320.
Clerk A, Bogoyevitch MA, Anderson MB, Sugden PH (1994) J Biol Chem 269: 32848328-57
Cook SJ, Rubinfeld B, Albert I, and McCormick F (1993) EMBO J 12: 3475-3485.
Dudley DT, Pang L, Decker SJ, Bridges AJ, and Saltiel AR (1995) Proc Natl Acad Sci USA 92: 7686-7689.
Duff, J. L., Marrero, M. B., Paxton, W. G., Schieffer, B., Bernstein, K. E., and Berk, B. C. (1995) Cardiovasc. Res. 30, 511-517.
Feig LA and Cooper GM (1998) Mol Cell Biol 8:3235-3243.,
Force, T., and Bonventre, J. V. (1998) Hypertension. 31, 152-161.
Fuller, S. J., Gillespie-Brown, J., and Sugden, P. H. (1998) J. Biol. Chem. 273,
18146-18152
Haywood GA, Tsao PS, von der Leyen HE, Mann MJ, Keeling PJ, Trindade PT, Lewis NP, Byrne CD, Rickenbacher PR, Bishopric NH et al. (1996) Circulation. 93: 1087-1094.
Jones LG, Gause KC and Meier KE (1995) Ann NY Acad Sci 766:484-486. Jun CD, Choi BM, Lee SY, Kang SS, Kim
Biophys Res Commun 204: 105-111. Kamata, H., Tanaka, C., Yagisawa, H., Matsuda, S., Gotoh, Y., Nishida, E., and Hirata, H. (1996) J. Biol. Chem. 271, 33018-33025
Kanno K, Hirata Y, Tsujino M, Imai T, Shichiri M, Ito H and Marumo F (1993) Biochem Biophys Res Commun. 194: 1282-1287.
Kim, H., Shim, J., Han, P. L., and Choi, E. J. (1997) Biochemistry. 36, 13677-13681. Komuro I and Yazaki Y (1993) Annu Rev
Physiol 55, 55-75.
Kotch W, Heidecker G, Lloyd P and Rapp UR (1991) Nature 349:426-428.
Lander, H. M., Ogiste, J. S., Teng, K. K., and Novogrodsky, A. (1995) J. Biol. Chem. 270, 21195-21198.
Lander HM, Hajjar DP, Hempstead BL, Mirza UA, Chait BT, Champbell S and Quilliam LAA (1997) J Biol Chem 272: 4323-4326.
Nathan C and Xie Q (1994) Cell 78: 915-918.
Post GR, Goldstein D, Thuerauf DJ, Glembotski CC, Brown JH. J Biol Chem 1996;271:8452-8457.
Powis, G., Gasdaska, J. R., and Baker, A. (1997) Adv. Pharmacol. 38, 329-359. Rizvi MA and Myers PR (1997) J Mol Cell
Cardiol 29: 1779-1789.
Saklatvala J, Rawlinson LM, Marshall CJ, Kracht M. FEBS Lett 1993;334:189-192. Schmidt HW, Lohmann SM and Walter U
(1993) Biochem Biophys Acta 1178: 153-175.
Schmidt, H. H. H. W., and Walter, U. (1994) Cell 78, 919-925
Schorb, W., Conrad, K. M., Singer, H. A., Dostal, D. E., and Baker, K. M. (1995) J. Mol. Cell. Cardiol. 27, 1151-60. Studer RK, DeRubertis FR and Craven PA (1996) J Am Soc Nephrol 7:999-105. Suhasini M, Li H, Lohmann SM, Boss GR
and Pilz RB (1998) Mol Cell Biol 18: 6983-6994.
Sundaresan, M., Yu, Z. X., Ferrabs, V. J., and Finkel, T. (1995) Science 270,
296-299.
Tsukahara H, Ende H, Magazine HI, Bahou WF and Goligorsky MS (1994) J Biol Chem 269: 21778-21785.
Wang DL, Chen JJ, Shih NL, Kao YC, Hsu KH, Huang WY and Liew CC (1992) Biochem Biophys Res Commun 183: 1260-1265.
Yamazaki T, Komuro I, Kudoh S, Zou Y, Shiojima I, Hiroi Y, Mizuno T, Maemura K, Kurihara H, Aikawa R,
Takano H and Yazaki Y (1996) J Biol Chem 271: 3221-3228.
8