Cyclopropenation and Related Reactions of Ruthenium
Vinylidene Complexes
Pei-Chen Ting, Ying-Chih Lin,* Gene-Hsiang Lee, Ming-Chu Cheng, and Yu Wang
Contribution from the Department of Chemistry, National Taiwan UniVersity, Taipei, Taiwan 106, Republic of China
ReceiVed January 2, 1996X
Abstract: Facile deprotonation of a number of cationic ruthenium vinylidene complexes, followed by cyclopropenation,
is accomplished in acetone. The deprotonation of [Ru]dCd(Ph)CH2R+, ([Ru]
)(η
5-C5H5)(PPh3)2Ru) by
n-Bu4-NOH induces a novel cyclization reaction and yields the neutral cyclopropenyl complexes [Ru]-CdC(Ph)CHR
(3b, R)CN; 3c, R)Ph; 3d, R)CHdCH2; 3e, R)CHdCMe2). Complex [Ru]-CdC(C6H9)CHCN
+ (3k) is
similarly prepared. Protonation of 3b-3e regenerates the corresponding vinylidene complexes. Deprotonation of
[Ru]dCdC(Ph)CH2COOMe+
(2h) by n-Bu4NOH induces a different type of cyclization and yields the neutral furan
complex [Ru]-CdC(Ph)CHdC(O)OMe (4h). The cyclopropenyl complex containing a methoxy substituent cannot
be prepared from [Ru]dCdC(Ph)CH2OCH3+
(2i), but F
-of n-Bu4NF attacks the CRof 2i to produce the unstable
vinyl complex [Ru]C(F)dC(Ph)CH2OCH3(5). Complex [Ru]-CdC(Ph)C(CN)OCH3(9b) was indirectly prepared
from the addition of TCNQ to 3b, giving [Ru]dCdC(Ph)CH(CN)TCNQ (6b) followed by methanolysis. Unlike 3, complex 9b is not converted to vinylidene complex, instead, removal of the methoxy substituent by acid gives the
cationic cyclopropenylium complex [Ru]-CdC(Ph)C(CN)
+
(10b). Complex [Ru]-CdC(Ph)C(COOMe)
+ (10h) is similarly prepared from 4h via a TCNQ complex 6h followed by a methoxy-substituted complex 9h. In the presence of allyl iodide, opening of the three-membered ring of 3b, followed by a subsequent oxidative coupling
reaction, gives a dimeric dicationic product{[Ru]dCdC(Ph)-CHCN}2
2+
(11). Proton abstraction of 11 by
n-Bu4-NF gives the biscyclopropenyl complex {[Ru]-CdC(Ph)CCN}2(12). Molecular structures of complexes 3b, 3f,
4h, 6b, 9b, and 11 have been confirmed by X-ray diffraction analysis.
Introduction
Cyclopropene is believed to be the most highly strained cycloalkene, with the estimated substantial strain energy of more
than 50 kcal/mol.1 This molecule has hence been under intense
investigation2and has played a crucial role in the development
of important concepts such as aromaticity and chemical
reac-tivities.3 Three general methods are known for the synthesis
of cyclopropenes:4 viz., addition of carbene to alkyne,5 ring
closure of vinylcarbene,6 and 1,2-elimination of a suitable
precursor such as halocyclopropane.7 Two recent papers2a,bhave
suggested vinylidene (alkylidenecarbene) to be the intermediate in the thermal rearrangement of cyclopropene: i.e. when substituted cyclopropenes are heated or irradiated, complex
mixtures of 1,3-dienes, allenes, and acetylenes are formed.8This
strongly suggests that the formation of acetylenes involves
vinylidenes as intermediates.9 Some theoretical results also
suggest that the acetylenic products are formed from vinylidene
produced through bond breaking and hydrogen shift.10,11 It thus
appears that vinylidene is an important intermediate in the
thermal rearrangement of cyclopropene to acetylene.12
How-ever, organic vinylidene (R2CdC:) is thermodynamically un-stable and evidence for its existence has been derived mostly from the reaction products. Fortunately, vinylidene, among a variety of reactive organic species that can be stabilized by complex formation with transition metals, has been shown to form a plethora of stable organometallic compounds.
Particu-XAbstract published in AdVance ACS Abstracts, June 15, 1996.
(1) (a) Liebman, J. F.; Greenberg, A. Strained Organic Molecules; Wiley: New York, 1978; p 91. (b) Special issue on strained organic compounds: Chem. ReV. 1989, 89.
(2) (a) Maier, G.; Preiss, T.; Reisenauer, H. P.; Hess, B. A., Jr.; Schaad, L. J. J. Am. Chem. Soc. 1994, 116, 2014. (b) Hopf, H.; Plagens, A.; Walsh, R. J. Chem. Soc., Chem. Commun. 1994, 1467. (c) Maier, G.; Reisenauer, H. P.; Pacl, H. Angew. Chem., Int. Ed. Engl. 1994, 33, 1248.
(3) (a) Liebman, J. F.; Greenberg, A. Chem. ReV. 1976, 76, 311. (b)
Halton, B.; Banwell, M. G. In The Chemistry of the Cyclopropyl Group.
Part 2; Patai, S., Rappoport, Z., Eds.; Wiley: Chichester, 1987; Chapter
21, p 1223.
(4) Billups, W. E.; McCord, D. J. Angew. Chem., Int. Ed. Engl. 1994,
33, 1332.
(5) (a) Maier, G.; Hoppe, M.; Reisenauer, H. P.; Kru¨ger, C. Angew.
Chem., Int. Ed. Engl. 1982, 21, 437. (b) Dowd, P.; Garner, P.; Schappert,
R.; Irngartinger, H.; Goldmann, A. J. Org. Chem. 1982, 47, 4240. (c) Suvorova, G. N.; Komendantov, M. I. Zh. Org. Khim. 1982, 18, 1882; Chem.
Abstr. 1983, 98, 71511. (c) Doyle, M. P; Protopopova, M.; Muller, P.;
Ene, D.; Shapiro, E. A. J. Am. Chem. Soc. 1994, 116, 8492.
(6) (a) Baldwin, J. E.; Black, K. A. J. Am. Chem. Soc. 1984, 106, 1029. (b) Ando, W.; Hanyu, Y.; Takata, T.; Ueno, K. J. Am. Chem. Soc. 1984,
106, 2216. (c) Du¨rr, H.; Klauck, G.; Peters, K.; von Schnering, H. G. Angew. Chem., Int. Ed. Engl. 1983, 22, 332. (d) Kett, M. W.; Johnson, R.
P. Tetrahedron. Lett. 1983, 24, 2523. (e) Franck-Neumann, M.; Miesch, M. Tetrahedron. Lett. 1984, 25, 2909.
(7) (a) Baird, M. S.; Buxton, S. R.; Whitley, J. S. Tetrahedron. Lett. 1983, 24, 1509. (b) Padwa, A.; Pulwer, M. J.; Rosenthal, R. J. J. Org.
Chem. 1984, 49, 856. (c) Harnisch, J.; Baumgartel, O.; Szeimies, G.;
Meerssche, M. V.; Germain, G.; Declercq, J.-P. J. Am. Chem. Soc. 1979,
101, 3370.
(8) Gajewski, J. J. Hydrocarbon Thermal Isomerizations; Wiley: New York, 1981; pp 22-25.
(9) Walsh, R.; Wolf, C.; Untiedt, S.; de Meijere, A. J. Chem. Soc., Chem.
Commun. 1992, 421.
(10) Yoshimine, M.; Pacansky, J.; Honjou, N. J. Am. Chem. Soc. 1989,
111, 2785, 4198.
(11) Stng, P. J. In Carbene(oide); Regitz, M., Ed.; Georg Thieme: Stuttgart, 1989; p 84ff.
(12) Likhotvorik, I. R.; Brown, D. W.; Jones, M., Jr. J. Am. Chem. Soc. 1994, 116, 6175.
S0002-7863(96)00001-7 CCC: $12.00 © 1996 American Chemical Society
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
larly the mononuclear ruthenium(II) moieties, CpRu(PR3)2+(Cp
) η5-C5H5), play an important role in the stabilization of
[Ru]dCdCRR′derivatives.
Metal vinylidene complexes have also attracted a great deal of attention since they offer the possibility of development of new types of organometallic intermediates that may have unusual reactivity. Extensive reviews on this subject have
appeared recently.13 The best entry into the transition metal
vinylidene complexes is the addition of electrophiles to the
electron-rich carbon of metal alkynyl complexes.14 A theoretical
study of the vinylidene complex has revealed the localization
of electron density on Cβ (HOMO) or the MdC double bond
and electron deficiency at CR.
15 Thus the MdC double bond
and the Cβ atom are more susceptible to electrophilic attack
whereas the CRatom is prone to nucleophilic attack.16 Hence
the reactions of such compounds containing electron-rich metals
with electrophiles lead to formation of carbene complexes.17
On the other hand, their reactions with nucleophiles generally result in the formation of vinyl derivatives. Protonation of
vinylidene ligand at Cβ is known to readily form a carbyne
unless the ligand is present in a cationic form. With a more electron rich metal center, addition to the MdC bond yields an
η2-allene
- or heteroketene-metal complex.
18,19 Addition of
the acetylenic alcohols HCtC(CH2)nOH to CpRuL2Cl also
affords cyclic carbene complexes. The reaction proceeds via initial formation of the vinylidene complexes, followed by an
intramolecular attack of the terminal alcohol function on CR.
20 A study of the reaction of alcohols with Ru vinylidene complexes has shown that the electron-withdrawing groups on the acetylide unit or on the metal facilitate nucleophilic attack
at CR.21
Most surprisingly, with such a background, the relation between vinylidene and cyclopropene in the organometallic system has been mostly left unnoticed. We believed that electron-withdrawing functionality, such as the CN group, at
Cγmight play a role in enhancing the acidity of its neighboring
proton. Thus an intramolecular cycloaddition leading to the formation of the cyclopropenyl complex may be effected by a base. We have reported our preliminary results on one specific
compound in a recent communication.22 After thorough
ex-ploration, it has been observed that the method indeed leads to a number of cyclopropenyl complexes. Utilizing the above-mentioned reactivities, herein we report the unprecedented cyclopropenation reaction of the vinylidene ligands with various
substituents at Cγand limitations of this type of reaction. In
addition, a coupling reaction of the cyclopropenyl complex
leading to the synthesis of the first 2,2′-bicyclopropenyl metal
complex is also reported. Results and Discussion
Metal Vinylidene Complexes. Treatment of [Ru]-CtC -Ph (1a) with ICH2CN affords the cationic vinylidene complex
[Ru]dCdC(Ph)CH2CN+
(2b) with 72% yield. Similarly,
preparations of complexes [Ru]dCdC(Ph)CH2R+(2a, R
)H;
2c, R)Ph; 2d, R)CHdCH2; 2e, R)CHdCMe2; 2h, R)
COOCH3; 2i, R) COOC2H5; 2j, R )OCH3) have all been
achieved with high yields. The complex
[Ru]dCdC(C6H9)CH2-CN+
(2k, C6H9)1-cyclohexenyl) is also prepared from the
reaction of [Ru]-CtC-C6H9with ICH2CN. With the
excep-tions of 2h and 2i, the vinylidene complexes mentioned above
have been prepared in CH2Cl2either at room temperature or at
refluxing temperature. For the synthesis of 2h and 2i, a mixture
of CH2Cl2/CHCl3 (1:1 v/v) was used as solvent due to the
necessity of achieving higher reaction temperature. The most characteristic spectroscopic data of these vinylidene complexes
consist of strongly deshielded CR resonance as a triplet at δ
340 ( 5 in the
13C NMR spectrum and a single 31P NMR
resonance normally at around δ 42 ( 1 in CDCl3 at room
temperature, which is due to the fluxional behavior of the
vinylidene ligand.23
Deprotonation/Cyclopropenation of Vinylidene Com-plexes. Deprotonation of 2b by n-Bu4NOH induces a new cyclization reaction and yields a neutral cyclopropenyl complex
[Ru]-CdC(Ph)CHCN (3b) (Scheme 1). This reaction occurs
only in acetone. The light-orange-yellow crystalline precipitate forms directly in the reaction mixture and can be obtained in analytically pure form by a simple filtration. No cyclopropena-tion reaccyclopropena-tion is observed in CH3CN or MeOH. When the reaction is carried out at lower concentration, single crystals of 3b, suitable for X-ray diffraction analysis, are directly obtained. Reaction of 2b with n-Bu4NF (1 M in THF) or DBU (1,8-diazabicyclo[5.4.0]undecene) or KOH (dissolved in a minimum amount of H2O) also yields 3b. Complex 3b is stable in air and soluble in CH2Cl2, CHCl3, and THF but insoluble in diethyl ether, n-hexane, MeOH, and CH3CN.
The31P NMR spectrum of 3b displays resonances with the
expected two doublets pattern (δ 49.7 and 51.6 with2J
P-P ) 34.6 Hz) arising from the asymmetric three-membered ring. In
the1H NMR spetrum of 3b, the resonance of the methyne proton
appears atδ 1.40, and in the13C{1H}NMR spectrum, a triplet
atδ 126.2 with2J C-P
)23.5 Hz is assigned to the
ruthenium-bonded CRcarbon.
(13) (a) Bruce, M. R. Chem. ReV. 1991, 91, 197. (b) Bruce, M. I.;
Swincer, A. G. AdV. Organomet. Chem. 1983, 22, 59. (c) Davies, S. G.;
McNally, J. P.; Smallridge, A. J. AdV. Organomet. Chem. 1990, 30, 1.
(14) (a) Werner, H. Angew. Chem., Int. Ed. Engl. 1990, 29, 1077. (b) Werner, H.; Rappert, T.; Wolf, J. Isr. J. Chem. 1990, 30, 377. (c) Werner, H.; Hohn, A.; Schulz, M. J. Chem. Soc., Dalton Trans. 1991, 777. (d) Schafer, M.; Wolf, J.; Werner, H. J. Chem. Soc., Chem. Commun. 1991, 1341. (e) Schneider, D.; Werner, H. Angew. Chem. 1991, 103, 710. (f) Werner, H.; Dirnberger, T.; Hohn, A. Chem. Ber. 1991, 124, 1957. (g) Werner, H.; Weinhand, R.; Knaup, W. Organometallics 1991, 10, 3967. (h) Rappert, T. O.; Mahr, N.; Wolf, J.; Werner, H. Organometallics 1992,
11, 4156. (i) Nakanishi, S.; Goda, K.; Uchiyama, S.; Otsuji, Y. Bull. Chem. Soc. Jpn. 1992, 65, 2560. (j) Haquette, P.; Pirio, N.; Touchard, D.; Toupet,
L.; Dixneuf, P. H. J. Chem. Soc., Chem. Commun. 1993, 163. (k) Watatuski, Y.; Koga, N.; Yamazaki, H.; Morokuma, K. J. Am. Chem. Soc. 1994, 116, 8105.
(15) (a) Kostic, N. M.; Fenske, R. F. Organometallics 1982, 1, 974. (b) Werner, H.; Wolf, J.; Muller, G.; Kru¨ger, C. Angew. Chem., Int. Ed. Engl. 1984, 28, 431.
(16) (a) Espuelas, J.; Esteruelas, M. A.; Lahoz, F. J.; Oro, L. A.; Valero, C. Organometallics 1993, 12, 663. (b) Werner, H.; Meyer, U.; Esteruelas, M. A.; Sola, E.; Oro, L. A.; Valero, C. J. Organomet. Chem. 1989, 366, 187. (c) Andriollo, A.; Esteruels, M. A.; Meyer, U.; Oro, L. A.; Sanchez-Delgardo, R. A.; Sola, E.; Velcro, C.; Werner, H. J. Am. Chem. Soc. 1989,
111, 7431. (d) Espuelas, J.; Esteruelas, M. A.; Lahoz, F. J.; Oro, L. A.;
Ruiz, N. J. Am. Chem. Soc. 1993, 115, 4683.
(17) (a) Davison, A.; Selegue, J. P. J. Am. Chem. Soc. 1978, 100, 7763. (b) Davison, A.; Selegue, J. P. J. Am. Chem. Soc. 1980, 102, 2455. (c) Casey, C. P.; Miles, W. H.; Tukoda, H.; O’Connor, J. M. J. Am. Chem.
Soc. 1982, 104, 3761. (d) Kremer, K. A. M.; Kuo, G.; O’Connor, E. I.;
Helquist, P.; Kerber, R. C. J. Am. Chem. Soc. 1982, 104, 6119. (e) Bodner, G. S.; Smith, D. E.; Hatton, W. G.; Heah, P. C.; Georgiou, S.; Rheingold, A. L.; Geib, S. J.; Hutchinson, J. P.; Gladysz, J. A. J. Am. Chem. Soc. 1987, 109, 7688.
(18) (a) Werner, H.; Wolf, J.; Zolk, R.; Schubert, U. Angew. Chem., Int.
Ed. Engl. 1983, 22, 981. (b) Wolf, J.; Zolk, R.; Schubert, U.; Werner, H. J. Organomet. Chem. 1988, 340, 161.
(19) (a) Hoel, E. L.; Ansell, G. B.; Leta, S. Organometallics 1984, 3, 1633. (b) Consiglio, G.; Schwab, R.; Morandini, F. J. Chem. Soc., Chem.
Commun. 1988, 25.
(20) Bruce, M. I.; Swincer, A. G.; Thomson, B. J.; Wallis, R. C. Aust.
J. Chem. 1980, 33, 2605.
(21) Bruce, M. I.; Swincer, A. G. Aust. J. Chem. 1980, 33, 1471.
(22) Ting, P. C.; Lin, Y. C.; Cheng, M. C.; Wang, Y. Organometallics 1994, 13, 2150.
(23) (a) Allen, D. L.; Gibson, V. C.; Green, M. L.; Skinner, T. F.; Bashikin, J.; Grebenik, P. D. J. Chem. Soc., Chem. Commun. 1985, 895. (b) Consiglio, G.; Morandini, F. Chem. ReV. 1987, 87, 761.
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
The deprotonation/cyclopropenation in acetone is a general reaction for a number of vinylidene complexes, namely, similar reactions are also known to occur for 2c, 2d, and 2e, giving
[Ru]-CdC(Ph)CHR (3c, R)Ph; 3d, R)CHdCH2; 3e, R)
CHdCMe2), respectively. Unlike 3b, complexes 3c-e can be
obtained only by using n-Bu4NOH as proton abstractor and the
reactions generally take longer. Complexes 3b-e are stable in
THF, but in CHCl3compounds 3c, 3d, and 3e are less stable
than 3b. Furthermore, 3c decomposes in CDCl3 producing
Cp(PPh3)2RuCl and some unidentified organic products. De-composition of 3d and 3e produces a complicated mixture. The
stability of the cyclopropenyl complexes in CHCl3follows the
trend for the substituents of CN > Ph > CHdCH2 >
CHdCMe2. The phenyl group on the Cγis not essential since
deprotonation of 2k also gives [Ru]-CdC(C6H9)CHCN (3k),
which exhibits better solubility in common organic solvents. Facile deprotonation indicates the acidic nature of the methylene
protons of 2b-2e and 2k, which may be ascribed to the
combined effect of the cationic character, the electron-withdrawing substituent, and the benzylic/allylic property of the vinylidene complexes. 2a is inert toward n-Bu4NOH in acetone probably due to the lack of this acidic proton. It also appears
that the hybridization of the Cδshould either be sp or sp2for
the cyclopropenation to occur. However, the vinylidene
com-plex with a propargyl substituent at Cβis too reactive to yield
any isolable product. This is probably due to the presence of the acidic proton that complicates the outcome. When treated with nucleophiles, 2b fails to produce the intermolecular addition
product.24
Synthesis of metal cyclopropenyl derivatives in which the
metal bonds to C(sp3) of the cyclopropene ring (in this case the
three-membered ring can be viewed as an antiaromatic
cyclo-propenide ion) has been reported in the literature.25 However,
to our knowledge, only one example of such a derivative in
which the metal is bonded to the C(sp2) of the three-membered
ring has been reported.26 A few structurally different transition
metal cyclopropenylidene complexes, mostly prepared from
dichlorocyclopropene27and a number ofπ-cyclopropene
com-plexes,28are also known. The acidity of the aliphatic protons
on a coordinated dppe ligand in a cationic iron vinylidene
complex29has been employed for inducing the intramolecular
cyclization between the dppe and vinylidene ligand.
Electrophilic Additions of Ruthenium Cyclopropenyl Complexes 3. Additions of CF3COOH to 3b-e regenerate 2b
-e, respectively, indicating the basic character of the methyne carbon of the three-membered ring. Furthermore, 3k was converted to 2k in MeOH indicating even stronger basicity. This protonation is different from the acid-induced demethoxylation
of the iron cyclopropenyl complex.26 Attempts to remove
hydrogen bonded to the three-membered ring using Ph3C+
yielded an unexpected product. Treatment of 3b with Ph3C+
affords{[Ru]dCdC(Ph)CH(CPh3)CN}+
(2f) with 64% yield. In this reaction, 2b is also isolated as a minor product (yield
<30%, probably due to contamination of HPF6in Ph3CPF6).
Although Ph3C+
is commonly used as a hydride abstraction
reagent,30as is evident from its reaction with several organic
cyclopropenyl compounds,31it however serves as an electrophile
in the reaction with 3b. There are a few examples in the
literature in which electrophilic addition of Ph3C+
resulted in
the formation of the C-C bond.
32
Further deprotonation of the methyne proton of 2f by
n-Bu4-NF also affords [Ru]-CdC(Ph)C(CPh3)CN (3f) (Scheme 2).
The yield is only 38% which may be attributed to the steric effect of the trityl cation. This same effect prevents protonation
of 3f to yield 2f. As expected, the31P NMR spectra of 2f and
3f both display two doublet resonances. Treatment of 3b with
HgCl2 also produces a vinylidene product {
[Ru]dCdC(Ph)-CH(HgCl)CN}+
(2g), with 81% yield. The formation of these
(24) (a) Davison, A.; Solar, J. P. J. Organomet. Chem. 1978, 155, C8. (b) Bell, R. A.; Chisholm, M. H. Inorg. Chem. 1977, 16, 687.
(25) (a) Lo¨we, C.; Shklover, V.; Bosch, H. W.; Berke, H. Chem. Ber. 1993, 126, 1769. (b) Gompper, R.; Bartmann, E. Angew. Chem., Int. Ed.
Engl. 1978, 17, 456. (c) DeSimone, D. M.; Desrosiers, P. J.; Hughes, R.
P. J. Am. Chem. Soc. 1982, 104, 4842. (d) Hughes, R. P.; Donaldson, W. A. J. Am. Chem. Soc. 1982, 104, 4846. (e) Weiss, R.; Priesner, C. Angew.
Chem., Int. Ed. Engl. 1978, 17, 457.
(26) Gompper, R.; Bartmann, E. Angew. Chem., Int. Ed. Engl. 1985,
24, 209.
(27) (a) Kirchgassner, U.; Piana, H.; Schubert, U. J. Am. Chem. Soc. 1991, 113, 2228. (b) Miki, S.; Ohno, T.; Iwasaki, H.; Yoshida, Z. I. J.
Phys. Org. Chem. 1988, 1, 333. (c) Yoshida, Z. I. Pure Appl. Chem. 1982, 54, 1059.
(28) (a) Schrock, R. R. Acc. Chem. Res. 1986, 19, 342. (b) Hughes, R. P.; Reisch, J. W.; Rheingold, A. L. Organometallics 1985, 4, 1754. (c) Hughes, R. P.; Kla¨ui, W.; Reisch, J. W.; Mu¨ller, A.; Rheingold, A. L.
Organometallics 1985, 4, 1761. (d) Mealli, C.; Midollini, S.; Moneti, S.;
Sacconi, L.; Silvestre, J.; Albright, T. A. J. Am. Chem. Soc. 1982, 104, 59. (29) Adams, R. D.; Davison, A.; Selegue, J. P. J. Am. Chem. Soc. 1979,
101, 7232.
(30) (a) Casey, C. P.; Marder, S. R. Organometallics 1985, 4, 411. (b) Deeming, A. J.; Ullah, S. S.; Domingos, A. J. P.; Johnson, B. F. G.; Lewis, J. J. Chem. Soc., Dalton Trans. 1974, 2093.
(31) Zimmerman, H. E.; Aasen, S. M. J. Org. Chem. 1978, 43, 1493. (32) (a) Lewis, J.; Parkins, A. W. J. Chem. Soc. A 1967, 1150. (b) Schrock, R. R.; Johnson, B. F. G.; Lewis, J. J. Chem. Soc., Dalton Trans. 1974, 951. (c) Harris, P. J.; Knox, S. A. R.; McKinney, R. J.; Stone, F. G. A. J. Chem. Soc., Dalton Trans. 1978, 1009.
Scheme 1 Scheme 2
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
vinylidene complexes occurs by selective cleavage of the cyclopropenyl single bond near the metal center. This selectivity is similar to what has been reported for the unsymmetrical cyclopropenes where the methyl-substituted single bond is
cleaved.33 Attempts to carry out cyclopropenation of 2g by
using n-Bu4NOH, n-Bu4NF, and DBU result in cleavage of the
C-Hg bond yielding 3b.
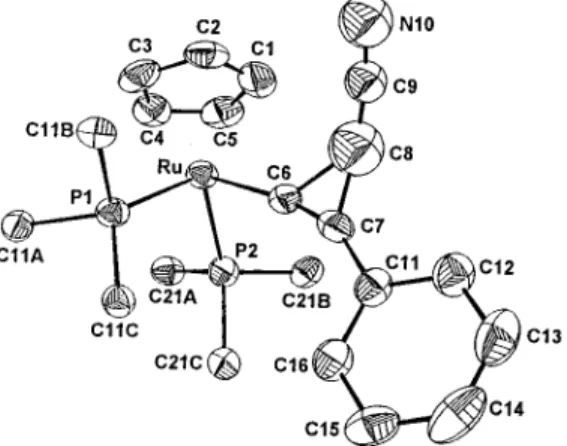
Structures of Two Ru Cyclopropenyl Complexes. The molecular structures of 3b and 3f have been determined by X-ray diffraction studies. The two optical isomers of 3b have been observed to crystallize together. An ORTEP drawing of
one isomer of 3b is shown in Figure 1. The Ru-C(6) bond
length of 2.034(5) Å is typical for a Ru-C single bond and the
C(6)-C(7) bond length of 1.289(8) Å is a double bond,
indicating the coordination of the sp2carbon of the
cyclopro-penyl ligand. The bond angles Ru-C(6)-C(7) and C(6)
-C(7)-C(11) of 169.7(4)°and 156.2(5)°, respectively, are both
far greater than that of an idealized C(sp2) hybridization. The
C(6)-C(8) and C(7)-C(8) bond lengths of 1.58(1) and
1.45-(1) Å, respectively, are significantly different, conforming with
the favorable cleavage of the C(6)-C(8) bond described above.
The phenyl group on the three-membered ring is approximately coplanar with the cyclopropene and lies far away from the Cp.
An ORTEP drawing of 3f is shown in Figure 2. The C(6)
-C(7) and -C(7)-C(9) bond lengths of 1.59(2) and 1.50(2) Å,
respectively, again differ significantly. The phenyl ring on Cβ
is no longer parallel to the three-membered ring, probably due
to the steric hindrance between the CPh3unit and the phenyl
group on the cyclopropenyl moiety. This also indicates that formation of the three-membered ring does not require the
presence of the phenyl group on Cβ.
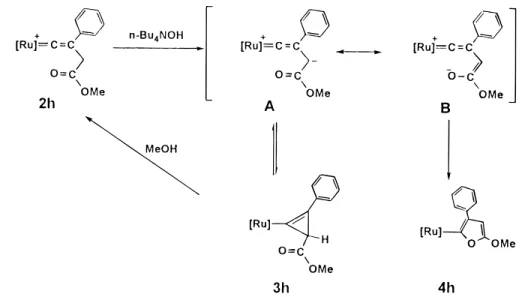
Another type of Cyclization Induced by Base.
Deproto-nation of [Ru]dCdC(Ph)CH2COOMe+, 2h, by n-Bu4NOH at
room temperature induces a different type of cyclization yielding
the neutral furan complex [Ru]-CdC(Ph)CHdC(OMe)O (4h)
(Scheme 3). Similar to cyclopropenation, this reaction also occurs only in acetone. 4h is additionally obtained if n-Bu4NF
or DBU is used. The most characteristic feature in the31P NMR
spectrum of 4h is a singlet resonance atδ 51.3 indicating lack
of an asymmetric center. Also noticeable in the 13C NMR
spectrum is the presence of a triplet resonance atδ 154.6 (JC-P
)19.0 Hz) assignable to CR. By monitoring the reaction using
31P NMR spectroscopy, [Ru]
-CdC(Ph)CHCOOMe (3h) was
also observed at the initial stage of the reaction which gets converted to 4h in acetone within 30 min at room temperature.
The reaction, if carried out at 5°C, yields 3h as a major product
and 1a as a minor product, without formation of 4h. Complex 3h in MeOH is susceptible to protonation whereas no reaction is observed between 4h and MeOH. However, protonation of 4h by acetic acid regenerates 2h quantitatively.
Owing to high strain energy of the cyclopropene ring, a more stable five-membered furan ring is expected to be the thermo-dynamic product. The fact that formation of 3h can be observed may imply that the deprotonation step yields a zwitterionic transition state with two resonance forms A (keto ester) and B (enol ester) (Scheme 3), which subsequently produce 3h and 4h, respectively. Lack of 4h in the products at 5°C can be interpreted in terms of the absence of enol form B at this temperature. The formation of 3h is favored by the proximity
of CRand Cγof the vinylidene ligand in 2h as well as lower
mobility of the ester group at low temperature.
The thermal or photochemical ring opening of substituted cyclopropenes affords vinylcarbene intermediates in a reversible manner. Numerous examples of trapping of these species have
been reported.34 Cyclizations of alkynol and epoxyalkyne
catalyzed by Mo complex have also been recently reported.35
In these reactions, vinylidene and epoxyvinylidene have been proposed as intermediates. The effect of substituents on the selectivity of vinylcarbene formation depends upon whether thermal or photochemical activation is used, which is
exam-(33) Padwa, A.; Blocklock, T. J.; Getman, D.; Hatanaka, N.; Loza, R. J.
Org. Chem. 1978, 43, 1481.
(34) (a) Davis, J. H.; Goddard, W. A., III; Bergman, R. G. J. Am. Chem.
Soc. 1976, 98, 4015. (b) Streeper, R. D.; Gardner, P. D. Tetrahedron Lett.
1973, 767. (c) York, E. J.; Dittmar, W.; Stevenson, J. R.; Bergman, R. G.
J. Am. Chem. Soc. 1972, 94, 2882; 1973, 95, 5680.
(35) McDonald, F. E.; Schultz, C. C. J. Am. Chem. Soc. 1994, 116, 9363. (36) (a) Pincock, J. A.; Moutsokapas, A. Can. J. Chem. 1977, 55, 979. (b) Komendantov, M. I.; Domnin, I. N.; Bulueheva, E. V. Tetrahedron 1975,
31, 2495.
(37) (a) Trost, B. M.; Flygare, J. A. J. Org. Chem. 1994, 59, 1078. (b) Katritzky, A. R.; Li, J.; Gordeev, M. F. J. Org. Chem. 1993, 58, 3038. (c) Tani, K.; Sato, Y.; Okamoto, S.; Sato, F. Tetrahedron Lett. 1993, 34, 4975. (d) Arcadi, A.; Cacchi, S.; Larock, R. C.; Marinelli, F. Tetrahedron Lett. 1993, 34, 2813. (e) Marshall, J. A.; DuBay, W. J. J. Am. Chem. Soc. 1992,
114, 1450. (f) Fukuda, Y.; Shiragami, H.; Utimoto, K.; Nozaki, H. J. Org. Chem. 1991, 56, 5816. (g) Takai, K.; Tezuka, M.; Kataoka, Y.; Utimoto,
K. J. Org. Chem. 1990, 55, 5310. Figure 1. An ORTEP drawing (50% thermal ellipsoid) of 3b with
some of the phenyl groups on the phosphine ligands and hydrogen atoms eliminated for clarity. Selected bond distances (Å) and angles follow (deg): Ru-C(6), 2.034(5); C(6)-C(7), 1.289(8); C(6)-C(8),
1.579(10); C(7)-C(8), 1.452(10); C(8)-C(9), 1.215(16); C(9)-N(10),
1.102(18); Ru-C(6)-C(7), 169.7(4); Ru-C(6)-C(8), 130.4(4); C(7)
-C(6)-C(8), 59.8(4); C(6)-C(7)-C(8), 70.1(5); C(6)-C(8)-C(7),
50.1-(4); C(8)-C(9)-N(10), 170.1(13).
Figure 2. An ORTEP drawing (33% thermal ellipsoid) of 3f with some
of the phenyl groups on the phosphine ligands and hydrogen atoms eliminated for clarity. Selected bond distances (Å) and angles follow (deg): Ru-C(6), 2.069(14); C(6)-C(7), 1.589(18); C(6)-C(9),
1.332-(20); C(7)-C(9), 1.503(19); C(7)-C(8), 1.584(19); C(7)-C(10),
1.522-(19); C(10)-N, 1.129(18); Ru-C(6)-C(9), 157.1(11); Ru-C(6)-C(7),
139.1(10); C(7)-C(6)-C(9), 61.2(9); C(6)-C(7)-C(9), 51.0(8); C(6)
-C(9)-C(7), 67.9(10); C(7)-C(10)-N, 175.7(14).
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
plified by the reactions of ester resulting in the production of
furans.36 Several methods have recently been developed for
furan synthesis.37 Other middle and late transition metal
complexes react with terminal alkynols to give cyclic
oxacar-benes.38
Structure of the Ru Furan Complex. The molecular structure of 4h has been determined by X-ray diffraction analysis. The crystal is found to contain two independent molecules, but with no essential structural difference between them. An ORTEP drawing of one molecule is shown in Figure
3. The Ru-C(1A) bond length of 2.076(7) Å indicates a Ru-C
single bond and the C(1A)-C(2A) and C(9A)-C(10A) bond
lengths of 1.370(9) and 1.33(1) Å, respectively, are typical CdC double bonds. As for the similar bonds in the three-membered
ring of 3b and 3f, the C(1A)-O(1A) bond length of 1.442(8)
Å near the Ru center in the five-membered ring is significantly
longer than the C(10A)-O(1A) bond length of 1.347(8) Å. This
is consistent with the result of protonation reaction in which
the bond cleavage occurs at the C-O bond near the Ru center.
Interestingly, the phenyl ring is near the Cp unit in the solid state.
Unstable Vinyl Complex via Fluoride Attack at the
r-Carbon. No deprotonation was observed in the reaction of
[Ru]dCdC(Ph)CH2OCH3+
(2j) with n-Bu4NOH or DBU in acetone. With a donor oxygen atom in 2j it is not unexpected that the above-mentioned methodology is not suitable for the preparation of the methoxy-substituted cyclopropenyl complex even though the iron cyclopropenyl complex with a methoxy
substituent has been reported previously.26 Upon adding
n-Bu4-NF to 2j, a different but more conventional reaction pattern is observed. Namely the reaction produces a yellow metal vinyl
complex [Ru]-C(F)dC(Ph)CH2OCH3(5). In this case about
80% conversion occurred in acetone at 10°C. Complex 5 is
soluble in CHCl3and THF. However, upon dissolution at room
temperature, complex 5 immediately converts back to 2j.
Therefore the spectroscopic data are obtained at-40°C. In
the13C NMR spectrum of 5, a doublet resonance (3J
C-F )21.8
Hz) atδ 70.8 (inverted in the DEPT-135 experiment) is assigned
to the methylene carbon. The coupling constant JP-F
)47 Hz
of the doublet resonance atδ 50.2 in the31P NMR spectrum is
consistent with that of the triplet resonance in the19F NMR
spectrum.
The importance of ionic fluorides as proton abstractors in
base-assisted reactions,39and also as a source of fluorine atoms
in the synthesis of orgnofluorine derivatives,40has been well
documented. It can thus be expected that there should be factors other than the basicity and nucleophilicity associated with the ionic fluoride that govern the reactions of 2b and/or 2j with n-Bu4NF. These factors associated with n-Bu4NF are not yet clear.
(38) (a) Quayle, P.; Rahman, S.; Ward, E. L. M.; Herbert, J. Tetrahedron
Lett. 1994, 35, 3801. (b) Hinkle, R. J.; Stang, P. J.; Arif, A. M.
Organometallics 1993, 12, 3510. (c) Stang, P. J.; Huang, Y. H. J.
Organomet. Chem. 1992, 431, 247. (d) Le Bozec, H.; Ouzzine, K.; Dixneuf,
P. H. Organometallics 1991, 10, 2768. (e) O’Connor, J. M.; Pu, L.; Rheingold, A. L. J. Am. Chem. Soc. 1990, 112, 6232. (f) Ditz, K. H.; Sturm, W.; Alt, H. G. Organometallics 1987, 6, 1424. (g) Parlier, A.; Rudler, H.
J. Chem. Soc., Chem. Commun. 1986, 514. (h) Curtis, P. J.; Davies, S. G. J. Chem. Soc., Chem. Commun. 1984, 747.
(39) (a) Clark, J. H. Chem. ReV. 1980, 80, 429. (b) Jakobson, G. G.;
Akmentova, N. E. Synthesis 1983, 169. (c) Clark, J. H. J. Chem. Soc.,
Chem. Commun. 1978, 789. (d) Landini, D.; Maia, A.; Rampoldi, A. J. Org. Chem. 1989, 54, 328.
(40) (a) Chi, D. Y.; Kilbourn, M. R.; Katzenellenbogen, J. A. J. Org.
Chem. 1987, 52, 658. (b) Haas, A.; Lieb, M. Chimia 1985, 39, 134. (c)
Cox, D. P.; Terpinsky, J.; Lawrynowicz, W. J. Org. Chem. 1984, 49, 3216. Scheme 3
Figure 3. An ORTEP drawing (50% thermal ellipsoid) of 4h with
some of the phenyl groups on the phosphine ligands and hydrogen atoms eliminated for clarity. Selected bond distances (Å) and angles follow (deg): Ru-C(1A), 2.076(7); C(1A)-C(2A), 1.370(9); C(1A)
-O(1A), 1.442(8); C(2A)-C(9A), 1.454(10); C(9A)-C(10A),
1.330-(10); C(10A)-O(1A), 1.347(8); C(10A)-O(2A), 1.358(9); O(2A)
-C(11A), 1.395(11); Ru-C(1A)-C(2A), 140.0(5); Ru-C(1A)-O(1A),
115.7(4); O(1A)-C(1A)-C(2A), 104.3(5); C(1A)-C(2A)-C(9A),
109.5(6); C(2A)-C(9A)-C(10A), 105.7(6); C(9A)-C(10A)-O(1A),
111.5(6); C(10A)-O(1A)-C(1A), 109.0(5).
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
Electrophilic Addition of TCNQ to Cyclopropenyl Com-plexes. By comparing the protonation reactions of our neutral cyclopropenyl complexes, which lead to formation of cationic vinylidene complexes, with the same type of reaction of a similar
complex reported in the literature,26it can be noted that the
complex consisting of a methoxy substituent, which leads to cyclopropenylium complex upon protonation, behaves very differently from those without such a group. It is thus clear
that the sp3 carbon center of the cyclopropenyl complexes 3
without an alkoxy group is an electron-rich center. Thus it would be impossible to use the simple nucleophilic substitution
reaction for direct addition of groups such as-CN or-OMe
to the three-membered ring. However, by using TCNQ
[(NC)2C(C6H4)C(CN)2], it becomes viable to first add
nucleo-philes to the cyclopropenyl CRand then transfer to the Cβcarbon
leading to formation of various MeO-substituted complexes. The following section describes the chemical reactivity of various complexes involving TCNQ.
Addition of TCNQ to 3b yielded the zwitterionic complex [Ru]dCdC(Ph)CH(CN)(TCNQ) (6b) (Scheme 5). One termi-nus of TCNQ probably acts as an electrophile, adding to the
methyne carbon and resulting in the formation of a C-C bond.
An alternative pathway would be a single electron transfer (SET)
process41followed by a subsequent fast C
-C bond formation
in the solvent cage. Complex 6b, a light orange colored solid, displays a characteristic dark violet-red color in solution, and its spectroscopic data display the feature of a vinylidene
complex. The pattern of two-doublet resonances atδ 40.6, 38.8
with JP-P
)26.6 Hz in the31P NMR spectrum arises from the
asymmetric Cγcenter. Localization of the negative charge at
the free terminus of TCNQ causes the Ru center to display the cationic feature which is evidenced by chemical shift in these
31P NMR resonances in the same region as that of other cationic
complexes. The structure of 6b has also been determined by X-ray diffraction analysis. An ORTEP drawing is shown in
Figure 4. The newly formed C(9)-C(10) bond is rather weak
as indicated by its extensively long bond length (1.60(1) Å). Addition of TCNQ to 4h also opens up the five-membered ring and produces the zwitterionic complex [Ru]dCdC(Ph)CH-(COOMe)(TCNQ) (6h) with 88% yield. Complex 6h has been
characterized by spectroscopic methods. The31P NMR
spec-trum of 6h exhibits two doublets atδ 40.0 and 38.7 which are
very close to that of 6b.
The reaction of TCNQ with 3d produces a different zwitte-rionic vinylidene complex [Ru]dCdC(Ph)CHdCHCH2(TCNQ), (7d) with TCNQ attached to the terminal carbon atom of the allylic unit (Scheme 5). This reaction has to be carried out at
-40°C because of the higher reactivity of 3d. The relatively
more electron-rich vinyl group, instead of the sp3carbon of the
three-membered ring, of 3d serves as a better nucleophilic
center. This causes a shift of the double bond to Cγ-Cδ.
Spectroscopic data clearly reveal the site of electrophilic
addition. The doublet resonance atδ 2.64, assignable to the
CH2group, in the1H NMR spectrum of 7d and the
correspond-ing inverted resonance atδ 46.4 in the13C NMR DEPT-135
clearly indicate an aliphatic CH2 unit in the molecule. A
terminal vinyl group would give an inverted13C resonance for
the dCH2 unit at a much lower field region. The coupling
constant JH-Hof 15.1 Hz between the olefinic protons indicates
a trans configuration at the double bond. In the 31P NMR
spectrum, only a singlet resonance atδ 41.2 was observed.
Cyclopropenyl Complexes with a Methoxy Substituent. Attempted deprotonation of 6b using n-Bu4NOH did not result in the formation of the expected cyclopropenyl complex (41) (a) Tanko, J. M.; Drumright, R. E.; Suleman, N. K.; Brammer, L.
E. J. Am. Chem. Soc. 1994, 116, 1785. (b) Tolbert, L. M.; Sun, X. J.; Ashby, E. C. J. Am. Chem. Soc. 1995, 117, 2681.
Scheme 4
Scheme 5
Figure 4. An ORTEP drawing (50% thermal ellipsoid) of 6b with
some of the phenyl groups on the phosphine ligands and hydrogen atoms eliminated for clarity. Selected bond distances (Å) and angles follows (deg): Ru-C(1), 1.811(10); C(1)-C(2), 1.328(14); C(2)-C(9), 1.542(15); C(9)-C(10), 1.602(14); C(10-C(11), 1.546(15); C(11) -C(12), 1.379(15); C(12)-C(13), 1.358(16); C(13)-C(14), 1.416(16); C(14)-C(15), 1.426(16); C(14)-C(16), 1.393(16); C(16)-C(17), 1.365(16); C(11)-C(17), 1.380(15); C(9)-C(18), 1.486(15); C(10) -C(19), 1.490(15); C(10)-C(20), 1.442(15); C(15)-C(21), 1.397(16); C(15)-C(22), 1.390(17); C(18)-N(1), 1.116(14); C(19)-N(2), 1.120-(14); C(20)-N(3), 1.111(15); C(21)-N(4), 1.143(15); C(22)-N(5), 1.147(16); Ru-C(1)-C(2), 173.7(8); C(1)-C(2)-C(9), 117.8(9); C(2)-C(9)-C(10), 114.6(8).
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
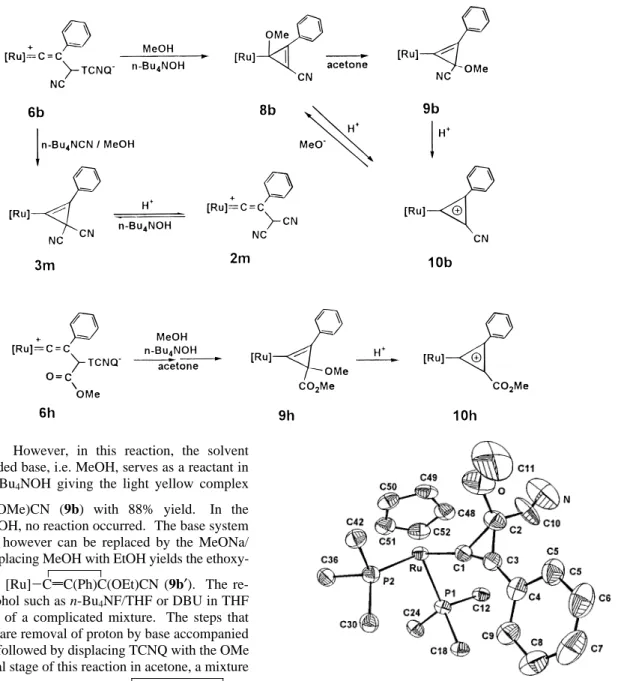
containing TCNQ. However, in this reaction, the solvent molecule of the added base, i.e. MeOH, serves as a reactant in the presence of n-Bu4NOH giving the light yellow complex
[Ru]-CdC(Ph)C(OMe)CN (9b) with 88% yield. In the
absence of n-Bu4NOH, no reaction occurred. The base system n-Bu4NOH/MeOH however can be replaced by the MeONa/ MeOH system. Replacing MeOH with EtOH yields the
ethoxy-substituted product [Ru]-CdC(Ph)C(OEt)CN (9b′). The
re-agents without alcohol such as n-Bu4NF/THF or DBU in THF result in formation of a complicated mixture. The steps that lead to the product are removal of proton by base accompanied by the cyclization, followed by displacing TCNQ with the OMe group. At the initial stage of this reaction in acetone, a mixture
of two isomeric products 9b and [Ru]-C(OMe)C(Ph)dCCN
(8b), i.e. the methoxy group at CR, is observed when the reaction
is monitored by the 31P NMR spectra (Scheme 6). Pure 8b
can, however, be obtained by a different method which is
described below. Complex 8b is stable in CDCl3or in THF,
but converts to 9b in acetone. In the31P NMR spectrum of 9b
the characteristic two doublet resonances atδ 51.7, 49.6 with
JP-P
)36.0 Hz are observed whereas C
Rappears as a triplet
resonance at δ 136.2 with JP-C
) 19.8 Hz in the
13C NMR
spectrum. For 9b′, in addition to the two-doublet31P
reso-nances, the 1H NMR spectrum displays resonances with two
multiplet patterns which may be assigned to the OCH2group
and arise due to the chiral center of the three-membered ring. The fact that base reagents without alcohol produce a complicated mixture probably indicates that the deprotonation is followed by various decomposition pathways. Furthermore, the fact that the reaction requires the presence of base leads us to believe that the deprotonation step may still be the first step
in the formation of 9b. Cleavage of the weak Cγ-C(TCNQ)
bond accompanying the attachment of the MeO group initially
to CRfollowed by a shift to Cβsatisfactorily accounts for the
formation of 9b. In the 31P NMR spectrum of 8b the
two-doublet (atδ 51.2 and 50.7 with JP-P
)29.6 Hz) pattern arises
due to the chiral center at the ring. The ethyl analogue 8b′is
kinetically more stable, i.e. at the initial stage of reaction only
8b′was observed. In order to firmly establish the location of
the methoxy group, the crystal structure of 9b has been determined. An ORTEP drawing of 9b is shown in Figure 5.
The phenyl group on Cβis again approximately coplanar with
the three-membered ring. Interestingly, a longer bond length
of C(1)-C(2) (1.541(4) Å) as compared to that of C(2)-C(3)
(1.447(5) Å) is also observed.
Protonation of 8b or 9b removes the methoxy group and
produces the cyclopropenylium complex, [Ru]-CC(Ph)CCN
+ (10b), with 78% yield (Scheme 6). The symmetrical planar structure of the three-membered ring of 10b is revealed by the
31P NMR spectrum, which shows only a singlet resonance atδ
Scheme 6
Figure 5. An ORTEP drawing (50% thermal ellipsoid) of 9b with
some of the phenyl groups on the phosphine ligands and hydrogen atoms eliminated for clarity. Selected bond distances (Å) and angles follow (deg): Ru-C(1), 2.036(3); C(1)-C(2), 1.541(4); C(1)-C(3), 1.319(4); C(2)-C(3), 1.447(5); C(2)-O, 1.474(4); C(2)-C(10), 1.429-(5); C(10)-N, 1.098(5); O-C(11); 1.178(6); Ru-C(1)-C(2), 132.1-(2); Ru-C(1)-C(3), 167.7(3); C(2)-C(1)-C(3), 60.2(2); C(1)-C(2) -C(3), 52.3(2); C(1)-C(3)-C(2), 67.5(2); C(2)-O-C(11), 130.9(5); C(2)-C(10)-N, 156.0(5).
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
46.8. In the13C NMR spectrum the resonance attributed to the
CRappears atδ 213.0 with JC-P
)17.2 Hz. This reactivity is
very different from opening of the three-membered ring of 3, yet similar to the reactivity of organic cyclopropene with a
methoxy substituent.42 Reaction of MeONa with 10b in THF
yields pure 8b which converts to 9b in acetone in about 40 min.
Similarly a suspension of complex 6h in acetone undergoes
methanolysis to yield [Ru]-CdC(Ph)C(CO2Me)(OMe) (9h),
another MeO-substituted cyclopropenyl complex with 60% yield (Scheme 6). The high solubility of 9h in acetone, however, hinders direct precipitation. The complex is hence purified by
hexane extraction. The31P NMR (two doublets atδ 53.6 and
48.0) and the1H NMR (two methyl resonances atδ 3.63 and
3.29) spectra of 9h are consistent with its formulation. Unlike 3h which converts to 4h, complex 9h stabilized by the methoxy group does not convert to a substituted furan. The
three-membered ring of 9h remains unchanged even at 45 °C in
acetone. The effect of the MeO group in stabilizing the
cyclopropenyl ring is consistent with what has been observed
in many analogous organic compounds.42 With the TCNQ
group present at a distant carbon atom, complex 7d is inert in n-Bu4NOH/MeOH. Protonation of 9h again removes the methoxy group giving the cationic cyclopropenylium complex
[Ru]-CC(Ph)C(CO2Me)
+
(10h). The31P NMR spectrum of
10h displays only a singlet atδ 47.6, characteristic of a cationic
cyclopropenylium complex, where only one methyl resonance atδ 3.80 is observed in the1H NMR spectrum.
However, the presence of a stronger nucleophile prohibits formation of the MeO-substituted complex 9. For example, the reaction of n-Bu4NCN in the presence of MeOH with 6b does not yield 9b but brings about addition of the CN group with
removal of TCNQ, giving [Ru]-CdC(Ph)C(CN)2, (3m).
Ac-companied by deprotonation, the stronger nucleophile CN
-displaces TCNQ to form the product. Further protonation of 3m, lacking the MeO substituent, produces the vinylidene
complex [Ru]dCdC(Ph)CH(CN)2+(2m) instead of the
cyclo-propenylium complex (Scheme 6). This result further reveals the unique influence of the methoxy group present in the three-membered ring which effectively controls the protonation reaction of the cyclopropenyl complexes.
Oxidative Coupling Reactions of Metal Cyclopropenyl Complexes. On the basis of successful addition of the trityl
group to 3b, we attempted to induce a C-C bond formation in
3b by using organic halides and found the formation of a new coupling product in the presence of allyl iodide. Treatment of 3b with a 20-fold excess of allyl iodide affords the dimeric
dicationic vinylidene complex{[Ru]dCdC(Ph)CHCN}22+(11)
with 49% yield (Scheme 7). The yield of 11 depends on the amount of allyl iodide used. If only 1 equiv of allyl iodide is used, this reaction slowly produces 2b as a major product and only a trace amount of 11. Using other organic iodides such as methyl iodide, ethyl iodide, and iodobenzene produces no coupling product. The reactions of 3c or 3d with allyl iodide also do not produce the coupling product.
Complex 11 is insoluble in most of the organic solvents and only sparingly soluble in DMSO wherein it forms an orange solution. The mass spectrum of 11 is consistent with the
formulation {[Ru]dCdC(Ph)CHCN}2I+
. In the 31P NMR
spectrum of 11, the chemical shift of the resonances atδ 41.3
and 42.3 is close to that observed for 2. The molecular structure of 11 has also been determined by X-ray diffraction analysis.
Interestingly, the counterions in the solid state are two I3
-anions. A view of one molecule of 11 is shown in Figure 6.
The center of the central C-C bond lies on a center of
symmetry, thus half of the molecule is symmetry-generated from
the other. The Ru-C(6) bond length of 1.83(2)Å is consistent
with the RudC double bond formulation and the Ru-C(6)
-C(7) bond angle of 174(2)°is similar to that in related vinylidene
complexes.
The formation of 11 probably involves the cationic ruthenium
vinylidene radical43induced from the reaction of 3b with C3H5I.
The coupling of the allyl radical resulting in the formation of
the bicyclopropyl molecule44 and radical annulations of allyl
iodomalononitriles45 have been reported in the literature.
Oxidative carbon-carbon coupling of the cationic iron
vi-nylidene complex [Cp(dppe)FedCdCHMe]+
leading to
forma-tion of [Cp(dppe)FedCdCMe]22+
has been reported,46whereas
(42) (a) Breslow, R.; Chang, H. W. J. Am. Chem. Soc. 1961, 83, 2367. (b) Krebs, A. W. Angew. Chem., Int. Ed. Engl. 1965, 4, 10. (c) Closs, G. L.; Boll, W. A.; Heyn, H.; Dev, V. J. Am. Chem. Soc. 1968, 90, 173.
(43) (a) Rabier, A.; Lugan, N.; Mathieu, R.; Geoffroy, G. L.
Organo-metallics 1994, 13, 4676. (b) Antinolo, A.; Otero, A.; Fajardo, M.;
Garcia-Yebra, C.; Gil-Sanz, R.; Lopez-Mardomingo, C.; Martin, A.; Gomez-Sal, P. Organometallics 1994, 13, 4679.
(44) Holtzhauer, K.; Cometta-Morini, C.; Oth, J. F. M. J. Phys. Org.
Chem. 1990, 3, 219.
(45) Curran, D. P.; Seong, C. M. Tetrahedron 1992, 48, 2175. (46) Lyer, R. S.; Selegue, J. P. J. Am. Chem. Soc. 1987, 109, 910. (47) Le Narvor, N.; Toupet, L.; Lapinte, C. J. Am. Chem. Soc. 1995,
117, 7129.
(48) Connelly, N. G.; Gamasa, M. P.; Gimeno, J.; Lapinte, C.; Lastra, E.; Maher, J. P.; Le Narvor, N.; Rieger, A. L.; Rieger, P. H. J. Chem. Soc.,
Dalton Trans. 1993, 2575.
Scheme 7
Figure 6. An ORTEP drawing (33% thermal ellipsoid) of 11 with
some of the phenyl groups on the phosphine ligands and hydrogen atoms eliminated for clarity. Selected bond distances (Å) and angles follow (deg): Ru-C(6), 1.826(20); C(6)-C(7), 1.34(3); C(7)-C(8),
1.52(3); C(8)-C(8a), 1.49(3); C(8)-C(9), 1.56(3); C(9)-N(10),
1.12-(3); Ru-C(6)-C(7), 174.4(16); C(6)-C(7)-C(8), 120.6(17); C(7)
-C(8)-C(8a), 114.7(16); C(8)-C(9)-N(10), 178.1(20).
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
another very similar coupling47 has been attributed to the
presence of 17-electron species, confirmed by ESR.48
Unsub-stituted vinylidene complex Cp(PPh3)2RudCdCH2also
under-goes oxidative coupling by MeI and produces a similar dimer.49
There are also few examples of metal acetylide couplings.50The
possible role of azavinylidene51in the conversion of nitriles to
diimido-bridged dimer in tantalum and niobium complexes52
has been recently addressed. These examples are, nevertheless, different from what is observed in 3b, namely in our system
the oxidative coupling at Cγresults in formation of a C6bridge
between the Ru metal centers. Complex 11 undergoes two deprotonation/cyclopropenation in the presence of excess
n-Bu4-NOH to give the neutral 2,2′-bicyclopropenyl complex{[Ru]
-CdC(Ph)CCN}2(12). Complex 12 displays the typical light
yellow orange color of the cyclopropenyl complexes. Product analyses included elementary analysis, mass spectroscopy, and
1H NMR spectroscopy which shows characteristic Cp absorption
at δ 4.87. Similar to 11, complex 12 also displays the
characteristic AB pattern in its 31P NMR spectrum. The
unsubstituted 2,2′-bicyclopropene has been prepared53and its
structure has been determined by X-ray diffraction analysis at
103 K.54
Conclusion. The facile preparation of neutral Ru cyclopro-penyl complexes has been achieved by deprotonation of a CH
or CH2 unit at Cγ of the cationic vinylidene complexes in
acetone. Successful accomplishment of the preparation of complexes with various substituents such as CN, Ph, and vinyl
groups at CH or CH2 renders this preparation a potentially
versatile synthetic method. The deprotonation of vinylidene complexes consisting of an ester group yields the five-membered furan moiety as thermodynamic products. Protonation of both of the cyclization products, yielding back the vinylidene
complexes, shows the nucleophilic nature of the antecedent Cγ
carbon of the vinylidene ligand. Thus other electrophiles could
also be added to this same Cγ site by reaction with
cyclopro-penyl or furan complex. However, when TCNQ was employed for this purpose, the addition could be modified leading eventually to the formation of cyclopropenyl complex with a methoxy substituent, which displays higher stability of the three-membered ring and shows particular reactivity. Thus in the present system, use of TCNQ appears to serve as an entry to the cyclopropenylium complex. A cyclopropenyl complex with a methoxy group behaves differently from that without such a unit.
Experimental Section
General Procedures. All manipulations were performed under
nitrogen using vacuum-line, dry box, and standard Schlenk techniques. CH3CN and CH2Cl2were distilled from CaH2and diethyl ether and THF from Na/ketyl. All other solvents and reagents were of reagent grade and were used without further purification. NMR spectra were recorded on Bruker AC-200 and AM-300WB FT-NMR spectrometers at room temperature (unless states otherwise) and are reported in units
ofδ with residual protons in the solvent as an internal standard (CDCl3, δ 7.24; CD3CN,δ 1.93; C2D6CO,δ 2.04). FAB mass spectra were recorded on a JEOL SX-102A spectrometer. Complexes 1a, [Ru]
-CtC-C6H9,
551k, and [Ru]dCdC(Ph)CH
2R+(2c, R
)Ph; 2d, R)
CHdCH2)56 were prepared following the methods reported in the literature. Elemental analyses and X-ray diffraction studies were carried out at the Regional Center of Analytical Instrument located at the National Taiwan University.
Synthesis of [[Ru]dCdC(Ph)CH2CN][PF6] (2b). A Schlenk flask was charged with complex 1a (0.475 g, 0.60 mmol) and NH4PF6(0.123 g, 0.75 mmol) and CH2Cl2(20 mL) were added after the atmosphere was replaced with nitrogen. The resulting solution was stirred at room temperature and ICH2CN (0.1 mL, 1.5 mmol) was added. The clear solution was stirred for 18 h, then the solvent was reduced to about 5 mL. This mixture was slowly added to 60 mL ofVigorously stirred
diethyl ether. The pale red precipitate thus formed was filtered off and washed with diethyl ether and hexane. The product was recrystal-lized from CH2Cl2/hexane (1:5) and identified as 2b (0.42 g, 0.43 mmol, 72%). Spectroscopic data of 2b: 1H NMR CD 3COCD3: 8.16-7.03 (Ph); 5.61 (s, 5H, Cp); 3.56 (s, 2H, CH2). 13C NMR CD3COCD3: 345.6 (t, JP-C )17.9 Hz, C R); 134.8 -128.4 (Ph); 123.0 (Cβ); 118.5 (CN); 95.6 (Cp); 14.5 (CH2). 31P NMR CD3COCD3: 42.4 (s). MS FAB m/z: 834 (M+, Ru )104), 572 (M + -PPh3), 431 (M + -PPh3, C2
-PhCH2CN). Anal. Calcd for C51H42NP3F6Ru: C, 62.70; H, 4.33; N, 1.43. Found: C, 62.90; H, 4.15; N, 1.96.
Complex [[Ru]dCdC(Ph)CH2CHdCMe2][PF6] (2e) (0.84 g, 0.81 mmol, 77% yield from 0.85 g of 1a) was similarly prepared from BrCH2CHdCMe2. Spectroscopic data of 2e: 1H NMR CDCl3: 7.38
-6.85 (m, 35H, Ph); 5.04 (s, 5H, Cp); 4.92 (m, 1H, dCH); 2.90 (d, JH-H ) 6.5 Hz, 2H, CH2); 1.58, 1.11 (s, 6H, 2 CH3). 13C NMR CDCl3: 348.9 (t, JP-C )15.8 Hz, C R); 134.7 -124.7 (Ph); 119.8 (Cβ); 94.1 (Cp); 25.8, 25.6 (2 CH3); 17.6 (CH2). 31P NMR CDCl3: 42.7 (s). MS FAB m/z: 863 (M+), 601 (M+ -PPh3), 431 (M + -PPh3, C2
-PhCH2CHCMe2). Anal. Calcd for C54H49P3F6Ru: C, 64.47; H, 4.91. Found: C, 64.80; H, 4.65.
Complex [[Ru]dCdC(Ph)CH2COOMe][PF6] (2h) was prepared using the following method. A mixture of complex 1a (1.15 g, 1.45 mmol) and BrCH2COOMe (0.5 mL, 5.1 mmol) in 40 mL of CH2Cl2/ CHCl3(3:1) was heated to refulx for 8 h, then NH4PF6(0.25 g, 1.53 mmol) was added and the mixture was stirred at room temperature for 4 h. The workup procedure was the same as that for 2b. Purification by recrystallization from CH2Cl2/hexane (1:5) gave 2h (0.91 g, 0.90 mmol, 62% yield). Spectroscopic data of 2h: 1H NMR CD
3COCD3: 7.50-7.06 (m, 35H, Ph); 5.52 (s, 5H, Cp); 3.65 (s, 3H, CH3); 3.10 (s, 2H, CH2). 13C NMR CDCl3: 347.8 (JP-C )14.6 Hz, C R); 171.7 (s, CO2); 134.4-128.3 (Ph); 125.1 (Cβ); 90.7 (Cp); 52.2 (CH3); 32.1 (CH2). 31P NMR CDCl 3: 42.0 (s). MS FAB m/z: 867 (M+ ), 721 (M+ -C2 -PhCH2COOMe+CO), 693 (M + -C2PhCH2COOMe), 431 (M +
-PPh3, C2PhCH2COOMe). Anal. Calcd for C52H45O2P3F6Ru: C, 61.84; H, 4.49. Found: C, 62.23; H, 4.71.
Complex [[Ru]dCdC(Ph)CH2COOEt][PF6] (2i) was prepared in 68% isolated yield using the same procedure as that for 2h. Spectro-scopic data of 2i: 1H NMR CDCl
3: 7.40-6.88 (m, 35H, Ph); 5.22 (s, 5H, Cp); 4.08 (q, JH-H )7.13 Hz, 2H, OCH2); 3.00 (s, 2H, CH2); 1.15 (t, JH-H )7.13 Hz, 3H, CH3). 13C NMR CDCl 3: 347.8 (JP-C )15.1 Hz, CR); 171.2 (s, CO2); 134.3 -128.3 (Ph); 125.1 (Cβ); 94.8 (Cp); 61.2 (CH2CO2); 32.3 (OCH2); 14.1 (CH3). 31P NMR CDCl3: 42.1 (s). MS FAB m/z: 882 (M+ ), 619 (M+ -PPh3), 431 (M + -PPh3, C2PhCH2
-COOEt). Anal. Calcd for C53H47O2P3F6Ru: C, 62.17; H, 4.63. Found: C, 62.62; H, 4.50.
Synthesis of [[Ru]dCdC(Ph)CH2OCH3][PF6] (2j). The synthetic procedure was similar to that used for the preparation of 2b: A solution of 1a (0.923 g, 1.16 mmol) in 20 mL of CH2Cl2, ICH2OCH3(0.15 mL, 1.17 mmol) (Caution: Free ICH2OCH3is a potential carcinogen), and NH4PF6 (0.27 g, 1.66 mmol) were used. The reaction was completed immediately upon mixing of the reactants. The product was recrystallized from CH2Cl2/hexane (1:5) and identified as 2j (0.87g, 0.88 mmol, 76%). Spectroscopic data of 2j: 1H NMR CD
3CN: 7.93
-6.94 (Ph); 5.32 (s, 5H, Cp); 3.95 (s, 2H, CH2); 3.09 (s, 3H, CH3).13C
(49) Bruce, M. I.; Koutsantonis, G. A. Aust. J. Chem. 1991, 44, 207. (50) (a) Shih, K.-Y.; Schrock, R. R.; Kempe, R. J. Am. Chem. Soc. 1994,
116, 8804. (b) Evans, W. T.; Keyer, R. A.; Ziller, J. W. Organometallics
1993, 12, 2618. (c) Shutowski, D. G.; Stucky, G. D. J. Am. Chem. Soc. 1976, 98, 1376.
(51) Feng, S. G.; White, P. S.; Templeton, J. L. J. Am. Chem. Soc. 1994,
116, 8613.
(52) (a) Finn, P. A.; King, M. S.; Kilty, P. A.; McCarley, R. E. J. Am.
Chem. Soc. 1975, 97, 220. (b) Cotton, F. A.; Hall, T. W. Inorg. Chem.
1978, 17, 3525. (c) Roskamp, E. J.; Pedersen, S. F. J. Am. Chem. Soc. 1987, 109, 3152.
(53) Billups, W. E.; Haley, M. M. Angew. Chem., Int. Ed. Engl. 1989,
28, 1711.
(54) Bordalla, D.; Mootz, D.; Boese, R.; Osswald, W. J. Appl.
Crystal-logr. 1985, 18, 316.
(55) Bruce, M. I.; Hinterding, P.; Tiekink, E. R. T.; Skeleton, B. W.; White, A. H. J. Organomet. Chem. 1993, 450, 209.
(56) Bruce, M. I.; Humphrey, M. G. Aust. J. Chem. 1989, 42, 1067.
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
NMR CD3CN: 348.5 (t, JC-P )16.1 Hz, C R); 135.9 -128.8 (Ph); 118.2 (Cβ); 95.7 (Cp); 67.5 (CH3); 57.8 (CH2). 31P NMR CD3CN: 42.5 (s). MS FAB m/z: 839 (M+), 577 (M+ -PPh3), 431 (M + -PPh3, C2
-PhCH2OMe). Anal. Calcd for C51H45OP3F6Ru: C, 62.38; H, 4.62. Found: C, 62.11; H, 4.98.
Complex [[Ru]dCdC(C6H9)CH2CN]I (2k) (0.80 g, 0.82 mmol, 75% yield) was prepared from 1k (0.87 g, 1.09 mmol) and ICH2CN using the same procedure as for 2b. Spectroscopic data of 2k: 1H NMR CDCl3: 7.44-6.96 (m, 30H, Ph); 5.77 (br, 1H, dCH); 5.18 (s, 5H, Cp); 2.85 (s, 2H, CH2CN); 2.15, 1.84, 1.62, 1.53 (br, 8H, 4 CH2).13C NMR CDCl3: 349.0 (t, br, CR); 134.4 -128.8 (Ph); 125.0 (CH of C6H9); 124.2 (Cβ); 118.5 (CN); 95.0 (Cp); 30.9, 28.0, 22.8, 21.6 (4 CH2); 12.7 (CH2CN). 31P NMR CDCl3: 41.1 (s). MS FAB m/z: 836 (M+ ), 574 (M+ -PPh3), 431 (M + -C2(C6H9)CH2CN).
Synthesis of [Ru]CdC(Ph)C(CN)H (3b). To a solution of 2b (0.40
g, 0.41 mmol) in 15 mL of acetone was added a solution of n-Bu4 -NOH (4.5 mL, 1 M in MeOH). The mixture was stirred overnight yielding the light yellow microcrystalline precipitate which was filtered off and washed with 2× 5 mL of acetone, 2 × 10 mL of diethyl ether, and 10 mL of n-hexane, then dried under vacuum. The product was analytically pure and was identified as 3b (0.27 g, 0.33 mmol, 80%). When 2b was treated with n-Bu4NF (1 M in THF) or DBU, instead of
n-Bu4NOH, the same product was obtained. Single crystals suitable for X-ray diffraction analysis were grown from the same reaction mixture with lower concentration. Spectroscopic data of 3b: 1H NMR CDCl3: 7.20-6.61 (m, 35H, Ph); 4.54 (s, 5H, Cp); 1.40 (s, 1H, CH). 13C NMR CDCl 3: 134.8-128.4 (Ph); 126.2 (t, JC-P)23.0 Hz, CR); 113.8 (CN); 86.3 (Cp); 7.96 (CH). 31P NMR CDCl 3: 51.7, 49.6 (AB, JP-P )34.6 Hz). MS FAB m/z: 834 (M + +1), 572 (M + -PPh3), 430 (M+
-PPh3, C2PhCHCN). Anal. Calcd for C51H41NP2Ru: C,
73.72; H, 4.97; N, 1.69. Found: C, 73.63; H, 4.55; N, 1.42.
Synthesis of [Ru]-CdC(Ph)C(Ph)H (3c). Complex 3c (0.14 g,
0.16 mmol, 55% yield) was similarly prepared from 2c (0.30 g, 0.29 mmol) and n-Bu4NOH (3.0 mL) in 15 mL of acetone. Spectroscopic data for 3c: 1H NMR CDCl 3: 8.19-6.61 (m, 40H, Ph), 4.22 (s, 5H, Cp), 2.54 (s, 1H, CH). 13C NMR CDCl 3: 140.8-119.3 (Ph), 137.7 (t, JC-P )19.7 Hz, C R), 85.2 (Cp), 32.9 (CH). 31P NMR CDCl 3: 54.7, 47.8 (d, JP-P )34.9 Hz, 2 PPh3). MS FAB m/z: 885 (M + ), 721 (M+ +CO-C15H11), 693 (M +
-C15H11). Anal. Calcd for C56H46P2Ru:
C, 76.26; H, 5.26. Found: C, 76.56; H, 4.98.
Synthesis of [Ru]-CdC(Ph)C(C2H3)H (3d). Complex 3d was
similarly prepared from 2d (0.44 g, 0.45 mmol) and 5.0 mL of n-Bu4 -NOH in 10 mL of acetone. The product was obtained in 53% yield (0.20 g, 0.24 mmol). Spectroscopic data for 3d: 1H NMR CDCl
3: 7.45-6.63 (m, 35H, Ph), 5.84 (ddd, JH -H )17.0, 10.1, 9.2 Hz, 1H, dCH), 5.24 (dd, JH-H )17.0, 2.5 Hz, 1H of dCH2), 4.78, (dd, JH -H )10.1, 2.5 Hz, 1H of dCH2), 4.49 (s, 5H, Cp), 2.02 (d, 1H, JH -H ) 9.2 Hz, CH). 13C NMR CDCl 3: 153.8 (dCH), 138.4 (t, JC-P )19.3 Hz, CR), 135.5 -123.7 (Ph), 105.9 (dCH2), 85.7 (Cp), 32.8 (CH). 31P NMR CDCl3: 53.2, 49.9 (AB, JP-P )35.5 Hz, 2 PPh3). MS FAB m/z: 835 (M+ +1), 795 (M + +1-C3H4), 721 (M + +CO-C11H9), 693 (M+ -C11H9), 431 (M +
-C11H9,PPh3). Anal. Calcd for C52H44P2
-Ru: C, 75.07; H, 5.33. Found: C, 75.01; H, 5.22.
Synthesis of [Ru]-CdC(Ph)C(CHdCMe2)H (3e). Complex 3e
in 48% yield (0.17 g, 0.20 mmol) was similarly prepared from 2e (0.43 g, 0.41 mmol) in 10 mL of acetone and n-Bu4NOH (4.5 mL). Spectroscopic data for 3e: 1H NMR CDCl
3: 7.61-6.62 (m, 35H, Ph), 4.94 (d, JH-H )9.4 Hz, 1H, CH), 4.39 (s, 5H, Cp), 1.94 (d, 1H, JH -H )9.4 Hz, dCH); 1.89, 1.70 (s, 2 CH3). 31P NMR CDCl 3: 53.0, 50.2 (d, JP-P )35.7 Hz, 2 PPh3). MS FAB m/z: 863 (M + +1), 601 (M + +1-PPh3), 431 (M +
-C13H13,PPh3). Anal. Calcd for C54H48P2Ru:
C, 75.42; H, 5.63. Found: C, 75.23; H, 5.87. Protonation of 3b by CF3COOH in CDCl3was carried out in a NMR tube and the reaction cleanly yielded 2b. The yield is>95% based on the integration of the
Cp resonances relative to an internal standard. Similarly protonation of 3c, 3d, and 3e gave 2c, 2d, and 2e, respectively, all with>95%
NMR yields.
Synthesis of [Ru]CdC(C6H9)C(CN)H (3k). The cyclopropenyl complex with a cyclohexenyl group on the Cβwas soluble in acetone
thus a slightly modified procedure is used. To a solution of 2k (0.45 g, 0.47 mmol) in 15 mL of acetone was added a solution of n-Bu4 -NOH (2.0 mL). The solution was stirred for 3 h. Then the workup procedure was the same as that for 3b. This product was identified as
3k (0.30 g, 0.36 mmol, 77% yield) which gave 2k quantitatively in
the presence of MeOH. Replacing n-Bu4NOH by n-Bu4NF or DBU gave the same product with slightly lower yield. Spectroscopic data for 3k: 1H NMR CDCl 3: 7.44-6.97 (m, 30H, Ph), 5.41 (t, br, 1H, dCH), 4.53 (s, 5H, Cp); 2.01, 1.63, 1.43, 1.35 (br, 4 CH2); 1.08 (s, 1H, CHCN). 13C NMR CDCl 3: 140.0-127.0 (Ph), 126.2 (dCH in C6H9), 116.2 (CN), 86.0 (Cp), 26.9, 25.6, 22.8, 22.3 (CH2in C6H9), 7.7 (CHCN). 31P NMR CDCl 3: 51.7, 49.0 (AB, JP-P )36.4 Hz, 2 PPh3). MS FAB m/z: 838 (M+ + 1), 693 (M + - cyclopropenyl moiety), 576 (M+ +1 -PPh3), 431 (M + -cyclopropenyl moiety,
PPh3). Anal. Calcd for C54H48P2Ru: C, 75.42; H, 5.63. Found: C, 75.23; H, 5.87.
Synthesis of [[Ru]dCdC(Ph)CH(CN)CPh3][PF6] (2f). To a solid mixture of 3b (0.76 g, 0.91 mmol) and Ph3CPF6(0.36 g, 0.93 mmol) at 0°C was added by syringe 25 mL of CH2Cl2. The mixture was stirred for 40 min, and then the solvent was removed under vacuum. The residue which contained 2f and 2b was washed with 3× 20 mL of benzene to remove 2b then with 2× 10 mL of diethyl ether and dried to give 2f (0.71 g, 0.58 mmol, 64%). The solvent of a portion of 2b was removed and the residue was redissolved in CH2Cl2and poured into a stirred diethyl ether to give 2b (0.20 g, 0.21 mmol, 28% yield). Spectroscopic data of 2f: 1H NMR CD
3CN: 7.49-6.58 (Ph); 5.29 (s, 5H, Cp); 5.03 (s, 1H, CH). 13C NMR CD 3CN: 340.3 (t, JC-P )16.5 Hz, CR); 135.9-128.8 (Ph); 125.3 (Cβ); 122.6 (CN); 96.2 (Cp); 60.1 (CPh3); 36.0 (CH). 31P NMR CD3CN: 41.3, 38.6 (d, JP-P)26.5 Hz). MS FAB m/z: 1076 (M+), 834 (M+ - CPh3), 571 (M +
-CPh3,PPh3). Anal. Calcd for C70H56NP3F6Ru: C, 68.96; H, 4.63; N, 1.15. Found: C, 68.70; H, 5.03; N, 1.09.
Synthesis of [[Ru]dCdC(Ph)CH(CN)HgCl]Cl (2g). To a mixture
of 3b (0.47 g, 0.56 mmol) and HgCl2(0.19 g, 0.70 mmol) at 0°C was added by syringe 25 mL of CH2Cl2. The mixture was stirred for 40 min. The workup procedure was the same as that in 2f and no 2b was observed. The product identified as 2g was obtained (0.55 g, 0.45 mmol, 81%). Spectroscopic data for 2g: 1H NMR CDCl
3: 7.45 -6.76 (m, 35H, Ph), 5.32 (s, 5H, Cp), 3.62 (s, 1H, CH). 13C NMR CDCl3: 344.3 (t, JC-P )13.1 Hz, C R), 134.5 -127.1 (Ph), 125.2 (Cβ), 120.9 (CN), 95.2 (Cp), 26.2 (CH). 31P NMR CD 3CN: 42.4, 40.3 (AB, JP-P )26.4 Hz, 2 PPh3). MS FAB m/z: 1070 (M +), 833 (M+ -HgCl), 693 (M+ -HgCl, C2PhCHCN), 571 (M + -HgCl, PPh3).
Reaction of 2h with Bu4NOH. To a suspension of complex 2h (0.94 g, 0.93 mmol) in 15 mL of acetone at room temperature was added a 2.5-mL solution of n-Bu4OH. The solution gave orange precipitate after being stirred overnight. The precipitate was filtered and washed with 10 mL of MeOH, 2× 5 mL of acetone, and 10 mL of hexane and then dried under vacuum. Recrystallization from a mixture of C6H12/CHCl3(1:1) yielded [Ru]-CdC(Ph)CHdC(O)OCH3
(4h) (0.64 g, 0.74 mmol, 80% yield). Spectroscopic data for 4h: 1H NMR CDCl3: 7.32-6.97 (m, 35H, Ph); 4.92 (s, 1H, CH); 4.05 (s, 5H, Cp); 3.04 (s, 3H, CH3). 13C NMR CDCl3: 164.0 (CO2); 154.6 (t, JC-P )19.0 Hz, C R); 140.5 -125.3 (Ph); 86.6 (Cγ); 83.9 (Cp); 58.0 (CH3). 31P NMR CDCl 3: 51.3 (s). MS FAB m/z: 867 (M+ +1), 721 (M +
-C2PhCH(CO2Me)+CO), 693 (M
+
-C2PhCH(CO2Me)), 431 (M
+
-C2PhCH(CO2Me),PPh3). Anal. Calcd for C52H44O2P2Ru: C, 72.29; H, 5.13. Found: C, 74.49; H, 5.75 (the deviation might be due to the solvent trapped in the solid during recrystallization).
When the same reaction was carried out at 5 °C, [Ru]-
CdC-(Ph)CHdCOOCH3(3h) and 1a with a ratio of 2:1 were isolated in 75% total yield. At this temperature, 4h was not observed. No attempt was made to separate 3h and 1a. Spectroscopic data of 3h: 1H NMR CDCl3: 7.50-6.54 (m, 35H, Ph); 4.40 (s, 5H, Cp); 3.72 (s, 3H, CH3);
2.12 (s, 1H, CH). 31P NMR CDCl
3: 52.7, 48.0 (AB, JP-P
)35.5 Hz).
Complex 3h was completely converted to 4h in CDCl3 at room temperature for 4 h.
Complex [Ru]-CdC(Ph)CHdC(O)OEt (4i) (0.301 g, 0.340 mmol)
was similarly prepared from 2i (0.450 g, 0.440 mmol, 78% yield) and
n-Bu4NOH. Spectroscopic data for 4i: 1H NMR CDCl3: 7.34-6.91
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009