Quantum Mechanical Study on the Facial Selectivity of Dioxa and
Trioxa Cage Molecules
Ito Chao,*,†J. H. Shih,†and Hsien-Jen Wu*,‡
Institute of Chemistry, Academia Sinica, Nankang, Taipei, Taiwan, and Department of Applied Chemistry, Chiao Tung University, Hsinchu, Taiwan
ichao@chem.sinica.edu.tw Received June 7, 2000
High facial selectivity (>99%) of nucleophilic addition to the carbonyl groups of the title compounds (1 and 2) has been achieved for the novel trioxa cage 2, but not for the dioxa 1. Similar experimental observations were made for the carbene addition to the double bonds of cage compounds, 3 and 4. Calculations were carried out for the cage compounds and their reaction transition structures, with LiH as a nucleophile and :CCl2 as an attacking carbene. The calculated facial preference for nucleophilic and carbene addition agreed well with experimental results. The origins of facial selectivity are examined from the viewpoints of structure, frontier orbitals, and molecular electrostatic potential of the reactants, as well as strain, electrostatic, and hyperconjugation effects in the transition state. For dioxa cages, the structural facial difference around the reaction center is minor, but the electronic difference of syn and anti faces generated by the two remote oxygen atoms is clearly demonstrated via frontier orbital and MEP analyses. For trioxa cages, the close proximity of the third ether oxygen (Os) to the reaction center brings large structural and electronic changes around the reaction center. The calculated electrostatic and strain energy differences of syn and anti transition structures are significantly larger for trioxa cages than for the dioxa cages. Therefore, they both contribute to the enhanced facial selectivity of trioxa compounds. Finally, analysis of hyperconjugative stabilization in transition structures reveals the danger of relying solely on Cieplak or Anh models in rationalization of facial selectivity, especially when nonequivalent steric and electrostatic effects as those present in the trioxa systems are involved.
Introduction
Since the first rule proposed by Cram1 in 1952 to explain the diastereofacial selectivity in the addition of nucleophiles to R-chiral carbonyl compounds, organic chemists have not ceased to construct models that provide rationalizations and predictions for preferential addition to one π-face over the other of a trigonal carbon center. In Cram’s rule, the largest group attached to the R-carbon should be anti to the carbonyl group and the nucleophile approaches the carbonyl group from the side with less steric congestion. Similar to Cram’s rule, the Felkin-Anh model2,3also considers the conformation of substituents of the R-carbon and the steric effect encoun-tered by an incoming nucleophile. However, in this model the largest group is perpendicular to the plane of the carbonyl group and is antiperiplanar to the nucleophile. In addition, Anh and Eisenstein stressed the importance of electronic effect in the transition state. They suggested a heteroatom such as Cl should be placed antiperiplanar to the nucleophile since the low lying σC-X* could lower the πCO* and stabilized the transition state.3,4 Another way of putting it, the transition state can be stabilized
by the hyperconjugation between the incipient bond, which involves the nucleophile and the πCO*, and the vacant antiperiplanar antibonding orbital of a vicinal sigma bond. For cyclic ketones, where the conformation around the R-carbon is relatively rigid comparing to acyclic ketones, the Felkin model also identified the so-called “torsional strain”2factor. This factor has been used as one of the reasons why nonbulky hydrides attack unhindered cyclohexanones from the axial side rather than the equatorial. In the axial approach the incipient bond may suffer from 1,3 diaxial-type interactions, but this forming bond in the transition structure is basically staggered with groups attached to the R-carbon. On the other hand, in the equatorial approach the incipient bond is nearly eclipsed to the vicinal bonds and hence this approach is unfavorable in terms of torsional strain. Houk et al. developed an empirical force field (modified MM2) to model transition states of nucleophilic additions to carbonyls.5 The calculated product ratios were in excellent agreement with experiment for acyclic and cyclic ketones and provided strong support to the Felkin model.
In the early 1980s, Cieplak provided another model which is also of electronic origin.6 Contrary to the hyperconjugation in the Felkin-Anh model between the * To whom correspondence should be addressed. Phone:
++886-2-2789-8530. Fax: ++886-2-2783-1237.
†Academia Sinica. ‡Chiao Tung University.
(1) (a) Cram, D. J.; Elhafez, F. A. A. J. Am. Chem. Soc. 1952, 74, 5828. (b) Mengel, A.; Reiser, O. Chem. Rev. 1999, 99, 1191.
(2) Che´rest, M.; Felkin, H.; Prudent, N. Tetrahedron Lett. 1968, 18, 2199.
(3) (a) Anh, N. T. Top. Curr. Chem. 1980, 88, 145. (b) Anh, N. T.; Eisenstein, O. Nouv, J. Chim. 1977, 1, 61. (c) Anh, N. T.; Eisenstein, O. Tetrahedron Lett. 1976, 3, 155.
(4) Anh, N. T.; Eisenstein, O.; Lefour, J.-M.; Tran Huu Dau, M. E. J. Am. Chem. Soc. 1973, 95, 6146.
(5) (a) Wu, Y.-D.; Houk, K. N. J. Am. Chem. Soc. 1987, 109, 908. (b) Wu, Y.-D.; Houk, K. N.; Trost, B. M. J. Am. Chem. Soc. 1987, 109, 5560.
(6) (a) Cieplak, A. S. J. Am. Chem. Soc. 1981, 103, 4540. (b) Cieplak, A. S.; Tait, B. D.; Johnson, C. R. J. Am. Chem. Soc. 1989, 111, 8447. 10.1021/jo000872j CCC: $19.00 © 2000 American Chemical Society
incipient bond and the vacant antibonding orbital of an antiperiplanar vicinal bond, Cieplak emphasized the importance of hyperconjugation between the filled orbital of a vicinal bond and the antibonding orbital of the incipient bond. In brief, the Cieplak model predicts that the addition reaction takes place on the face that is anti to the electron rich vicinal bonds. This model has been applied successfully to nucleophilic addition to carbonyl compounds, electrophilic addition to olefins, cycloaddi-tions and many other reaccycloaddi-tions.7Nevertheless, just as other aforementioned models, the Cieplak model does not always lead to correct predictions and has been chal-lenged on the basis of theoretical and experimental results.8 One of the effects that are often invoked to provide an alternative explanation to the Cieplak-type hyperconjugation is the electrostatic effect.9,10 Computa-tionally, this effect has been estimated by placing point charges next to the reaction center of a ground-state reactant11or a reaction transition structure with removal of the nuclephile.10Experimentally, a good correlation between logarithm of the product ratio and electrostatic field parameters (σF) has been taken as an indication for the presence of electrostatic effect.12
Analyses based on ground-state reactant properties have also been shown useful in predicting π-facial selectivity. Gung and others studied the structure distor-tion of reactants such as heteroatom-substituted cyclo-hexanones and admantanones.13Mehta et al. illustrated that the facial molecular electrostatic potential of meth-ylenenorsnoutanes was affected by remote substitution.14 Klein,15Fukui,16Frenking,17and Ohwada18pointed out the unequal facial distribution of frontier orbitals. Li-otta,19Dannenberg,20and Tomoda21developed methods attempting to quantify the orbital distortion, unequal facial electron distribution, or accessible space. Adcock measured the NMR chemical shifts of reactants or their analogues to shed light on the origin of facial selectivity.22 Overall, there are many different opinions and debates on the issue of origins of facial selectivity.8Recently, one issue of Chemical Review was entirely devoted to models and physical organic investigations of diastereofacial selectivity and synthetic strategies for achieving selectiv-ity.23
One of us has synthesized dioxa and novel trioxa cage compounds 1-4. Ketones 1 and 2 were subjected to nu-cleophilic addition by NaBH4and CH3MgBr and alkenes 3 and 4 were treated with dichlorocarbene. For dioxa cages (1 and 3), the addition reactions took place on both sides of the trigonal carbons, with a preference at the face syn to the oxygen atoms in the cage. For trioxa compounds (2 and 4), additions took place essentially at the face anti to the oxygen atoms (product ratio >99: <1).24Hydride (NaBH4) reduction of 1 has been reported
by Mehta et al.25 They demonstrated the correlation between electrostatic effect and syn attack preference with charge replacement ab initio calculations25aand σF deduced from 13C NMR chemical shift data.25b Other effects such as distortion of structure and frontier orbit-als, torsional strain and hyperconjugation were not addressed. If electrostatic factor is important in the hydride reduction of dioxa cage 1, which involves highly polar nucleophilic attacking species, is it also important for the addition reaction of dioxa cage 3 with a relative neutral electrophilic dichlorocarbene?26If hyperconjuga-tion, be it of Cieplak-type or Anh-type, does play a role, does it help to achieve the excellent facial selectivity observed for addition reactions of trioxa cages 2 and 4? Does the filled nonbonded lone pair orbital of the ad-ditional oxygen atom in 2 and 4 also interacts with the antibonding of the incipient bond? Conceivably, introduc-tion of oxygen atoms in the cage should generate certain structure distortion around the reaction center. How does this distortion correlate with facial selectivity?
In this paper, we report theoretical studies of hydride reduction of 1 and 2 and carbene addition of 3 and 4. It has been shown that calculations with LiH for ketone reduction reproduce the trends observed experimentally in NaBH4reduction.8,27Therefore, LiH was used in our calculations for hydride reduction for the sake of com-putational efficiency. In terms of carbene addition, dichlorocarbene, a singlet carbene in the ground state,26,28 is used in calculations as in experiments. Nevertheless, the methyl groups on the terminal alkene carbon of 3 and 4 are replaced with hydrogens in calculations and labeled as 3H and 4H. The controversial Cieplak-type hyperconjugation, as well as the Anh-type, are evaluated by the Natural Bond Orbital (NBO) analysis of Weinhold et al.29The unsymmetrical facial distribution of frontier
(7) Kaselj, M.; Chung, W.-S.; le Noble, W. J. Chem. Rev. 1999, 99, 1387 and references therein.
(8) Gung, B. W. Tetrahedron 1996, 52, 5263.
(9) (a) Wong, S. S.; Paddon-Row, M. N. J. Chem. Soc., Chem. Commun. 1991, 327. (b) Wong, S. S.; Paddon-Row, M. N. Aust. J. Chem. 1991, 44, 765. (c) Wu, Y -D.; Tucker, J. A.; Houk, K. N. J. Am. Chem. Soc. 1991, 113, 5018.
(10) Paddon-Row, M. N.; Wu, Y.-D.; Houk, K. N. J. Am. Chem. Soc.
1992, 114, 10638.
(11) Ganguly, B.; Chandrasekhar, J.; Khan, F. A.; Mehta, G. J. Org. Chem. 1993, 58, 1734.
(12) Adcock, W.; Cotton, J.; Trout, N. A. J. Org. Chem. 1994, 59, 1867.
(13) (a) Gung, B. W.; Wolf, M. A.; Mareska, D. A.; Karipides, A. J. Org. Chem. 1994, 59, 4899. (b) Gung, B. W. Chem. Rev. 1999, 99, 1377. (c) Gung, B. W.; Wolf, M. A. J. Org. Chem. 1996, 61, 232.
(14) Mehta, G.; Ravikrishna, C.; Gadre, S. R.; Suresh, C. H.; Kalyanaraman, P.; Chandrasekhar, J. J. Chem. Soc., Chem. Commun.
1998, 975.
(15) Klein, J. Tetrahedron Lett. 1973, 4307.
(16) Fukui, K. Theory of Orientation and Stereoselection; Springer-Verlag: Heidelberg, 1979.
(17) Frenking, G.; Kohler, K. F.; Reetz, M. T. Angew. Chem., Int. Ed. Engl. 1991, 30, 1146.
(18) Ohwada, T. Chem. Rev. 1999, 99, 1337 and references therein. (19) (a) Liotta, C. L. Tetrahedron Lett. 1975, 519. (b) Liotta, C. L.; Burgess, E. M.; Eberhardt, W. H. J. Am. Chem. Soc. 1984, 106, 4849. (20) (a) Huang, X. L.; Dannenberg, J. J.; Duran, M.; Bertra´n, J.J. Am. Chem. Soc. 1993, 115, 4024. (b) Huang, X. L.; Dannenberg, J. J. J. Am. Chem. Soc. 1993, 115, 6017.
(21) (a) Tomoda, S. Chem. Rev. 1999, 99, 1243 and references therein. (b) Tomoda, S.; Senju, T. J. Chem. Soc., Chem. Commun. 1999, 621.
(22) Adcock, W.; Trout, N. A. Chem. Rev. 1999, 99, 1415 and references therein.
(23) Gung, B. W.; le Noble, B., Eds, Chemical Reviews, 1999, 5. (24) Wu, H. J.; Wu. C. Y.; Lin, H. C.; Chao, C. S.; Chao, I.; Shih, J. H. Manuscript in preparation.
(25) (a) Mehta, G.; Khan, F. A.; Ganguly, B.; Chandrasekhar, J. J. Chem. Soc., Perkin Trans. 2 1994, 2275. (b) Mehta, G.; Khan, F. A.; Adcock W. J. Chem. Soc., Perkin Trans. 2 1995, 2189.
orbitals or electrostatic potential are investigated quali-tatively by mapping the values of the specific property onto the electron density isosurface of the reactant.30,31
Computational Methods
All geometry optimization calculations were performed with GAUSSIAN 9432or GAUSSIAN 9833program suites at the theory level of HF/6-31G*. This level has been shown to correctly predict facial selectivity of related systems.10,25aEach stationary point was characterized as a minima or a transition state by frequency calculations. All the transition states were further characterized by analysis of the vibrational modes corresponding to their imaginary frequencies. Frontier molec-ular orbitals (HOMO and LUMO) or molecmolec-ular electrostatic potential (MEP) of a substrate were mapped onto its isosurface with electron density of 0.002 au with the Spartan 4.1.1 program.34 Natural bond orbital (NBO) analysis and the second-order perturbative stabilization energy of transition states were carried out and evaluated with the program NBO 4.0.35
Results and Discussion
Activation Energies. The product ratio observed experimentally for NaBH4reduction of dioxa cage 1 is 85:15 (syn/anti) and of trioxa cage 2 is <1:>99; for dichlorocarbene addition of dioxa 3 is 60:40 and of trioxa 4 is <1:>99. In other words, the syn attack is favored for dioxa cages and the anti attack is favored for trioxa cages. The calculation results at HF/6-31G* level are listed in Table 1, and they correlate well with the observed facial selectivity. For example, the energy differences of syn and anti transition structures of dioxa cages 1 and 3H are 1.26 and 0.43 kcal/mol, respectively, leading to higher activation energies for the anti attack and thus higher percentage of the syn product. For trioxa cages 2 and 4H, the energy differences of syn and anti
transition structures (more than 3.8 kcal/mol) are sig-nificantly higher than that of dioxa cages 1 and 3H. Therefore, the effectively exclusive anti addition in the trioxa systems is successfully modeled by our calcula-tions.
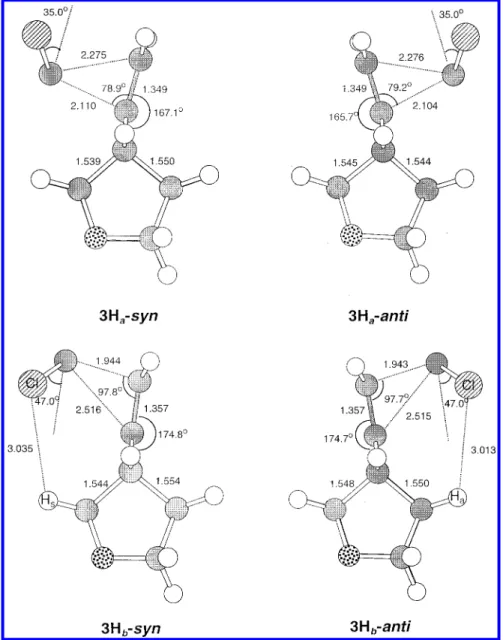
It is noted that in dichlorocarbene addition, four different transition structures were located. This is because the transition state is asynchronous in terms of the two forming bonds. We define the trajectory in which the bond with larger degree of formation involves the inner ene carbon as “trajectory a” and that involves the terminal ene carbon as “trajectory b”. For 3H, both syn and anti attacks favor trajectory a (see 3Ha-syn, 3Ha
-anti, 3Hb-syn, 3Hb-anti in Table 1; transition structures are shown in Figure 7). However, for 4H trajectory a is lower in energy for the anti attack, but trajectory b is lower for the syn attack (transition structures are shown in Figure 8). We will discuss this change of preferred trajectory in more details in a later section.
On the basis of satisfactory predictions of facial selec-tivity by calculated energy differences of transition structures, results at the HF/6-31G* level are used for further analyses.
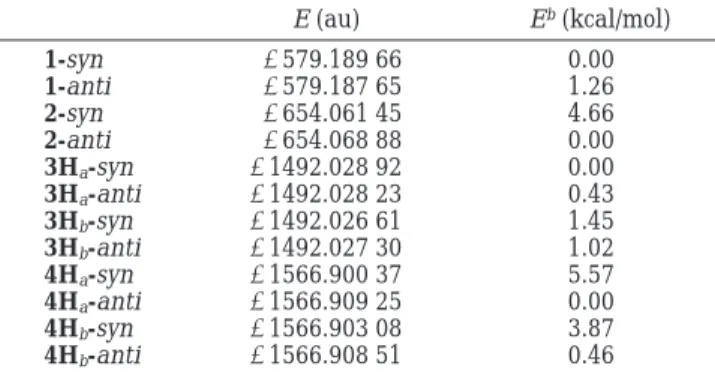
Structures and Properties of Dioxa and Trioxa Cages. Calculated structures and atom labels of the cage compounds are shown in Figure 1. The effect of the two oxygen atoms on structural distortion of the dioxa cages is rather minor. It is shown in Table 2 that hydrogens (Hb1 and Hb2) on the bridgehead carbons (Cb1 and Cb2) twist out of the carbonyl plane by less than 1° (see dihedral angles Y-Ct-Cb1-Hb1 and Y-Ct-Cb2-Hb2 in Table 2); for 1 the bridgehead hydrogens lean toward the syn side and for 3H toward the anti side. The difference between dihedral angles Y-Ct-Cb1-Cs1and Y-Ct-Cb1 -Ca1is smaller than 2°. Meanwhile, the C-C bonds (Cb1 -Cs1, Cb2-Cs2, Cb1-Ca1, and Cb2-Ca2) which may involve in hyperconjugation with the forming bond in the transi-tion state differ by 0.007 and 0.008 Å for 1 and 3H, respectively. The Cssymmetry of the dioxa cages 1 and 3H is clearly shown with data presented in Table 2. The plane bisecting the angle Cb1-Ct-Cb2is the Cssymmetry
plane.
With the presence of a third oxygen atom (labeled as Os) in the cage, trioxa cages 2 and 4H distort slightly (27) Mehta, G.; Ravikrishna, C.; Ganguly, B.; Chandrasekhar, J. J.
Chem. Soc., Chem. Commun. 1997, 75.
(28) (a) Rondan, N. G.; Houk, K. N.; Moss, R. A. J. Am. Chem. Soc.
1980, 102, 1770. (b) Moss, R. A. Acc. Chem. Res. 1989, 22, 15. (c) Sakai,
S. Int. J. Quantum Chem. 1998, 70, 291.
(29) (a) Reed, A. E.; Weinhold, F. J. Chem. Phys. 1983, 78, 4066. (b) Reed, A. E.; Curtiss, L. A.; Weinhold, F. Chem. Rev. 1988, 88, 889. (30) Meyers, A. I.; Seefeld, M. A.; Lefker, B. A.; Blake, J. F. J. Am. Chem. Soc. 1997, 119, 4565.
(31) Wu, Y.-D.; Li, Y.; Na, J.; Houk, K. N. J. Org. Chem. 1993, 58, 4625.
(32) Gaussian 94, Revision E.2: Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Gill, P. M. W.; Johnson, B. G.; Robb, M. A.; Cheeseman, J. R.; Keith, T. A.; Petersson, G. A.; Montgomery, J. A.; Raghavachari, K.; Al-Laham, M. A.; Zakrzewski, V. G.; Ortiz, J. V.; Foresman, J. B.; Cioslowski, J.; Stefanov, B. B.; Nanayakkara, A.; Challacombe, M.; Peng, C. Y.; Ayala, P. Y.; Chen, W.; Wong, M. W.; Andres, J. L.; Replogle, E. S.; Gomperts, R.; Martin, R. L.; Fox, D. J.; Binkley, J. S.; Defrees, D. J.; Baker, J.; Stewart, J. P.; Head-Gordon, M.; Gonzalez, C.; Pople, J. A. Gaussian, Inc., Pittsburgh, PA, 1995.
(33) Gaussian 98, Revision A.5: Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Zakrzewski, V. G.; Montgomery, J. A., Jr.; Stratmann, R. E.; Burant, J. C.; Dapprich, S.; Millam, J. M.; Daniels, A. D.; Kudin, K. N.; Strain, M. C.; Farkas, O.; Tomasi, J.; Barone, V.; Cossi, M.; Cammi, R.; Mennucci, B.; Pomelli, C.; Adamo, C.; Clifford, S.; Ochterski, J.; Petersson, G. A.; Ayala, P. Y.; Cui, Q.; Morokuma, K.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Cioslowski, J.; Ortiz, J. V.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Gomperts, R.; Martin, R. L.; Fox, D. J.; Keith, T. A.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Gonzalez, C.; Challacombe, M.; Gill, P. M. W.; Johnson, B. G.; Chen, W.; Wong, M. W.; Andres, J. L.; Head-Gordon, M.; Replogle, E. S.; Pople, J. A. Gaussian, Inc., Pittsburgh, PA, 1998.
(34) Hehre, W. J. et al. SPARTAN, Version 4.1.1, Wavefunction, Inc, 18401 Von Karman Ave., #370, Irvine, CA 92715.
(35) NBO 4.0. Glendening, E. D.; Badenhoop, J. K.; Reed, A. E.; Carpenter, J. E.; Weinhold, F. Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 1996.
Table 1. Calculated Energies of Addition Transition Structuresawith Full Geometry Optimization at the
HF/6-31G* Level
E (au) ∆Eb(kcal/mol)
1-syn -579.189 66 0.00 1-anti -579.187 65 1.26 2-syn -654.061 45 4.66 2-anti -654.068 88 0.00 3Ha-syn -1492.028 92 0.00 3Ha-anti -1492.028 23 0.43 3Hb-syn -1492.026 61 1.45 3Hb-anti -1492.027 30 1.02 4Ha-syn -1566.900 37 5.57 4Ha-anti -1566.909 25 0.00 4Hb-syn -1566.903 08 3.87 4Hb-anti -1566.908 51 0.46
aLabels syn and anti represent the additions from the syn and
anti faces, respectively. Transition states of LiH addition to ketones 1 and 2 and that of dichlorocarbene addition to enes 3H and 4H are calculated. Labels a and b represent the addition trajectories with the carbon lone pair of dichlorocarbene pointing to the inner ene carbon, Ct, and the outer one, Y, respectively.
out of the Cssymmetry. Therefore, geometry parameters
related to the atoms in the front (e.g., Cb1, Hb1, Cs1) are inequivalent to those in the rear (Cb2, Hb2, Cs2). The bridgehead hydrogens lean toward the anti face by ca. 10° (see dihedral angles Y-Ct-Cb1-Hb1and Y-Ct-Cb2 -Hb2in Table 2). More importantly, dihedral angles that are related to the accessibility of different faces of the reaction center, Y-Ct-Cb1-Cs1, Y-Ct-Cb2-Cs2, Y-Ct -Cb1-Ca1, and Y-Ct-Cb2-Ca2, show obvious inequivalently of the two faces. According to these angles (Table 2), the anti face of 2 and 4H is more open than the syn by nearly 20°. Meanwhile, the third oxygen atom, Os, is 2.6 Å away from the trigonal carbon, Ct, and the Y-Ct-Osangle is 130°. Therefore, Osnot only cause different accessibility of syn and anti faces (defined by Y-Ct-Cb-Csand Y-Ct -Cb-Ca), it also blocks the bottom part of the syn face. Another change brought by Osto the atoms next to the reaction center is that C-C bonds which are candidates for hyperconjugation with the incipient bond are slightly longer in the syn face (Cb1-Cs1, Cb2-Cs2) than in the anti face (Cb1-Ca1and Cb2-Ca2). This is contrary to what are observed in dioxa cages 1 and 3H. Finally, it is also noted that we have located structures of Cssymmetry for 2 and
4H (Table 2, 2-sym and 4H-sym). These structures have very small imaginary frequency and the energies are virtually identical to that of the “ground state”. As can be seen in Table 2, the structural parameters of sym-metry and unsymmetrical trioxa cages are similar. All structural features described above for the unsym-metrical structures also apply to the symunsym-metrical ones. Although the inferior accessibility of the syn face of 2 and 4H can be used to rationalize the high facial selectivity for anti attack, it is important to have a full understanding of other factors influencing the selectivity. Otherwise, one will not have an integrated view about facial difference of dioxa and trioxa cages 1-4. First, we look at frontier orbitals of these cages. Structural asym-metry of dioxa ketone 1 is minor in the vicinity of the carbonyl group as shown in Table 1, nevertheless, the unequal facial distribution of LUMO coefficient is obvious as shown in Figure 2(a). Comparison of syn and anti faces around reaction center atom Ctshows that there is more significant LUMO coefficient (darker color marked with a “+”) found for the syn face. For the more distorted trioxa ketone 2, there is larger LUMO coefficient found for the anti face, contrary to that of 1. Other than frontier Figure 1. Structures and atom labels of calculated cage compounds. Geometry optimized structures at the theory level of HF/ 6-31G* are shown. Gray, white, and mosaic circles represent carbon, hydrogen, and oxygen atoms, respectively; (Y) the ke-tone oxygen or terminal alkene carbon, (t) trigonal carbon, (b) bridgehead, (s) syn, (a) anti, (1) front, and (2) rear positions, respectively. For example, Cs1represents the carbon atom located at front position of syn face and Hs1is the hydrogen attached
to the carbon Cs1.
Table 2. Selected Structural Parameters of Substrates Optimized at the HF/6-31G* Levela
1 2 2-sym 3H 4H 4H-sym
dihedral angleb (deg)
Y-Ct-Cb1-Hb1 0.6(s) 9.7(a) 10.1 0.5(a) 8.7(a) 9.2
Y-Ct-Cb2-Hb2 0.6(s) 10.4(a) 10.1 0.5(a) 9.8(a) 9.2
Y-Ct-Cb1-Cs1 131.0 116.2 115.8 129.3 117.5 116.8 Y-Ct-Cb2-Cs2 131.0 115.3 115.8 129.3 116.1 116.8 Y-Ct-Cb1-Ca1 129.5 136.5 136.8 130.7 134.7 135.2 Y-Ct-Cb2-Ca2 129.5 137.2 136.8 130.7 135.9 135.2 bond length (Å) Cb1-Cs1 1.540 1.550 1.549 1.540 1.549 1.547 Cb2-Cs2 1.540 1.548 1.549 1.540 1.545 1.547 Cb1-Ca1 1.547 1.540 1.540 1.548 1.543 1.544 Cb2-Ca2 1.547 1.541 1.540 1.548 1.545 1.544 angle (deg) Y-Ct-Os 130.0 130.0 131.7 131.8 nonbonded distance (Å) Ct-Os 2.588 2.588 2.622 2.621
a2-sym and 4H-sym are the structures of C
ssymmetry for 2 and 4H.bAbsolute values of dihedral angles are shown in the table; symbols, s and a, in parentheses indicate the bridgehead hydrogens lean toward syn and anti faces, respectively.
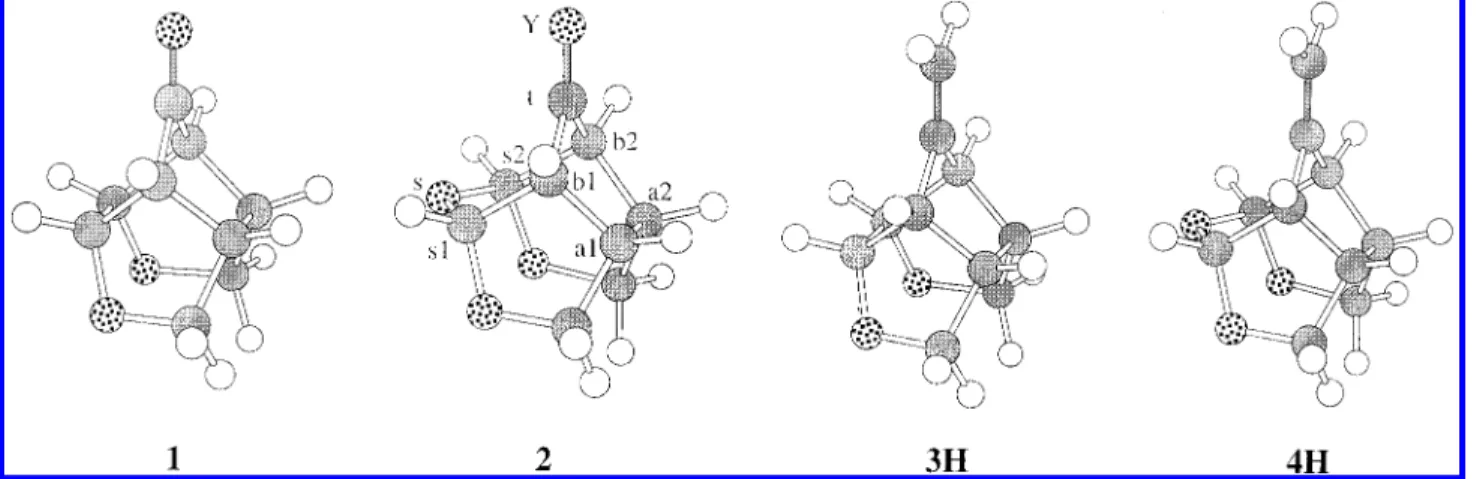
orbitals, molecular electrostatic potentials (MEPs) have also been found useful in predicting reaction sites.36For 1, the more electron deficient nature (darker color marked with a “+”) of the syn face than the anti face of the carbonyl carbon atom can be clearly seen in Figure 3a. However, for 2 the syn face MEP above the carbonyl carbon atom is less positive than that of the anti face because of the oxygen atom (Os) ca. 2.6 Å away. On the basis of LUMO and MEP of the reactant ground state, one would predict the hydride reduction to take place in the syn face for 1 and the anti face for 2, consistent with experimental findings.
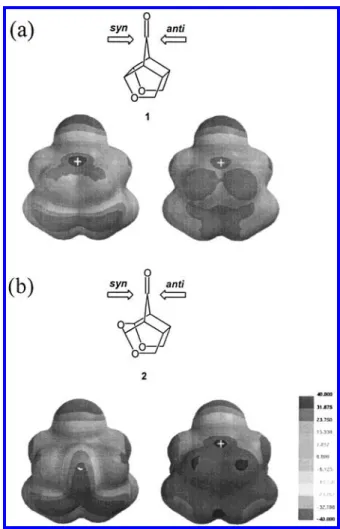
For the ene addition, the unsymmetrical facial distri-bution of HOMO coefficient of dioxa ene 3H and trioxa 4H (Figure S1) is not so obvious as the frontier orbitals (LUMOs) of 1 and 2 (Figure 2), but the facial difference of LUMO coefficient is more prominent (Figure 4). The LUMO coefficient above the inner ene carbon atom has a higher value in the syn face than the anti for 3H and the reverse is true for 4H. This leads to the prediction that in trajectory a, where the nonbonded lone pair orbital of the carbon atom of dichlorocarbene is pointing
at the empty orbital above the inner ene carbon atom, 3H should favor syn attack and 4H favor anti attack. This is in line with the energy trends listed in Table 1 for trajectory a.
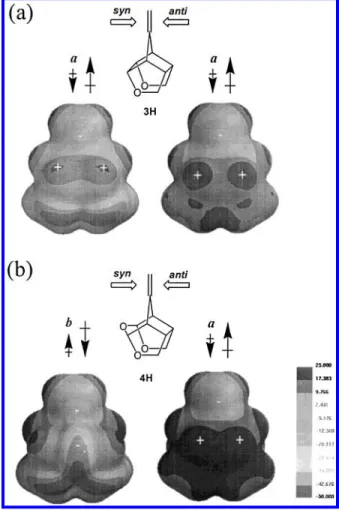
Although dichlorocarbene is not so ionic as metal hydrides, the MEPs of dichlorocarbene and ene com-pounds can be used to rationalize the energy trends of trajectories a and b for each facial attack. It is evident from the MEP of dichlorocarbene (Figure S2) that this molecule can be considered as a dipole with its negative end at the nonbonded lone pair electrons of the carbon atom (this direction is confirmed by calculation). The MEPs of 3H and 4H (Figure 5) show positive electrostatic potentials on the isodensity surfaces in the area of cage hydrogen atoms Hs1, Hs2, Ha1, and Ha2(e.g., see gray and black areas in the middle of 3H in Figure 5) and negative electrostate potentials in the area of CdC, except for the syn face of 4H. The presence of Os makes the MEP of the syn face of 4H largely negative. Based on an electrostatic argument, one would predict that trajectory
a is preferred over b for 3H and for anti attack of 4H.
This is because the negative end of dichlorocarbene is pointing to the positive area of 3H/4H(anti) in trajectory
a. For syn attack of trioxa cage 4H the negative end of
dichlorocarbene will be in the vicinity of an oxygen atom (Os) in trajectory a, so trajectory b is preferred instead. (36) (a) Molecular Electrostatic Potentials: Concepts and
Applica-tions; Murray, J. S., Sen, K., Eds.; Elsevier: Amsterdam, 1996. (b) Chemical Applications of Atomic and Molecular Electrostatic Poten-tials: Reactivity, Structure, Scattering, and Energetics of Organic, Inorganic, and Biological Systems; Politzer, P., Truhlar, D. G., Eds.; Plenum Press: New York, 1981.
Figure 2. Lowest unoccupied molecular orbital (LUMO) mapped onto the isosurface with an electron density of 0.002 au for 1 (upper-half, (a)) and 2 (lower-half, (b)), respectively. Pictures on the left and right are images viewed from syn and anti faces, respectively. The position of the carbonyl carbon (Ct) is labeled with a plus sign.
Figure 3. Molecular electrostatic potential (MEP) mapped onto the isosurface with an electron density of 0.002 au for 1 (upper-half, (a)) and 2 (lower-half, (b)). The positions of the most positive and negative values are indicated with plus and minus signs, respectively.
The above deduction complies with the switch in attack-ing trajectory described in the previous section and in Table 1.
Transition Structure. For the nearly structurally unbiased dioxa cages 1 and 3H, the transition structures of syn and anti attacks are very similar (Figures 6 and 7). For trioxa ketone 2, the CdO and forming H--C bonds in the anti transition structure (1.236 and 1.978 Å) are longer and shorter than the counterparts in the syn transition structure (1.229 and 2.161 Å), respectively. Therefore, the transition structure of the anti attack is later than that of the syn attack. One interesting phenomenon of transition structures of hydride reduction of 2 is that 2-anti is symmetrical, whereas 2-syn is unsymmetrical. As mentioned previously, the potential energy difference between symmetrical and unsym-metrical 2 is negligible. Therefore, it is easy for 2 to adopt a structure that is most suitable for hydride attack. For the anti attack, because the number and types of atoms the hydride feels along the reaction path are the same in the front part (Cb1, Ca1, Hb1, Ha1) and in the rear (Cb2, Ca2, Hb2, Ha2), it is not surprising that 2-anti is of Cs
symmetry. For the syn attack, the H--C-O attacking angle (95°) is smaller than that of 2-anti, 1-syn and 1-anti (98-99°). The distance between H-and Os(2.768 Å) is not much longer than the sum of VDW radii of neutral H and O, 2.6 Å. Furthermore, Osdeviates from Hs1-Cs1-Cs2(or Hs2-Cs2-Cs1) plane in the syn transition structure by ca. 40°, which is 20° larger than that in the ground state. The Ct‚‚‚Osdistance, 2.774 Å, is also longer
than that of the ground state (2.588 Å). These structural features of 2-syn imply that the incoming hydride has experienced strong influence of Osand the two atoms are trying to avoid each other. The inferior accessibility of the syn face has been overcome by downward movement of Os and deformation of the Li-H...CdO transition structure. We believe that the trioxa cage distorts out of the Cssymmetry to minimize the repulsion between Os
and hydride. For example, the H-‚‚‚Os distance can be lengthened if the cage is distorted.
In Figure 6, all the C-C bonds antiperiplanar to the forming bond are longer than that of the corresponding bond in the ground-state reactant, as what would be predicted on the basis of a hyperconjugative model. For example, in 1-syn, the antiperiplanar Cb1-Ca1and Cb2 -Ca2bonds (1.553 Å) are 0.006 Å longer than that of the ground-state reactant (1.547 Å in Table 2). On the other hand, the periplanar C-C bonds (such as Cb1-Cs1 in 1-syn) of all hydride reduction transition structures are shorter in the transition state than in the ground state. Taking 1-syn again as an example, the Cb1-Cs1bond is 1.534 Å in the transition structure and 1.540 Å in the ground state. The shortening of periplanar bonds can be rationalized by hyperconjugation model as well. When Figure 4. Lowest unoccupied molecular orbital (LUMO) for
3H (upper-half, (a)) and 4H (lower-half, (b)), respectively. The position of carbonyl carbon (Ct) is labeled with a plus sign.
Figure 5. Molecular electrostatic potential (MEP) for 3H (upper-half, (a)) and 4H (lower-half, (b)). The positions of the most positive and negative values are indicated with plus and minus signs, respectively. The preferred directions of the dipole moment of dichlorocarbene (small arrows) are labeled for each facial attack (large arrows are for the local dipole direction in the vicinity of reaction centers of 3H and 4H). Labels a and b represent the addition trajectories with the carbon lone pair of dichlorocarbene pointing to the inner ene carbon, Ct, and
the hydride attacks the carbonyl CdO antibonding orbital, the carbonyl group CdO bents away from the face of addition, as shown in Figure 6. In this addition process, the CdO antibonding orbital develops into the H--C forming bond and overlaps less and less with the peripla-nar C-C bonds due to the bending motion of CdO. With loss of hyperconjugation with an empty orbital, the periplanar C-C bonds become shorter.
For dichlorocarbene addition, characteristics of transi-tion structures of trajectories a (in which carbene forms partial bonding with the inner alkene atom to a greater extent) and b (carbene forms partial bonding with the terminal carbon atom to a greater extent) are very different (Figures 7 and 8). For trajectory a, distances of the two newly forming bonds differ by less than 0.18 Å in both syn and anti attacks of 3H and 4H (3Ha-syn, 3Ha
-anti, 4Ha-syn, and 4Ha-anti). The attacking angles (formed by the major forming bond and the double bond) are ca. 79° and the tilt angles of carbene from the line parallel to the alkene double bond are ca. 35°. It is noted that in the presence of an additional oxygen atom in the
trioxa cage, the geometry of reaction center in 4Ha-syn does not differ significantly from that of 3Ha-syn, 3Ha
-anti, and 4Ha-anit, unlike the change of attacking angles in 2-syn (Figure 6). Nevertheless, Osstill moves downward in 4Ha-syn as in 2-syn. This implies that 1) the repulsion between Os and :CCl2 is smaller than between Osand H-and 2) movement of Osis relatively easy.
For trajectory b, the distance differences, attacking angles and tilt angles of 3Hb-syn, 3Hb-anti, and 4Hb
-anti are ca. 0.57 Å, 98°, and 47°, respectively. These
values are significantly larger than those in trajectory
a. The geometry around the reaction center of 4Hb-syn differs from the other three transition structures of trajectory b in that its forming bonds are slightly shorter (ca. 0.02 Å) and the tilt angle is larger by 3°. It is noted that a large distance difference between the two forming bonds has been reported for alkene with steric hindrance around one of the alkene carbon atom.37Examination of the Cl‚‚‚Hsand Cl‚‚‚Hadistances shows that they are all around 3.0 Å (Figures 7 and 8), equal to the sum of VDW Figure 6. Transition structures of LiH addition to substrates, 1 (upper-half) and 2 (lower-half) at the theory level of HF/6-31G*. Selected geometric parameters are shown with distance in unit of Å and angle in unit of degree. Geometric parameter of the rear part is shown in parentheses or not shown when it is the same as the front one. Labels of syn and anti represent the addition from syn and anti faces with respect to the oxygen atoms of heterorings, respectively.
radii of Cl and H atoms. These short Cl‚‚‚H distances and the long Ccarbene‚‚‚Ct distances (ca. 2.5 Å) imply steric requirement is playing a role in large difference of forming bonds, attacking angle and tilting angle in trajectory b.
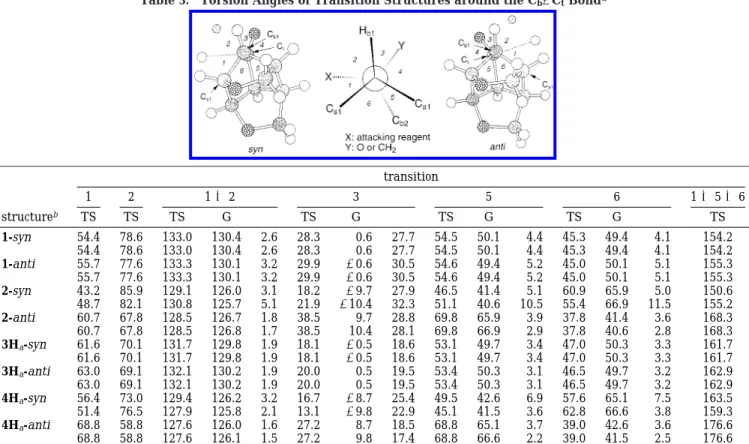
Torsional Strain Effect. Analysis of the torsion angles around the Newman projections with a view down to the Cb1-Ctand Cb2-Ctbonds can provide information related to the torsion strain effect. It can further afford as a criterion to judge which structure (syn or anti) is more favored. In general, a favored transition structure should prefer a good alignment between the forming bond and antiperiplanar bond, prefer a staggered attacking angle, and minimize the structural change from reactant to transition structure. This is because a good alignment gains more stabilization energy from hyperconjugation interaction, a staggered attacking angle minimizes the bond-bond repulsion, and minimal structure distortion requires less energy to deform the reactant.
The torsion angles around Cb1-Ct and Cb2-Ctbonds are listed in Table 3. Trajectory b of carbene addition is
not listed because in this trajectory dichlorocarbene basically attacks the terminal carbon atom. Therefore, torsional strain in trajectory b should not be as important as in all other transition structures where the reaction center is part of a ring. According to Table 3, the alignment between the forming bond and antiperiplanar bond can be judged by the sum of torsion angles 1, 5, and 6. As the ground state 1 and 3H are rather sym-metrical, the difference in alignment between transition structure of dioxa cages is minor. For trioxa cages, better alignment (1 + 5 + 6≈ 180°) between the forming and antiperiplanar bonds is observed in 2-anti (168.3°) than in 2-syn (150.6°, 155.2°). A similar observation is made for trajectory a of 4H (4Ha-anti: 176.6°; 4Ha-syn: 163.5°, 159.3°). Therefore, in our system the transition structure of the experimentally favored attacking face does provide better alignment for hyperconjugation.
Examination of angles 1 and 2 shows the attacking angles of 2-anti (60.7 for angle 1 and 67.8° for angle 2) and 4Ha-anti (68.8° and 58.8°) are more staggered than that of 2-syn (averaged 46.0° for angle 1 and 84.0° for angle 2) and 4Ha-syn (averaged 53.9° and 74.8°), respec-tively. As for distortion around the reaction center in the (37) Bernardi, F.; Bottoni, A.; Canepa, C.; Olivucci, M.; Robb, M.
A.; Tonachini, G. J. Org. Chem. 1997, 62, 2018.
transition structure relative to the ground state, we choose torsion angle 3 to represent the movement of Cd O and CdC upon addition reactions, sum of angles 1 and 2 to reflect the impact of the attacking reagent to atoms close to it, and angles 5 and 6 to detect the distortion of cage rings. The angle difference (∆) between the transi-tion structure and ground state is listed in Table 3. It is found that the difference of different facial attacks of dioxa cage is in general small, but the difference of trioxa cage is more significant. The angle differences ∆ of 2-anti for angles 3, 1 + 2, 5, and 6 are ca. 28°, 2°, 3°, and 3°, respectively. All are smaller than the corresponding values of 2-syn (ca. 30°, 4°, 8°, and 8°, respectively). The same trend is observed for 4Ha-anti and 4Ha-syn. This reflects that the trioxa cage suffers less structural distortion upon anti addition.
Although it is mentioned that geometries in the vicinity of the reaction center are rather similar in syn and anti transition structures for dioxa cages 1 and 3H, the strain experienced by the substrate may contribute differently to activation energies of the syn and anti attacks. The distorted energy Esublisted in Table 4 shows the energy differences of cage compounds frozen in their transition
structure without the inclusion of attacking reagent. It is found for 1 and 3H that the distorted energy of the syn attack is lower than that in the anti attack. On the other hand, for 2 and 4H, the Esubin the anti attack is lower. These trends echo the product ratios observed experimentally and structural analysis presented above. Therefore, not only the syn faces of 1 and 3H and anti faces of 2 and 4H are more reactive than their counter-parts based on the frontier orbital or MEP analyses, the skeletons of the cage compounds are also more apt to adopt distortion in the transition structures of the more reactive faces.
Electrostatic and Hyperconjugative Interactions. In Table 4 we list the energy differences between syn and anti faces from calculations in which the attacking reagents in the transition structure are replaced with point charges obtained from natural population analysis. In Charge Model I, all the atoms of the attacking reagent are replaced with charges. In Charge Model II, we followed Mehta,25a,27 Houk,10 and others to use the “charge replacement model” only for the hydride, but not for the metal ion. Mehta has also used hydride to illustrate the combined effect of electrostatic and hyper-Figure 8. Transition structures of dichlorocarbene addition to 4H at the theory level of HF/6-31G*.
conjugative interactions.27This is the Hydride Model in Table 4. We do not think electrostatic or hyperconjugation effects in the transition states can be quantified by any of the above model because a hard point charge is quite different from a relatively soft attacking reagent. Nev-ertheless, it should show the trend of facial difference of cage compounds under the influence of these two factors. Results in Table 4 clearly show that electrostatic or the combination of electrostatic and hyperconjugation effects help to achieve the experimentally observed facial dif-ferences.
Hyperconjugation of Cieplak and Anh types are based on the concept of donor-acceptor interactions of localized orbitals (lone pairs or bonds). The NBO analysis29,35 is
based on optimally transforming a given wave function into localized forms, which correspond to the one-center (“lone pair”) and two-center (“bond”) elements in the Lewis structure picture. Donor-acceptor interactions related to hyperconjugation can be estimated by the second-order perturbation analysis implemented in the NBO 4.0 program.35In Cieplak and Anh models, the focus is on the hyperconjugation between the orbitals of the forming bond and the vicinal bond, so only the Lewis resonance structures that have a forming bond are chosen for the NBO analysis. The NRT35,38module of the NBO 4.0 program determines resonance structures and weights on the basis of molecular electron density. Results of the NRT analysis shows that only one Lewis structure of each transition state bears the forming bond and the weights of these structures are 12.03%, 11.97%, 9.62%, and 25.40% for 1-syn, 1-anti, 2-syn, and 2-anti, respectively. The weight variance can be understood by inspection of geometry of the forming H--C bond in the transition structure. The weights of 1-syn and 1-anti are similar, which agree with the fact that transition structures 1-syn and 1-anti bear almost the same attacking angles and forming bond lengths (see Figure 6). For trioxa 2, the transition structure of 2-syn is earlier than 2-anti as mentioned in the transition structure section. Therefore, the weight of bond-forming Lewis structure of 2-syn is smaller than that of 2-anti.
The donor-acceptor orbital interactions related to the forming bond are collected in Table 5. It is noted
(38) (a) Gladening, E. D.; Weinhold, F. J. Comput. Chem. 1998, 19, 539. (b) Glendening, E. D.; Weinhold, F. J. Comput. Chem. 1998, 19, 610. (c) Glendening, E. D.; Badenhoop, J. K.; Weinhold, F. J. Comput. Chem. 1998, 19, 628.
Table 3. Torsion Angles of Transition Structures around the Cb-CtBonda
transition 1 2 1 + 2 3 5 6 1 + 5 + 6 structureb TS TS TS G ∆ TS G ∆ TS G ∆ TS G ∆ TS 1-syn 54.4 78.6 133.0 130.4 2.6 28.3 0.6 27.7 54.5 50.1 4.4 45.3 49.4 4.1 154.2 54.4 78.6 133.0 130.4 2.6 28.3 0.6 27.7 54.5 50.1 4.4 45.3 49.4 4.1 154.2 1-anti 55.7 77.6 133.3 130.1 3.2 29.9 -0.6 30.5 54.6 49.4 5.2 45.0 50.1 5.1 155.3 55.7 77.6 133.3 130.1 3.2 29.9 -0.6 30.5 54.6 49.4 5.2 45.0 50.1 5.1 155.3 2-syn 43.2 85.9 129.1 126.0 3.1 18.2 -9.7 27.9 46.5 41.4 5.1 60.9 65.9 5.0 150.6 48.7 82.1 130.8 125.7 5.1 21.9 -10.4 32.3 51.1 40.6 10.5 55.4 66.9 11.5 155.2 2-anti 60.7 67.8 128.5 126.7 1.8 38.5 9.7 28.8 69.8 65.9 3.9 37.8 41.4 3.6 168.3 60.7 67.8 128.5 126.8 1.7 38.5 10.4 28.1 69.8 66.9 2.9 37.8 40.6 2.8 168.3 3Ha-syn 61.6 70.1 131.7 129.8 1.9 18.1 -0.5 18.6 53.1 49.7 3.4 47.0 50.3 3.3 161.7 61.6 70.1 131.7 129.8 1.9 18.1 -0.5 18.6 53.1 49.7 3.4 47.0 50.3 3.3 161.7 3Ha-anti 63.0 69.1 132.1 130.2 1.9 20.0 0.5 19.5 53.4 50.3 3.1 46.5 49.7 3.2 162.9 63.0 69.1 132.1 130.2 1.9 20.0 0.5 19.5 53.4 50.3 3.1 46.5 49.7 3.2 162.9 4Ha-syn 56.4 73.0 129.4 126.2 3.2 16.7 -8.7 25.4 49.5 42.6 6.9 57.6 65.1 7.5 163.5 51.4 76.5 127.9 125.8 2.1 13.1 -9.8 22.9 45.1 41.5 3.6 62.8 66.6 3.8 159.3 4Ha-anti 68.8 58.8 127.6 126.0 1.6 27.2 8.7 18.5 68.8 65.1 3.7 39.0 42.6 3.6 176.6 68.8 58.8 127.6 126.1 1.5 27.2 9.8 17.4 68.8 66.6 2.2 39.0 41.5 2.5 176.6
aPictures show the definition of the torsion angles. The dotted line connects the attacking atom and the reaction center (carbon C t). Two sets of torsion angles are shown for each transition structure; the first one and the second one are viewed down to the Cb1-Ctand Cb2-Ctbonds, respectively.bTS, G, and ∆ represent the transition structure, state substrate, and the difference between ground-state substrate and transition structure, respectively.
Table 4. Calculated Relative Energies of Substrates in Transition States and the Relative Energies Evaluated
with the Charge Model and the Hydride Modela Esubb charge model Ic charge model IId hydride modele 1-syn 0.00 0.00 0.00 0.00 1-anti 0.82 2.89 1.52 1.91 2-syn 1.07 5.85 8.18 11.41 2-anti 0.00 0.00 0.00 0.00 3Ha-syn 0.00 0.00 3Ha-anti 0.11 0.28 4Hb-syn 2.73 4.28 4Ha-anti 0.00 0.00
aOptimized Structures at the theory level of HF/6-31G* are used; energy is in units of kcal/mol.bSingle-point energy of the substrate in the transition structure.cSingle-point energy of transition structure with the atoms of LiH and :CCl2replaced by natural population charges.dSingle-point energy of substrate and hydride in transition structure with hydride replaced by a negative charge of -0.5e.eSingle-point energy of substrate and hydride in transition structure.
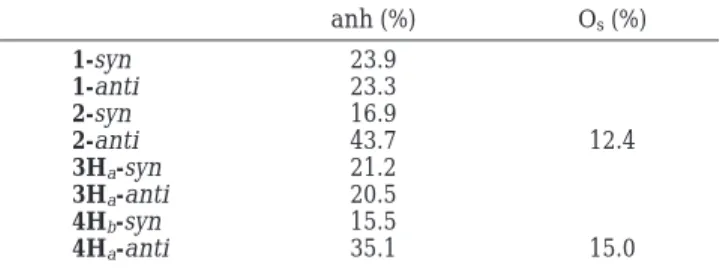
that the Lewis resonance structure that bears the idea of a fully formed bond cannot completely represent the transition structure which in fact has a partially formed bond. As a result, the hyperconjugative stabilization energies of the forming bond (Table S1) are overesti-mated. Moreover, since the resonance weights of syn and anti bond-forming Lewis structure are different, the degrees of overestimation are also different. This differ-ence in degrees of overestimation makes comparison of NBO hyperconjugative stabilization energies between syn and anti transition structures inappropriate. Therefore, we only consider the relative importance of different types of orbital interactions within a transition struc-ture. Within the framework of NBO the Cieplak effect is the major effect (Table S1), so it is taken as 100% to obtain the relative percentage of other hyperconjuga-tions related to the forming bond. For 1-syn and 1-anti, the magnitudes of the Anh effect are 23.9% and 23.3% of the Cieplak effect, respectively (Table 5). This indicates that no matter how the Cieplak effect favors the syn or anti addition, the Anh effect will give the same facial preference with a magnitude of 23% of the Cieplak effect. In other words, the Cieplak and Anh effects contribute to the facial preference in a cooperative manner. The same argument is true for 3H. Here, the magnitudes of the Anh effect are 21.1% and 20.5% of the Cieplak effect for syn and anti attacks, respectively. The above results imply that if the transition struc-tures are similar for syn and anti attacks and the magnitude of the Anh effect is a fixed fraction of the Cieplak effect, one can make correct prediction of facial preference by solely examining the Cieplak-type (or Anh-type) interaction.
For trioxa 2, the lone pair of Os gives a noticeable contribution to the hyperconjugation with the antibond-ing orbital of the formantibond-ing bond in the anti attack, but smaller than Cieplak and Anh effects (Table 5). The magnitudes of the Anh effects are 16.9% and 43.7% of the Cieplak effects for syn and anti attacks, respectively. In other words, the Anh effect weights more in the anti than in the syn addition. It implies that if the Cieplak effect favors the anti attack, the Anh effect will also give the same facial preference. On the other hand, if the Cieplak effect favors syn and anti additions to the same extent or favors syn addition slightly more, the combina-tion of Anh effect and Os contribution may have the
chance to override the Cieplak effect to favor the anti addition. The same reasoning applies to trioxa 4H. It clearly demonstrates that if the transition structures are not similar, judging the attacking preference solely by the Cieplak effect (or Anh effect) may lead to a wrong prediction.
Conclusion
We have carried out ab initio calculations of addition reactions of dioxa and trioxa cage compounds 1, 2, 3H, and 4H. The calculated relative energies of transition structures reproduce the trend of facial selectivity ob-served in experiment. We then looked at facial selectivity from the points of view of structure, frontier orbitals and MEP of the reactants, as well as strain, electrostatic, and hyperconjugation effects in the transition state.
For dioxa cages 1 and 3H, the structural facial differ-ence around the reaction center is minor. Nevertheless, the electronic difference of syn and anti faces generated by the two remote oxygen atoms is clearly demonstrated via frontier orbital and MEP analyses. For trioxa cages 2 and 4H, the close proximity of the third ether oxygen (Os) to the reaction center brings large structural and electronic changes around the reaction center. The syn face, which is more reactive in dioxa cages, is less acces-sible and less reactive than the anti face in trioxa cages. Analyses based on transition structures reveal several common features of our cage compounds. First of all, electrostatic interactions approximated by the charge replacement calculations afford facial difference in line with the transition state energies. Secondly, the more reactive faces are more apt to adopt structural distortion in the transition state. Moreover, the calculated electro-static and strain energy differences of syn and anti transition structures are significantly larger for trioxa cages than for the dioxa cages. Therefore, they both contribute to the enhanced facial selectivity of trioxa compounds. Finally, analysis of hyperconjugative stabi-lization in the transition state reveals the danger of relying solely on Cieplak or Anh models in rationalization of facial selectivity, especially when non-equivalent steric and electrostatic effects as those present in the trioxa systems are involved.
Acknowledgment. This work was supported by the National Science Council and Academia Sinica, Taiwan (ROC). The granting of computing time from the Na-tional Center for High-performance Computing and Computing Center of Academia Sinica is acknowledged. The helpful comments from the reviewers are also acknowedged.
Supporting Information Available: HOMO of 3H and 4H (Figure S1) and MEP of :CCl2(Figure S2). Hyperconjuga-tive stabilization energies estimated by NBO second-order perturbation analysis (Table S1). This material is available free of charge via the Internet at http://pubs.acs.org. JO000872J
Table 5. Percentage of Different Types of Hyperconjugative Stabilization Energies with Respect to
the Cieplak-Type Hyperconjugation
anh (%) Os(%) 1-syn 23.9 1-anti 23.3 2-syn 16.9 2-anti 43.7 12.4 3Ha-syn 21.2 3Ha-anti 20.5 4Hb-syn 15.5 4Ha-anti 35.1 15.0