L E T T E R S
nature cell biology VOLUME 13 | NUMBER 1 | JANUARY 2011
© 2011 Macmillan Publishers Limited. All rights reserved.
87
CDK1-dependent phosphorylation of EZH2 suppresses
methylation of H3K27 and promotes osteogenic
differentiation of human mesenchymal stem cells
Yongkun Wei
1, Ya-Huey Chen
2, Long-Yuan Li
2,3,4,9, Jingyu Lang
1, Su-Peng Yeh
5, Bin Shi
1, Cheng-Chieh
Yang
1, Jer-Yen Yang
1, Chun-Yi Lin
2, Chien-Chen Lai
6,7and Mien-Chie Hung
1,2,3,4,8,9 Enhancer of zeste homologue 2 (EZH2) is the catalytic subunitof Polycomb repressive complex 2 (PRC2) and catalyses the trimethylation of histone H3 on Lys 27 (H3K27), which represses gene transcription. EZH2 enhances cancer-cell invasiveness and regulates stem cell differentiation. Here, we demonstrate that EZH2 can be phosphorylated at Thr 487 through activation of cyclin-dependent kinase 1 (CDK1). The phosphorylation of EZH2 at Thr 487 disrupted EZH2 binding with the other PRC2 components SUZ12 and EED, and thereby inhibited EZH2 methyltransferase activity, resulting in inhibition of cancer-cell invasion. In human mesenchymal stem cells, activation of CDK1 promoted mesenchymal stem cell differentiation into osteoblasts through phosphorylation of EZH2 at Thr 487. These findings define a signalling link between CDK1 and EZH2 that may have an important role in diverse biological processes, including cancer-cell invasion and osteogenic differentiation of mesenchymal stem cells.
The Polycomb group (PcG) protein EZH2 is a histone lysine methyl- transferase associated with transcriptional repression. EZH2 functions in the multi-protein complex PRC2, which includes SUZ12 (suppres- sor of zeste 12) and EED (embryonic ectoderm development)1,2. EZH2 catalyses the addition of methyl groups to
histone H3 at Lys 27 (H3K27) in target gene promoters, leading to epigenetic silencing. EZH2 has an important role in controlling biological processes including X-chromosome inactivation, germline development, stem cell pluripo- tency and cancer metastasis3.
EZH2 is aberrantly overexpressed in aggressive solid tumours and overexpression of EZH2 has been implicated in cancer progression and metastases4–7. In addition, EZH2 knockdown leads to decreased
proliferation of cancer cells and a delay in the G2/M transition of the
cell cycle8. As EZH2 has a role in the G2/M transition and CDK1 is
one of the major G2/M kinases, and because both have a central function in controlling self-renewal and lineage specification of stem cells9, we investigated whether EZH2 is regulated by CDK1.
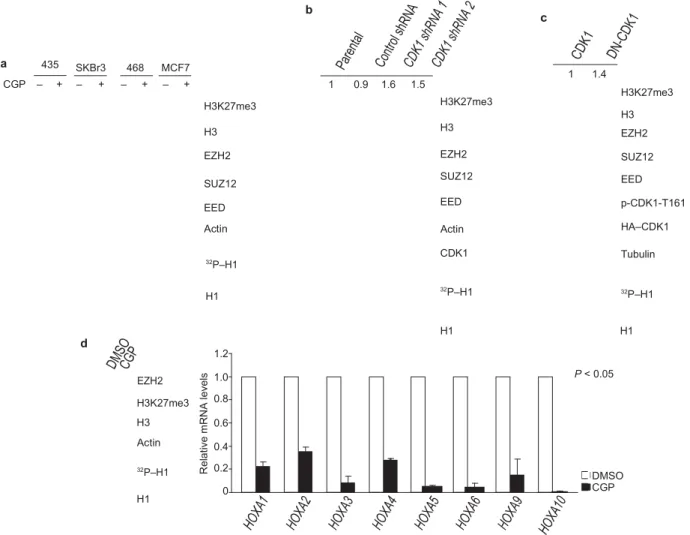
First, we investigated whether alteration of CDK1 activity affects H3K27 trimethylation. H3K27 trimethylation was increased in several cancer-cell lines after treatment with a CDK1 inhibitor, CGP74514A10, at a concentration (2 μM) that is specific to CDK1 in
these lines (Fig. 1a). The activity of CDK1 was inhibited by CGP74514A, as assessed by in vitro kinase assay of CDK1 using histone H1 as a substrate (Fig. 1a, botom). There was no change in EZH2, SUZ12 and EED protein level. In addition, H3K27 trimethylation level increased in accordance with CGP74514A in a dose- and time-dependent manner (Supplementary Information, Fig. S1a, b). Similar results were found when cells were treated with Roscovitine, a pan-CDK inhibitor (data not shown). Consistently, knockdown of endogenous CDK1 expression by two dif- ferent shRNA (to exclude potential off-target effects of the shRNA), or by small interfering RNA (siRNA), enhanced H3K27 trimethyla- tion (Fig. 1b and Supplementary Information, Fig. S1c). In addition, expression of a dominant-negative mutant CDK1 (DN-CDK1; ref. 11) also increased H3K27 trimethylation (Fig. 1c). As we had found that H3K27 trimethylation is increased with inhibition of CDK1 we next examined whether inhibition of CDK1 activity affects the expression of known EZH2-target genes. We tested gene expression of the HOXA family members using quantitative reverse transcription polymerase chain reaction (qRT–PCR). We found that expression of HOXA genes was suppressed by treatment with CGP74514A (Fig. 1d), indicating that inhibition of CDK1 affects the expression of EZH2-target genes.
As inactivation of CDK1 enhances trimethylation of H3K27, resulting in downregulation of EZH2-targeted gene expression, we next investi-gated whether CDK1, a serine/threonine kinase, might inhibit histone
1Department of Molecular and Cellular Oncology, The University of Texas M.D. Anderson Cancer Center, Houston, TX 77030, USA. 2Center for Molecular Medicine, China Medical University Hospital, Taichung 404, Taiwan. 3Graduate Institute of Cancer Biology, China Medical University, Taichung 404, Taiwan. 4Asia University, Taichung 413, Taiwan. 5Division of Hematology and Oncology, Department of Medicine, China Medical University and Hospital, Taichung 404, Taiwan. 6Graduate Institute of Chinese Medical Science, China Medical University, Taichung 404, Taiwan. 7Institute of Molecular Biology, National Chung Hsing University, Taichung
402, Taiwan. 8Program in Cancer Biology, The University of Texas Graduate School of Biomedical Sciences at Houston, Houston, TX 77030, USA. 9Correspondence should be addressed to L.-Y.L. or M.-C.H. (e-mail: [email protected] w or [email protected] g)
R el at iv e m R N A le ve ls b c a 435 SKBr3 468 MCF7 1 1.4 CGP – + – + – + – + H3K27me3 H3 EZH2 SUZ12 EED Actin 1 0.9 1.6 1.5 H3K27me3 H3 EZH2 SUZ12 EED Actin H3K27me3 H3 EZH2 SUZ12 EED p-CDK1-T161 HA–CDK1 32P–H1 H1 CDK1 32P–H1 Tubulin 32P–H1 d EZH2 H3K27me3 H3 Actin 32P–H1 H1 1.2 1.0 0.8 0.6 0.4 0.2 0 H1 H1 P < 0.05 DMSO CGP
Figure 1 CDK1 negatively regulates H3K27 trimethylation. (a) Top: 435, SKBr3, 468 and MCF7 cells were treated with CGP74514A as indicated, and the lysates were analysed by immunoblot using antibodies
against the specified proteins. Bottom: in vitro kinase assay. CDK1, immunoprecipitated from the cell lines treated with CGP74514A as indicated at the top, was incubated with H1 and [γ-32P]ATP. Reaction products were resolved by SDS–PAGE and visualized by
autoradiography
(equal loading of H1 was assessed by Coomassie-stained gel shown at the
bottom). (b) Lysates from MCF7 cells infected with lentiviruses expressing
control or two different CDK1 shRNA were immunoblotted with antibodies
against the indicated proteins. Relative intensities of the H3K27me3 bands are shown, normalized to the H3K27me3 band from parental MCF7 cells. Bottom: in vitro kinase assay, performed as in a, with CDK1
immunoprecipitated from cells treated as indicated at the top. (c) Lysates
of 293T cells transfected with plasmids encoding cyclin B, and CDK1 or dominant-negative mutant CDK1 (DN-CDK1) were immunoblotted
with antibodies against the indicated proteins. Relative intensities of the H3K27me3 bands are shown, normalized to the H3K27me3 band from 293T cells transfected with plasmid encoding CDK1. p-CDK1-T161; CDK1 phosphorylated at Thr 161. Bottom: in vitro kinase assay, performed as in a, with CDK1 immunoprecipitated from cells transfected as indicated at the top. (d) Left top: immunoblot of lysate from HEK293 cells treated with DMSO or CGP using antibodies against the indicated proteins. Left bottom: in vitro kinase assay, performed as in a, with CDK1 immunoprecipitated from cells treated as indicated at the top. Right: analysis of mRNA levels of HOXA families by qRT–PCR after treatment of HEK293 cells with DMSO or CGP74514A. Data are means ± s.e.m. (n = 3). Uncropped images of blots are shown in Supplementary Information, Fig. S6.
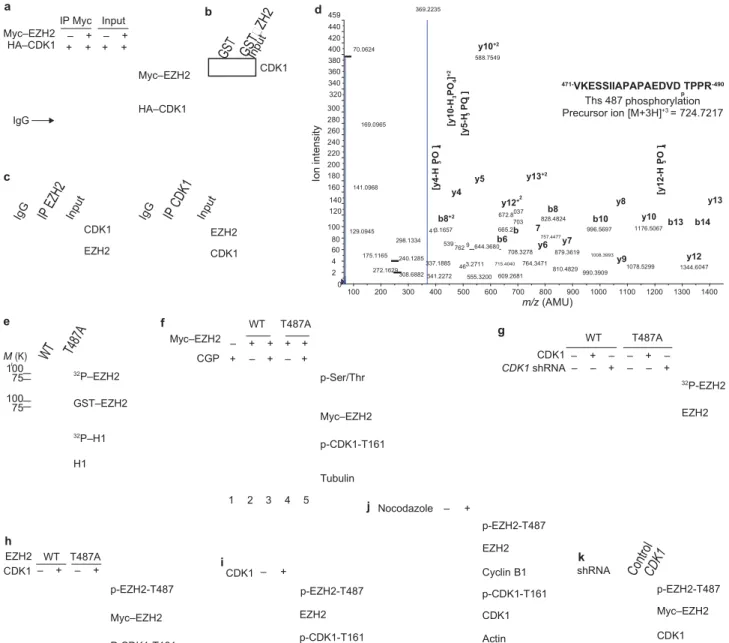
methyltransferase (HMTase) activity of EZH2 through phosphorylation of EZH2. We first examined whether CDK1 physically interacts with EZH2 by a co-immunoprecipitation experiment, which demonstrated an association between Myc-tagged EZH2 and haemmagglutinin (HA)- tagged CDK1 (Fig. 2a). We further validated the interaction using a GST pulldown assay to demonstrate that CDK1 binds to EZH2 (Fig. 2b). Importantly, this association was also detected with endogenous CDK1 and EZH2 by reciprocal immunoprecipitation, indicating that these two molecules interact in
vivo (Fig. 2c).
Next, we investigated whether CDK1 is able to phosphorylate EZH2. An in vitro kinase assay demonstrated that CDK1 catalysed the phos- phorylation of EZH2, but not glutathione S-transferase (GST; data not shown). Mass spectrometry analysis was used to identify which residue
in EZH2 is phosphorylated. We found that Thr 487 of EZH2 was phos-phorylated both in vivo (Supplementary Information, Fig. S2a), and
in vitro by CDK1 (Fig. 2d). When we replaced the Thr 487 residue
with alanine (EZH2T487A), CDK1 was no longer able to
phosphorylate the mutant EZH2, as assessed by in vitro CDK1 kinase assay (Fig. 2e). In cell culture, inhibition of CDK1 with CGP74514A also reduced the phos- phorylation of wild-type EZH2 (Fig. 2f; lanes 2 and 3) and again, the phosphorylation level of EZH2T487A mutant
was virtually undetectable in an immunoblot using antibody against phosphorylated serine/threo- nine (Fig. 2f; lanes 4 versus 2). We also compared the phosphorylation level of wild-type EZH2 and EZH2T487A
by in vivo 32P-labelling experi- ments. Phosphorylation of wild-type
EZH2 occurred in vivo, but was inhibited by expression of CDK1 shRNA (Fig. 2g). The phosphorylation
b8+2 3.1657 539 .762 46 2 037 703 b 047 r Io n in te ns ity [y 4-H P O ] 3 4 [y 10 -H P O ] + 2 3 4 [y 5-H P O ] 3 4 [y 12 -H P O ] 3 4 4 2 a Myc–EZH2 HA–CDK1 IP Myc Input – + – + + + + + b Myc–EZH2 CDK1 d 459 440 420 400 380 360 340 70.0624 369.2235 y10+2 588.7549 471-VKESSIIAPAPAEDVD TPPR-490 320 Ths 487phosphorylationp IgG c HA–CDK1 300 280 260 240 220 200 180 160 169.0965 141.0968 y5 y13+2 y4 Precursor ion [M+3H]+3 = 724.7217 140 120 y12 + 672.8 828.4824b8 y8 b10 y10 y13 b13 b14 CDK1 EZH2 100 80 129.0945 41 298.1334 665.2 b6 7 757.4477 996.5697 1176.5067 EZH2 CDK1 60 175.1165 9 644.3680 708.3278y6 y7879.3619 1008.3993 y12 240.1285 337.1885 272.1629 308.6882 341.2272 0 3.2711 555.3200 715.4040 764.3471 609.2681 810.4829990.3909 y9 1078.5299 1344.6 100 200 300 400 500 600 700 800 900 1000 1100 1200 1300 1400 m/z (AMU) e f WT T487A g Myc–EZH2 – + + + + WT T487A M (K) 100 75 32P–EZH2 CGP + – + – + p-Ser/Thr CDK1 – CDK1 shRNA – + – – + – + – – – + 32P-EZH2 100 75 GST–EZH2 32P–H1 H1 Myc–EZH2 p-CDK1-T161 Tubulin EZH2 h EZH2 WT T487A 1 2 3 4 5 i j Nocodazole – + p-EZH2-T487 EZH2 k CDK1 – + – + p-EZH2-T487 Myc–EZH2 P-CDK1-T161 CDK1 – + p-EZH2-T487 EZH2 p-CDK1-T161 Cyclin B1 p-CDK1-T161 CDK1 Actin shRNA p-EZH2-T487 Myc–EZH2 CDK1
Figure 2 CDK1 interacts with, and phosphorylates, EZH2 at Thr 487. (a) Lysates from 293T cells, transfected with plasmids encoding Myc–EZH2 and HA–CDK1 as indicated, were immunoprecipitated (IP) using anti-Myc and analysed by immunoblot using anti-Myc or anti-HA. IgG;
immunoglobulin
G. (b) Pulldowns. GST and GST–EZH2 were incubated with lysate from HeLa cells. Bound CDK1 was detected by immunoblotting. (c) Lysate from MCF7 cells was immunoprecipitated with antibodies against EZH2 (left)
or CDK1 (right), and analysed by immunoblotting. (d) Cyclin B, CDK1 and GST–EZH2 were subjected to an in vitro kinase assay and analysed by mass spectrometry. The spectrum of the charged ion (m/z 724.7217) shows
that Thr 487 is phosphorylated (lower case p) in the indicated peptide (top right). b ions, fragmentation ions containing the amino terminus of the peptide; y ions, fragmentation ions containing the carboxy terminus of the peptide. (e) In vitro kinase assay with CDK1, cyclin B, and wild-type GST–EZH2 (WT) or GST– EZH2T487A. Phosphorylation of EZH2 and H1
was visualized by autoradiography, and loading of GST–EZH2 and H1 was assessed by Coomassie-stained gel. (f) 293T cells were transfected with plasmids encoding wild-type Myc–EZH2 or Myc–EZH2T487A and treated with
CDK1 inhibitor CGP74514A or DMSO. Lysates were immunoprecipitated with anti-Myc and analysed by immunoblotting. (g) MCF7 cells stably expressing wild-type Myc–EZH2 or Myc–EZH2T487A were transfected with control vector or plasmids encoding CDK1 and cyclin B, and infected with lentivirus expressing CDK1 shRNA, as indicated. Cells were labelled with [32P]-orthophosphate, EZH2 was immunoprecipitated from lysates with anti- Myc and analysed by autoradiography. Immunoblotting was used to confirm equal loading of EZH2 (bottom). (h) HeLa cells expressing Myc– EZH2 or Myc–EZH2T487A were transfected with plasmid encoding CDK1 and cyclin B, or control vector. Cell lysates were immunoprecipitated with anti-Myc and immunoblotted. (i) Lysates of HeLa cells transfected with plasmid encoding CDK1 and cyclin B, or control vector, were
immunoprecipitated with anti- EZH2, and immunoblotted. (j) Immunoblot of lysates from HeLa cells
treated with Nocodazole as indicated. Cell lysates were immunoprecipitated with anti-EZH2. (k) MCF7 cells stably expressing wild-type Myc–EZH2 were infected with lentivirus expressing control or CDK1 shRNA. Cell lysates were immunoprecipitated with anti-Myc and immunoblotted. Uncropped images
of blots are shown in Supplementary Information, Fig. S6.
level of EZH2T487A was much lower than that of wild-type EZH2 and
was not altered by expression of CDK1 shRNA (Fig. 2g). These results suggested that Thr 487 is a major phosphorylation site of EZH2 in vivo
To confirm that CDK1 does indeed phosphorylate EZH2 at Thr 487 in vivo, we generated an antibody that specifically recognizes EZH2 phos- phorylated at Thr 487, but fails to detect an unphosphorylated EZH2 pep- tide (Supplementary Information, Fig. S2b). To confirm the specificity of
C el l n u m b er R el at iv e m R N A l ev el s C o n tr o l W T T 48 7A C el l n u m b er C o n tr o l W T T 48 7A C o n tr o l W T T 48 7A R el at iv e H O X A 7 m R N A l ev el s R el at iv e o cc u p an cy a t H O X A 7 C o n tr o l W T T 48 7A C o n tr o l W T T 48 7A R el at iv e H O X A 11 m R N A l ev el s C o n tr o l W T T 48 7A R el at iv e H O X A 9 m R N A l ev el s R el at iv e o cc u p an cy a t H O X A 9 C o n tr o l W T T 48 7A C o n tr o l W T T 48 7A C o n tr o l W T T 48 7A a CGP – + – +WT T487A b EZH2
WT T487A c Nickel-beads purification
WT T487A Input WT T487A Ratio 1 1.5 1.7 1.8 H3K27me3 Myc-EZH2 p-CDK1-T161 Actin 32P–H1 1 2 4 1 2 4 1 1.3 1.5 2.1 2.4 3.6 1 1.2 1.3 1.6 2.2 2.4 His–EZH2 SUZ12 EED CDK1 – + – + CDK1 – + – + His–EZH2 Ratio 1 0.6 1.7 1.7 SUZ12 Ratio 1 0.6 1.5 1.5 EED His–EZH2 SUZ12 EED p-CDK1-T161 HA–CDK1 Actin H1 1 2 3 4 1 1.8 3.1 1.9 3.0 5.2 1 2 3 4 4 6 3H-methyl Oligonucleosome Ratio 1 0.6 1.8 1.8 1 2 3 4 3H-methyl Oligonucleosome d e f 1.6 1.4 0.20 0.18 0.16 0.125 H3K27me3 H3 Myc-EZH2 Tubulin 1.2 1.0 0.8 0.6 0.4 0.2 0.0 0.14 0.12 0.10 0.08 0.06 0.04 0.02 0.00 0.100 0.075 0.050 0.025 0.000 g S G2/M – 0 2 4 7 8 10 h
HOXA7 HOXA9 HOXA10
h i
IgG EZH2 H3K27me3 IgG EZH2 H3K27me3
p-EZH2-T487 Myc–EZH2 Cyclin E Cyclin B1 Tubulin p-EZH2-T487 EZH2 Cyclin B1 Cyclin E Actin 2.00 1.75 1.50 1.25 1.00 0.75 0.50 0.25 0.00 G1 S G2/M G1 S G2/M 2.00 1.75 1.50 1.25 1.00 0.75 0.50 0.25 0.00 G1 S G2/M G1 S G2/M
j Control WT T487A k Control WT T487A WT T487A
1.25 WT T487A 50 P < 0.05 60 40 50 30 40 30 20 20 P < 0.01 1.00 0.75 0.50 0.25 0.00 G1 S G2/M G1 S G2/M 10 0 Control WT- EZH2 T487AEZH2 10 0 Control WT- EZH2 T487AEZH2
WT T487A
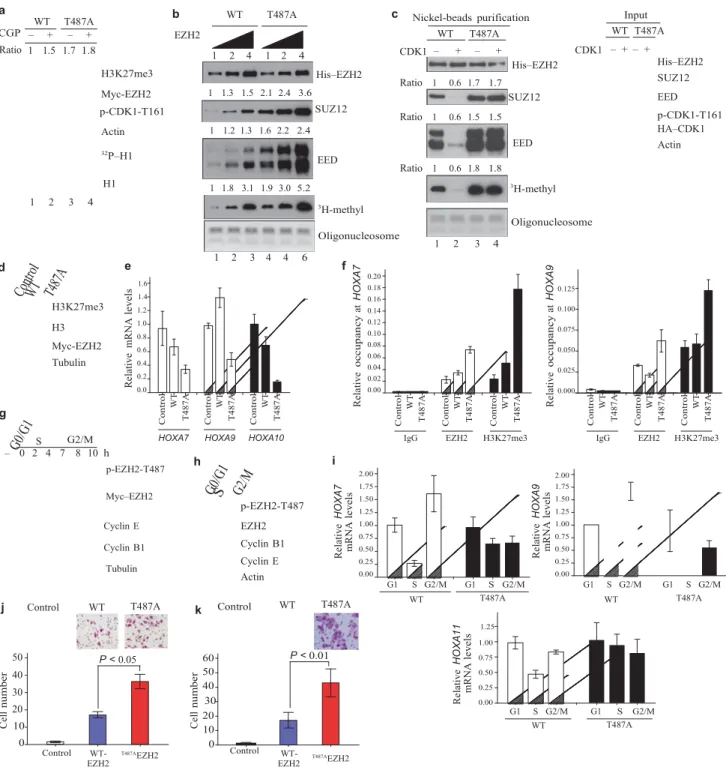
Figure 3 CDK1-mediated phosphorylation of EZH2 promotes disassociation of EZH2 from SUZ12 and EED and suppresses EZH2 HMTase activity. (a) HeLa cells were transfected with plasmids encoding wild-type EZH2 or
EZH2T487A and treated with CGP74514A as indicated. Top: immunoblot.
Relative intensity of the H3K27me3 bands is indicated. Bottom: Autoradiograph and immunoblot of in vitro kinase assay using H1 as substrate. (b) In vitro histone methyltransferase assay. PRC2 complexes were purified from MCF7 cell lines stably expressing wild-type Myc-His– EZH2 or Myc-His–EZH2T487A and 0.5,
1 and 2 μg were used in the assay. Top: immunoblots of purified proteins. Bottom: proteins were incubated with oligonucleosome and 3H-labelled
S-adenosylmethionine. Methylation was assessed by autoradiography and oligonucleosome loading by Coomassie-stained gel. Relative intensities of bands are indicated at the top of each panel. (c) MCF7 cell lines as in b were transfected with control vector or plasmids encoding HA–CDK1. Left: immunoblots of experiment performed as in b. Right: immunoblot of lysates
from MCF7 cells used in protein purification. (d) Immunoblot of lysates
stable MCF7 transfectants, established by transfection of cells with control vector or plasmids encoding Myc–EZH2 or Myc–EZH2T487A. (e) qRT–PCR of HOXA genes in MCF7 cell lines described in d. Data are means ± s.d. from three individual experiments. (f) Quantitative chromatin immunoprecipitation analysis on HOXA7 and HOXA9 promoters in MCF7 cell lines as described in d. Data are means ± s.d. from three individual experiments. (g) Immunoblot
of MCF7 cell lines stably expressing wild-type EZH2, treated by serum starvation (to collect cells at G0/G1 phase) or by double thymidine blockage and release (to collect cells at S and G2/M phases). (h) Immunoblot of
HeLa cells treated as in g. (i) qRT–PCR of HOXA gene expression in MCF7 stable cell lines treated as in g. Data are means ± s.d. from three individual experiments. (j) Images and quantification of cell migration of MCF7 stable cell lines. Data are means ± s.e.m. from three individual experiments. (k) Images and quantification of cell invasion of MCF7 stable cell lines. Data are means ± s.e.m. from three individual experiments. Uncropped images of blots are shown in Supplementary Information, Fig. S6.
nature cell biology VOLUME 13 | NUMBER 1 | JANUARY 2011
© 2011 Macmillan Publishers Limited. All rights reserved.
91 C o nt ro l C D K 1 hM S C O st e o bl a st h M S C O st e o bl a st Ig G hM S C O st e ob la st Ig G hM S C O st e ob la st
the antibody, it was incubated with wild-type Myc–EZH2 or non-phospho- rylatable Myc–EZH2T487A, which were resolved on a SDS–
PAGE gel and immunobloted. The signal from the phospho-EZH2-Thr 487 antibody alone was lowered when incubated with phosphorylated peptide, but not unphosphorylated petide (Supplementary Information, Fig. S2c). Using the phospho-EZH2-Thr 487 antibody, we immunoblotted lysates from HeLa cells co-transfected with plasmids encoding CDK1 and EZH2 or EZH2T487A,
which demonstrated that CDK1 stimulated the Thr 487 phosphorylation of wild-type EZH2, but not EZH2T487A (Fig. 2h). The
endogenous level of phosphorylated EZH2 at Thr 487 was also increased when CDK1 was overexpressed (Fig. 2i). Activation of CDK1 by treatment of cells with Nocodazole, which arrests cells in G2/M phase, can also induce
endog-a b p-CDK1-T161 CDK1 EZH2 p-EZH2-T487 H3K27me3 Tubulin A B C D E F A hMSC only B OM C OM + Control shRNA D OM + CDK1-A01 shRNA E OM + CDK1-B01 shRNA F OM + CDK1-DO1 shRNA
enous EZH2 phosphorylation at Thr 487 (Fig. 2j). Furthermore, depletion of endogenous CDK1 by transduction of cells with CDK1 shRNA reduced EZH2 phosphorylation (Fig. 2k), suggesting that endogenous CDK1 is required for EZH2 phosphorylation at Thr 487. These data indicate that
c OM – + + +
d Input IP: EZH2
EZH2 is phosphorylated at Thr 487 by CDK1 both in vitro and in vivo. Next, we investigated whether EZH2 phosphorylation mediates the ability of CDK1 to inhibit H3K27 trimethylation. Consistently, we
shRNA – – p-CDK1-T161 CDK1 1.0 0.4 SUZ12 EZH2
observed that the H3K27 trimethylation level was higher in HeLa cells
transfected with plasmids encoding the mutant EZH2T487A than in cells
transfected with plasmids encoding wild-type EZH2 (Fig. 3a; lanes 1 and
3). Additionally, inhibition of CDK1 with CGP74514A enhanced H3K27
trimethylation in cells transfected with plasmids encoding wild-type EZH2, but not in cells transfected with plasmids encoding EZH2T487A
(Fig. 3a; lanes 1–4 of Fig. 3a are from the same gel). Together, these
results suggest that phosphorylation of EZH2 at Thr 487 by CDK1 leads to a decrease in H3K27 trimethylation. 1 2 3 4 EZH2 p-EZH2-T487 H3K27me3 Runx2 Osteopontin Tubulin
Input IP: EZH2
EED Heavy chain EZH2
As CDK1 suppression of H3K27 trimethylation is mediated by EZH2, we investigated whether phosphorylation of EZH2 by CDK1 alters the HMTase activity of EZH2. An in vitro HMTase activity assay with oligo- nucleosome as the substrate showed that the EZH2T487A
mutant exhibited higher HMTase activity than that of wild-type EZH2 (Fig. 3b bottom, lanes 4–6 versus lanes 1–3; Fig. 3c, lane 3 versus 1). Activation of CDK1 by co-transfection of plasmids expressing cyclin B and CDK1 inhibited HMTase activity of wild-type EZH2 (Fig. 3c; lane 2 versus 1), but not EZH2T487A ( Fig. 3c; lane 4
versus 3). In addition, cells stably expressing EZH2T487A also exhibited
higher H3K27 trimethylation than that of wild- type EZH2 (Fig. 3d). Together, these results suggest that phosphorylation of EZH2 at Thr 487 by CDK1 inhibits HMTase activity of EZH2.
EZH2 forms a complex with SUZ12 and EED (PRC2) in vivo1,2 and
a stable complex is required for its HMTase activity. As CDK1-mediated EZH2 phosphorylation did not change the protein level of EZH2, but inhibited its HMTase activity, we next investigated whether phosphoryla- tion might interrupt the interaction of EZH2 with SUZ12 and EED. We examined the interaction of EZH2 with SUZ12 and EED through purifi- cation of Myc-His-tagged EZH2 complex from cell lines stably expressing wild-type EZH2 and EZH2T487A. There was
stronger binding between SUZ12, EED and EZH2T487A when
compared with wild-type EZH2 (Fig. 3b; lanes 4–6 versus lanes 1–3 and Supplementary Information, Fig. S3a and Fig. 3c; lanes 3 versus 1). Activation of CDK1 led to the inhibition of wild-type EZH2
binding to SUZ12 and EED (Fig. 3c; lane
2 versus 1), but did not affect the binding of EZH2T487A ( Fig. 3c; lane
4 versus 3). These data support the hypothesis that phosphorylation of EZH2 by CDK1 inhibits its HMTase activity through disruption of the binding between EZH2 and SUZ12 and EED.
nature cell biology VOLUME 13 | NUMBER 1 | JANUARY 2011
© 2011 Macmillan Publishers Limited. All rights reserved.
92 Figure 4 Phosphorylation of EZH2 by CDK1 promotes osteogenic
differentiation
of human mesenchymal stem cells. (a) Osteoblast differentiation medium induces activation of CDK1 in hMSCs. Immunoblot of lysates from undifferentiated cells or cells differentiated into osteoblasts or adipocytes. (b) hMSCs were left untreated or were treated with osteoblast differentiation medium (OM) and shRNA as indicated. Alizarin Red S staining was performed at day 7. Differentiated stem cells positive for Alizarin Red S are stained red. (c) Effect of CDK1 knockdown on the expression of osteogenic-specific genes in hMSCs. Cells were cultured in control medium or osteoblast differentiation medium, and infected with lentiviruses expressing control or CDK1 shRNA as indicated. Cell lysates were subjected to immunoblot analysis. (d) Disruption of PRC2 complex after osteogenic differentiation. Lysates from cells undifferentiated or differentiated into osteoblasts were immunoprecipitated with EZH2 antibody and subjected to immunoblot analysis as indicated.
To examine whether EZH2 phosphorylation mediated by CDK1 affects its transcriptional repressor function, we analysed expression levels of EZH2-target HOXA genes using qRT–PCR. We found that a number of genes in HOXA clusters are repressed by expression of EZH2T487A (Fig. 3e). Consistent with these results, we found that there
was high EZH2 and H3K27 trimethylation on the HOXA7 and
HOXA9 promoters, as detected by a quantitative chromatin
immunoprecipitation (qChIP) assay (Fig. 3f). These results suggest that phosphorylation of EZH2 at Thr 487 by CDK1 promotes dissociation of PRC2 and results in inhibition of its HMTase activity, which in turn derepresses genes silenced by EZH2.
As CDK1 functions as a cell-cycle kinase during G2/M phase, and also phosphorylates EZH2, we next investigated whether CDK1-mediated phosphorylation of EZH2 was regulated during the cell cycle (Fig. 3g, h). In cells with ectopically and endogenously expressed EZH2 phospho- rylation of EZH2 at Thr 487 was low in G0/G1 and S phase, but much higher in G2/M phase. This is consistent with the expression level of
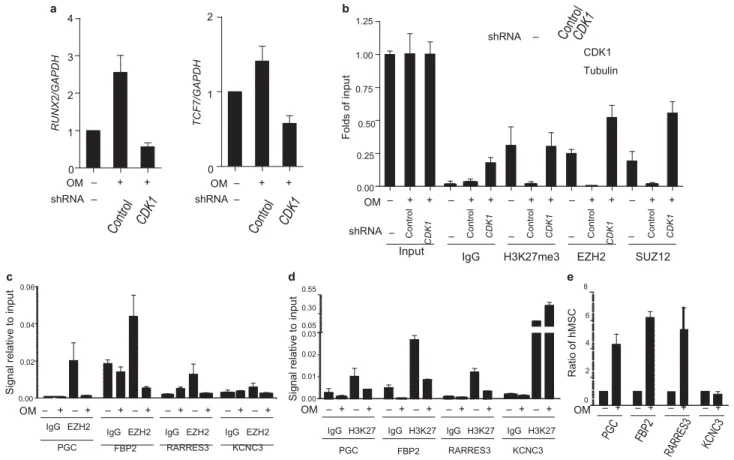
S ig na l r el at iv e to in p u t R U N X 2/ G A P D H T C F 7/ G A P D H S ig na l r el at iv e to in p u t F ol ds o f in p u t C o n tr o l C D K 1 C o n tr o l C D K 1 C o n tr o l C D K 1 R at io o f hM S C C o n tr o l C D K 1 C o n tr o l C D K 1 a b 4 2 1.25 3 1.00 2 1 0.75 shRNA – CDK1 Tubulin 0.50 1 0 OM – + + 0 OM – + + 0.25 0.00 shRNA – shRNA – OM – + + – + + – + + – + + – + + shRNA – – – – –
Input IgG H3K27me3 EZH2 SUZ12
c d e 0.06 0.55 8 0.04 0.02 0.30 6 0.05 0.03 4 0.02 2 0.01 0.00 OM – + – + – + – + – + – + – + – + 0.00 OM – + – + – + – + – + – + – + – + OM – +0 – + – + – + IgG EZH2
IgG EZH2 IgG EZH2 IgG EZH2 IgG H3K27 IgG H3K27 IgG H3K27 IgG H3K27
PGC FBP2 RARRES3 KCNC3 PGC FBP2 RARRES3 KCNC3
Figure 5 CDK1 regulates EZH2-target gene expression in human mesenchymal stem cells. (a) Effect of CDK1 knockdown on the expression of EZH2-target genes in hMSCs. Cells were cultured in control medium or osteoblast differentiation medium with or without CDK1 shRNA. mRNA
levels of RunX2 (left) and TCF7 (right) were measured by qRT–PCR, and are calculated relative to GAPDH expression. Data are means ± s.e.m. (n = 3).
(b) Effect of CDK1 knockdown on the binding of EZH2 to RunX2 gene promoter in hMSCs. Cells were cultured in control medium or osteoblast differentiation medium with or without CDK1 shRNA infection.
Quantitative
chromatin immunoprecipitation was performed using antibodies against the indicated proteins on the RunX2 promoter. Levels of CDK1 are shown by immunoblot (top). (c, d) Four genes shown to have differential EZH2 binding after osteogenic differentiation from a genome-wide ChIP-on-chip assay
were randomly selected for ChIP assay using antibodies against EZH2 (c) and H3K27me3 (d). hMSCs were cultured in control medium or osteoblast differentiation medium. (e) The expression of the four genes analysed in c and d was assessed by qRT–PCR from hMSCs cultured in control medium or osteoblast differentiation medium.
cyclin B1, which is known to activate CDK1 activity in G2/M phase. We found phosphorylation of EZH2 at Thr 487 occurred during the cell cycle in cells stably expressing wild-type EZH2, but not in cells express- ing EZH2T487A (Supplementary Information Fig. S3b).
In addition, the expression of several HOXA genes was regulated during the cell cycle, with much higher expression level in G2/M phase than in S phase, which correlated with the highest level of EZH2 phosphorylation and the lowest level of H3K27 trimethylation in G2/M phase (Fig. 3i and Supplementary Information Fig. S3b). These results suggest that CDK1 inhibits EZH2 activity and derepresses its target gene expression during cell cycle.
As EZH2 is known to enhance cell proliferation, migration and inva- sion4,5,6, and phosphorylation of EZH2 by CDK1 at Thr 487
inhibits EZH2 HMTase activity, we further investigated the effect of the T487A mutation on EZH2-mediated biological functions. Using stable trans- fectants expressing wild-type EZH2 and EZH2T487A, we
did not observe significant differences in cell proliferation (data not shown). However, expression of EZH2T487A significantly enhanced
cancer cell migration and invasion (Fig. 3j, k). These results suggest that phosphorylation of EZH2 at Thr 487 by CDK1 inhibits cancer cell migration and invasion.
EZH2 has an important role in regulating stem cell pluripotency and differentiation12–14. Human bone-marrow-derived mesenchymal
stem cells (hMSCs) are multipotent cells15. They differentiate into
adipogenic or osteogenic lineages when cultured under defined in
vitro conditions16–18. hMSCs differentiate into the osteogenic lineage
when cul- tured in osteoblast differentiation medium19. However, the
mechanisms governing hMSC differentiation are not well understood. Therefore, we investigated whether CDK1 regulation of EZH2 might have a role in hMSC differentiation. Treatment of hMSCs with osteoblast differentiation medium, but not adipocyte differentiation medium, resulted in sustained CDK1 activation (Fig. 4a). The activation of CDK1 was accompanied by an increase of EZH2 phosphorylation at Thr 487 and a decrease of H3K27 trimethylation (Fig. 4a). To determine whether the CDK1–EZH2 path- way is required for osteogenic differentiation, we used CDK1 shRNAs to knockdown endogenous CDK1 and measured the effects on hMSC osteogenic differentiation. Continuous incubation of hMSC with oste- oblast differentiation medium for 7 days resulted in positive Alizarin Red S staining (a standard marker for osteogenic differentiation; Fig. 4b, B), as well as increased expression of osteogenic marker genes, RunX2 and osteopontin (Fig. 4c, lanes 1 and 2), whereas knockdown of CDK1 by three different shRNAs blocked osteogenic differentiation (Fig. 4b, D–F). The knockdown of CDK1 by shRNA led to decreased EZH2 phosphorylation, increased H3K27 trimethylation and repression of the osteogenic mark- ers, Runx2 and osteopontin (Fig. 4c, lanes 3 and 4). Similar results were seen in multiple primary cultured hMSCs (Supplementary Information,
Fig. S4a, b and data not shown). Together, the results suggest that the phosphorylation of EZH2 by CDK1 is critical for the osteogenic differ- entiation of hMSC.
Next, we investigated whether activation of CDK1 by osteogenic dif-ferentiation also led to disruption of the PRC2 complex. Indeed, the interaction of EZH2 with SUZ12 and EED was decreased when hMSCs were induced to osteodifferentiation (Fig. 4d). These results support the hypothesis that activation of CDK1 promotes osteogenic differentiation through disruption of the PRC2 complex.
Several genes have been shown to be putative targets of EZH2, such as RunX2 and TCF7, which are known to be important for osteogenesis12. To further investigate whether activation of CDK1
promotes osteogenic differentiation through disruption of the PRC2 complex, we quantified the expression of EZH2-target genes during osteogenesis by qRT–PCR. The expression of RunX2 and TCF7 mRNAs significantly increased dur- ing osteogenesis (Fig. 5a). Knockdown of CDK1 by shRNA significantly inhibited the effect of osteoblast differentiation medium on the increased expression of
RunX2 and TCF7 (Fig. 5a). Consistently, a qChIP assay indicated
that osteogenic differentiation resulted in decreased binding of EZH2, SUZ12 and H3K27 trimethylation at the promoter of RunX2 (Fig. 5b). These changes were reversed with knockdown of CDK1 by shRNA (Fig. 5b). These results suggest that CDK1 regulates osteogenesis through suppression of methyltransferase activity of EZH2 and promotes derepression of target genes of EZH2 such as osteogenesis-related genes RunX2 and TCF7.
To further expand our findings, we carried out genome-wide screen-ing to identify EZH2-target genes in hMSCs before and after osteogenic differentiation. We performed ChIP using EZH2 antibody to immuno- precipitate EZH2 and its associated DNA, followed by hybridization with promoter microarrays (ChIP-on-chip). A large number of genes (> 4,000) bind to EZH2, but after osteogenic differentiation less than 30 genes bound (Supplementary Information, Tables S1 and S2), suggest- ing that EZH2 is disassociated from promoters of its target genes after osteogenic differentiation.
To validate the enrichment profiles of EZH2-target genes, we ran-domly selected nine EZH2-targeted genes that were bound by EZH2 in hMSC, but lost binding to EZH2 after osteodifferentiation, and quanti- fied for EZH2 and H3K27me3 enrichments in an independent experi- ment (Supplementary Information, Table S3). In eight of the nine genes the qChIP experiments confirmed the ChIP-on-chip results, with very similar decreased binding of EZH2 (Fig. 5c) and H3K27me3 (Fig. 5d) at these gene promoters after osteogenic differentiation. Consistently, the mRNA expression of these eight genes increased significantly dur- ing osteogenesis (Fig. 5e). The data from the three genes that were scored positive, PGC, RARRES3 and FBP2, are shown in Figure 5c–e. Similar results were observed for the other five genes (Supplementary Information, Table S3, Fig. S5a, b, c).
In this study, we demonstrated a novel signal pathway in which the protein kinase CDK1 regulates the histone methyltransferase EZH2. We have found that CDK1 phosphorylates EZH2 at Thr 487 thereby disrupt- ing EZH2 binding with SUZ12 and EED, and leading to suppression of H3K27 trimethylation and consequent derepression of target gene expression. This CDK1–EZH2 signalling pathway is important both in cell migration and invasion in cancer cells, and in the regulation of osteogenic differentiation of hMSCs through the modulation of EZH2- targeted osteogenic gene expression.
It is worthwhile to mention that while this manuscript was under revision, an interesting report was published that shows phosphorylation of EZH2 at Thr 350 by CDK1 and CDK2 is important for recruitment of EZH2 and maintenance of H3K27me3 levels at EZH2-target loci20. Blockage of Thr 350 phosphorylation
not only diminishes the global effect of EZH2 on gene silencing, but also mitigates EZH2-mediated cell proliferation and migration20. The
impact of EZH2 Thr 350 phospho- rylation on H3K27me3 levels in target-gene promoters is not because of changes in stability, formation or intrinsic HMTase activity of PRC2 (ref. 20). In our current study, we have identified a different phospho- rylation site of EZH2 by CDK1, Thr 487, which after phosphorylation not only inhibits binding of EZH2 to its targets on the promoter, but also inhibits HMTase activity of EZH2 through disruption of the PRC2 complex. We have further demonstrated that phosphorylation of EZH2 at Thr 487 by CDK1 stimulates mesenchymal stem cell osteogenic dif-ferentiation through derepression of putative EZH2-target genes. It is not clear whether a relationship exists between these two phosphorylation sites of EZH2. A systematic study may be required to further investigate the relationship between these two sites.
METHODS
Methods and any associated references are available in the online version of the paper at h t p:// w w w .n a t u r e .c o m/n a t u r e ce l l b i o lo g y/
note: Supplementary Information is available on the nature Cell Biology website ACknoWLedgeMentS
We thank S. A. Miller and J. Hsu for editorial assistance. We thank A. M. Labaff for characterization of EED antibody. This work was supported by grants from National Institutes of Health (grant R01 CA109311), Kadoorie Charitable Foundations, National Breast Cancer Foundation Inc. and the M. D. Anderson Cancer Center/China Medical University and Hospital Sister Foundation
Funds (to M.-C.H), NSC 96-3111-B-039 and NSC 97-3111-B-039 (to M.-C.H., L.-Y.L. and S.-P.Y), NHRI-EX98-9603BC, DOH97-TD-I-111-TM003, DOH98- TD-I-111-TM002 (to L.-Y.L.), DOH97-TD-G-111-041 (to M.-C.H.), DOH99- TD-C-111-005, NSC99-2632-B-039-001-MY3 (to L. –Y.L. and M. –C.H.). In memory of Mrs Serena Lin-Guo for her courageous battle against breast cancer.
AutHor ContriButionS
C.H., Y.W. and L.-Y.L. designed the project and wrote the paper. M.-C.H. and L.-Y.L. supervised the research. Y.W. performed most of the experiments in Figs 1–3. Y.-H.C. performed mesenchymal stem cell experiments in
Figs 4 and 5 and experiments investigating the interaction between EZH2 and CDK1. C.-C.L. performed the mass spectrometry analysis. C.-Y.L. generated and characterized the phospho-EZH2 antibody. S.-P.Y. collected and characterized primary human mesenchymal stem cells. J. L., B.S., C. -C.Y. and J.-Y.Y. assisted with experiments. All authors participated in interpreting the results.
CoMPeting FinAnCiAL intereStS The authors declare no competing financial interests. Published online at http://ww w .nature.com/naturecellbiolo g y
Reprints and permissions information is available online at http://npg.nature.com/ reprintsandpermission s /
1. Cao, R. et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing.
Science 298, 1039–1043 (2002).
2. Kuzmichev, A., Nishioka, K., Erdjument-Bromage, H., Tempst, P., Reinberg, D. Histone
methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 16, 2893–2905 (2002).
3. Cao, R., Zhang, Y. The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr. Opin. Genet. Dev. 14, 155–164 (2004).
4. Varambally, S. et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. nature 419, 624–629 (2002).
5. Kleer, C. G. et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic
transformation of breast epithelial cells. Proc. natl Acad. Sci. uSA 100, 11606–11611 (2003).
6. Bracken, A. P. et al. EZH2 is downstream of the pRB-E2F pathway, essential for
7. Bachmann, I. M. et al. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J. Clin. Oncol. 24, 268–273 (2006).
8. Gonzalez, M. E. et al. Downregulation of EZH2 decreases growth of estrogen receptor-neg-ative invasive breast carcinoma and requires BRCA1. Oncogene 28, 843–853 (2009).
9. Van Hoof, D. et al. Unraveling the human embryonic stem cell phosphoproteome. Cell
Stem Cell 5, 214–226 (2009).
10. Imbach, P. et al. 2, 6, 9-trisubstituted purines: optimization towards highly potent and selective CDK1 inhibitors. Bioorg. Med. Chem. Lett. 9, 91–96 (1999).
11. van den Heuvel, S. and Harlow, E. Distinct roles for cyclin-dependent kinases in cell cycle control. Science 262, 2050–2054 (1993).
12. Bracken, A. P. et al. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 20, 1123–1136 (2006).
13. Boyer, L. A. et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. nature 441, 349–353 (2006).
14. Lee, T. I. et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell 125, 301–313 (2006). 15. Barry, F. P. & Murphy, J. M. Mesenchymal stem cells: clinical applications and biological characterization. Int. J. Biochem. Cell Biol. 36,
568–584 (2004).
16. Bruder, S. P. et al. Mesenchymal stem cells in bone development, bone repair and skeletal regeneration therapy. J. Cell Biochem. 56, 283–294 (1994).
17. Mackay, A. M. et al. Chondrogenic differentiation of cultured human mesenchymal stem cells from marrow. Tissue Eng. 4, 415–428 (1998).
18. Pittenger, M. F. et al. Multilineage potential of adult human mesenchymal stem cells.
Science 284, 143–147 (1999).
19. Jaiswal, N., Haynesworth, SE., Caplan, A. I. and Bruder, S. P. Osteogenic differentiation of purified, culture-expanded human mesenchymal stem cells in vitro. J. Cell. Biochem.
64, 295–312 (1997).
20. Chen, S. et al. Cyclin-dependent kinases regulate epigenetic gene silencing through phosphorylation of EZH2. nat. Cell Biol. 12, 1108–1114 (2010).