Iterative Refinement Techniques for Improving
Constrained Feature Based Multiple Structure Superposition
Tien-Yu Chen, Yen-Chu Hsu, Hsin-Wei Wang, and Tun-Wen Pai
*Dept. of Computer Science and Engineering, National Taiwan Ocean University
*[email protected]
Abstract
-As large amount of protein tertiary structures are available, an efficient and effective tool for protein structural alignment becomes important for the detection and classification of protein function and fold recognition. In this paper, we present a novel iterative refinement algorithm for improving the multiple protein structure superposition based on constrained features of protein sequences. This algorithm is achieved according to its initial alignment from conserved motifs, and an updated virtual center structure is created iteratively followed by optimal pairwise alignments between each query protein and the updated virtual center structure. The refined alignment converges to its best alignment results based on the measurement and identification of the minimal average root mean square deviation (RMSD) and maximum number of average aligned residues. The effectiveness of the proposed algorithm was verified by comparing the performance between the constrained features based multiple structure alignment with or without adding the extra iterative refinement techniques. Keywords: multiple structure alignment, iterativerefinement, constrained feature, virtual center structure
1. Introduction
Protein structure comparison is an important task in structural molecular biology since it can facilitate identifying functional and evolutionary relationship of proteins. Up to now, there are more than 52,000 protein structures in Protein Data Bank (PDB) [1]. Therefore, how to develop an efficient and effective structure alignment tool becomes an important research topic. Now, various multiple structure alignment tools have been proposed, such as MultiProt which can find the common geometrical cores and detect high scoring partial multiple alignments for all possible number of molecules from the input molecules [2];
CE utilizes the combinatorial extension and Monte Carlo optimization techniques to align multiple protein structures [3]; 3DSS finds the invariant and common water molecules present in the superposed homologous protein structures [4]; COMPARER employs the DiCE structural alignment program to superpose selected structures and gives output file in the JOY format; CMSFA is a fast alignment algorithm for multiple protein structure alignment based on the important conserved features from one-dimensional primary sequences [5].
In general, the problem of superposing two protein structures is considered as a rigid body transformation problem. Hence, determine an optimal transformation of the query protein (slave) to the target protein (master) is necessary. Any rigid body motion in 3D-space can be decomposed into a translation and a rotation. The translation operation can be achieved by shifting the barycenters of both proteins coincided at the origin. Hence, the rigid body transformation problem can be restated as determining an optimal rotation matrix for superposing both query and target proteins.
For a multiple structural superposition problem, to find its optimal alignment is computationally prohibitive. One categorized approach employed by many previous methods is to align short fragments pairly from all of the proteins against each other optimally, and the final alignment concatenates these fragments based on dynamic programming techniques in a geometrically consistent way. The other class of approach can be achieved by employing the center-star mechanism where the most similar structure is designated as the center structure, and all other structures can be mutually or progressively aligned with the center structure through an optimal alignment pairwisely. Therefore, an efficient and heuristic result can be obtained through such processes but not guaranteed as an optimal result. Taking an example for the second type of method, if there are three structures A, B, and C for alignment, without
losing generality, the A can be selected as the target structure and B and C are defined as the query structures. To heuristicly obtain the multiple structure alignment, the structure B is firstly superposed onto the target structure A and the structure C is also independently superposed onto the target structure A simultaneously. However, in general, the structure B could not be optimally superposed onto the structure C. To overcome such dilemma, we have proposed an iterative refinement algorithm on the multiple protein superposition to improve the alignment performance and maintain the satisfaction of its computational requirements. In this paper, we employed the method of singular value decomposition (SVD) to determine an optimal rotation matrix for pairwise alignment [6]. About the initial aligned conditions, we have utilized the constrained multiple structure feature alignment (CMSFA) algorithm which is achieved by aligning the conserved motifs as its key anchor features. Once the initially aligned positions of the multiple protein structures were obtained, according to these coordinates, a virtual target protein can be built. Then, all protein structures were superimposed onto the virtual target protein structure through an optimal rotation transformation. After the optimized transformation of all protein structures with respect to the virtual center, a decreased average RMSD value can be expected while the number of aligned residues may be increased. At a newcome stage, an updated virtual center structure can then be constructed and all the similar processes are performed iteratively until both the average RMSD value and the number of aligned residues converged. All details of the proposed method are introduced in the following sections.
2. Materials and Methods
2.1. Problem definitionsFor pairwise structure alignment, an algorithm is designed to find an appropriate transformation to superpose one structure onto the other. In general, the numbers of Cα atoms of both protein structures are not necessarily equal. To guarantee both proteins can be optimally superimposed, we utilize the method of optimal structural superposition which is only suitable for structure alignment of two proteins within an identical number of Cα atoms. It is the most adaptive methodology adapted to superimpose two protein structures within the assumption of equal number of amino acids. However, for multiple structure alignment,
the number of proteins and amino acids in each protein reveal more complicated situation and an optimal superposition becomes unapproachable as described in previous section. Hence, to employ a heuristic alignment algorithm is inevitable. In this paper, an initial alignment of multiple structures is assumed to be able to be obtained and satisfies at least 50% of average residues of all structures within a limited distance range. The default criterion of distance range in this paper is set as 3 angstrom. With the initial alignment, the common aligned Cα atoms can be identified and the number of residues in each protein is equal for all structures. According to the results of initial alignment, an optimal structural superposition between two proteins (query protein and virtual center protein) can be described in the following sections.
Given two sets of elements {x1, x2,…, xn} and
{y1,y2,…,yn} in three dimensional space which are
defined as the Cα atoms of X (master protein) and Y(slave protein), and all the element xicorresponds
to the element yi within a certain limited range.
Assuming the centroids of two proteins coincide at the origin, the next step is to find an optimal rotation matrix R from Y to X. The distance metric D for measuring is defined as follows [7]:
(
)
∑
= − = n i i i Ry x D 1 2In this paper, we employ the method of singular value decomposition (SVD) to obtain the optimal rotation matrix. This method includes following consecutive steps:
(1) Let C = XYTbe the correlation matrix of X and Y.
(2) Perform singular value decomposition on C. C = USVTwhere UUT = VVT = I, S = diag(si),
s1≧s2≧s3≧0
(3) The minimum value of D with respect to R is ( )2 ( )2 2(1 2 3) minD xi yi s s s λ + + − + = , where λ = sign(det(C))
(4) The optimal rotation matrix R is then given by R U VT = λ 0 0 0 1 0 0 0 1
aligned residues by calculating their spatial centers of each aligned residue group, and the generated set of centers is defined as a virtual center structure. Hence, all protein structures are considered as slave proteins and perform the optimal pairwise alignment individually. Once all slave proteins are transformed into newly optimized positions, an updated new virtual center structure can be formulated for next refinement procedures.
2.2. Algorithm
All various multiple structure alignment methods can employ the proposed iterative refinement algorithm for a better achievement. In this paper, the initial all-to-all pairwise structure alignments are generated using constrained multiple structural feature alignment (CMSFA) which takes the conserved motifs as the key anchors for geometrical rotation operation. It is a method of sequence based multiple structural alignment, and only successfully aligned cases are considered in this study. The successful alignment represents at least 50% of residues for each protein could be aligned within the specified range limitation. With this initial aligned information, the iterative refinement algorithm includes the following four steps.
(1) Create a virtual target structure:
If there are N structures P1, P2,…, PN possessing various size of amino acids and which were aligned by CMSFA initially, the Cα atoms of corresponding aligned residues
from each protein are considered as a groups of size N residues. Assuming that CP1, CP2,…, CPN are the partial structures which contain the correspondingly aligned residues only. The arithmetic average in spatial domain for each group form CP1, CP2,…, CPN is calculated and denoted as VC as the virtual center structure.
(2) Optimal superposition:
Perform optimized superimposing operation from CPi onto VC individually. The optimal rotation matrices R1, R2,…,RN for each protein can be calculated. Apply these rotation matrices to P1, P2,…, PN respectively as the following formula: N 1,2,..., i where , P R Pi′= i i =
(3) Average aligned residue calculation: TakeP′1as the target structure to calculate
the number of average aligned residues between P′1 andP′j where j=2,…,N by the
following formula :
∑
= = N 2 j j 1 idues ligned_res pairwise_a 1 -N 1 (aa) dues igned_resi average_al , 1 ) ( (4) Average RMSD calculation:Calculate the value of average RMSD on aligned residues within 3 angstrom range limitation betweenP′1andP′jwhere j=2,…,N
according to the following form :
∑
= = N 2 j j 1 MSD pairwise_R 1 -N 1 (ar) SD average_RM , 1 ) ( (5) Convergence verification:For each refined alignment, the average
number of aligned residues (aa)k and
average RMSD value (ar)k for the kth iterative process can be obtained. If (aa)k+1
≦ (aa)k ± (aa)k ×ε% and (ar)k+1 ≧ (ar)k
± (ar)k ×δ %, it represents that the algorithm is converged to its local optimal of minimum RMSD and maximum number of aligned residues. The tolerance ofε% andδ% can be considered as an adjustable parameter which will affect the iteration number of the proposed algorithm. On the other hand, if the results of newly aligned average residues and RMSD values from the updated iterative refinement can not satisfied the previous requirements, then the processes will continue. The pseudo codes of the main iteration is described as follows :
; ; 1 ) 1 %) ) ( ) (( & %) ) ( ) ( ) (( ; 0 1 1 k return end k k ; (4) to ( step do ar ) ar ( ar aa aa aa while k k k k k k k + = × ± ≤ × ± ≥ = + + ε δ
2.3. System description
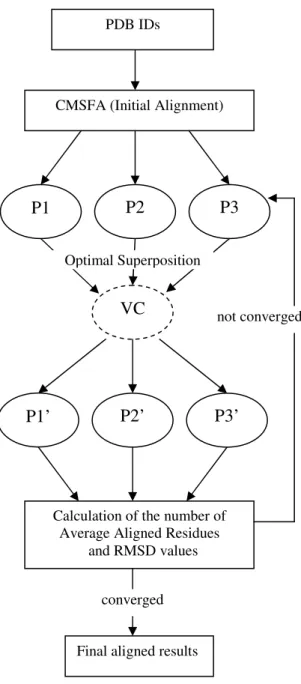
Figure 1 depicts the system configuration. The system requires importing protein structures of an interested set in PDB format. In this system, the corresponding sequence information will be extracted from PDB files for structural alignment. If three PDB sequences were inputted, the system employed CMSFA algorithm to align the multiple proteins based on sequence information initially. It provides three rotated structures P1, P2, and P3 as the initially aligned results. Next, a virtual center structure VC was created and the superimposition of P1, P2, and P3 onto VC were performed individually. Accordingly, P1’, P2’, and P3’ were obtained as the newly transformed structures through the first iteration. After comparing the number of average aligned residues and RMSD values of these two computations. If both conditions were converged and satisfied the pre-defined conditions, it would provide the final aligned results. Otherwise, the P1’, P2’, and P3’ will be updated according to a newly created virtual center structure. The iterative refinement processes will be performed until the best conditions could be achieved. It is obvious that the different initial alignment conditions will lead to different locally optimal alignment results. Therefore, a good initial alignment is quite important and dominates the performance in this study.
Figure 2 demonstrates the configuration of CMSFA system for its efficient multiple structure alignment. Again, the system requires importing IDs of a set of protein structures in PDB format. There are two main phases in CMSFA. The first phase focuses on sequence analysis. The consensus motifs among sequences were searched before hierarchical clustering operations. If the sequences under analysis contained the near neighboring proteins in addition to target protein family, the system will suggest using clustering operations to divide the near neighboring proteins into several subgroups for better performance.
The second phase includes key residue analysis, constrained multiple structure feature alignment (CMSFA). The key residues will be retrieved based on the characteristics of homologous, charged, and hydrophilic degree from the aligned consensus segments. Afterward, all protein be
structures will superimposed together rapidly by the geometry centers of those key residues. By means of the RMSD values between the target protein and the others, related biological applications can be performed.
CMSFA (Initial Alignment)
VC
P1
P2
P3
Optimal Superposition PDB IDs
P2’
P3’
P1’
Calculation of the number of Average Aligned Residues
and RMSD values
not converged
converged
Final aligned results
3. Result and Discussion
To demonstrate the convergence of the iterative refinement algorithm, we illustrated several protein superfamilies as examples.
(1) Human Ribonuclease A-like (RNase A-like):
1e21:a, 1gqv:a, 1dyt:a, 1rnf:a, 1b1i:a. (2) Serine Proteases (Subtilisin-like) :
1cse:e, 1sbn:e, 1pek:e, 3prk:e, 3tec:e. (3) Parvalbumin (EF-hand) :
1rtp, 1pva:a, 5cpv, 1pal, 5pal (4) Subtilases (Subtilisin-like) :
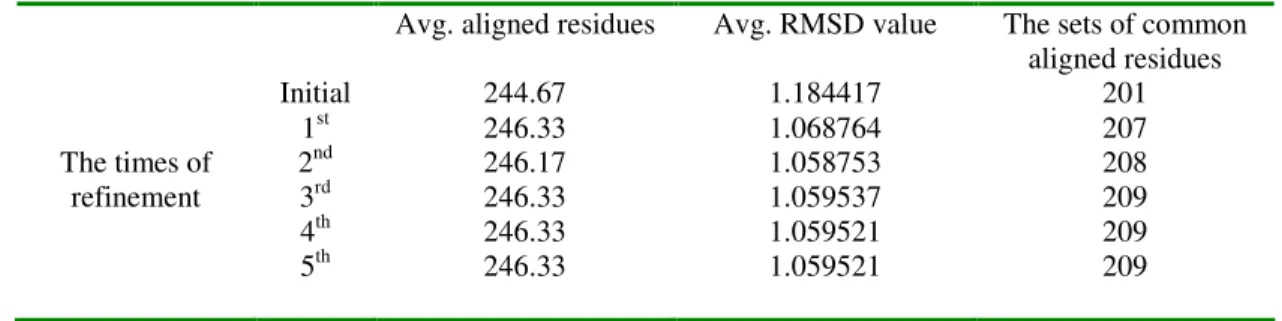
1dbi:a, 1thm, 1bh6:a, 1mee:a 1sup, 1gci, 2prk From Table 1 to 4, the number of average aligned residues and average RMSD values were shown and the common aligned residues in each cases were displayed in the last column. The second column represented the times of iterative refinement, while the third and the fourth columns denoted the increasing number of average aligned residues and decreasing values of average RMSDs respectively.
In Table 1, it can be discovered that the number of average aligned residues increased rapidly at its first refinement processes from 96.75 to 99.25, and the variation of the average number of aligned residues became obscure after the second refinement processes. The same situation occurs as the average RMSD values, the first refinement iteration decreased rapidly from 1.363832 to 1.228669, and the variation became obscure after the second refinement trial. Taking the last column for discussion, a total of 73 sets of common aligned residues was obtained from the initial alignment. After applying the iterative refinement module, there were eight more sets of common aligned residues obtained when it converged to a stable result. Both average RMSD values and number of aligned residues converged after the 5th refinement. The finally stabilized values were 98.75 and 1.197453 for the number of average aligned residues and average RMSD value.
Similarly, from Table 2 to 4, it converged to stable results as well. The iterative refinement techniques indeed accomplished our goal which facilitated to increase the number of average aligned residues and decrease the average RMSD values.
In the near future, the proposed module will be applied to various initial alignments obtained by different methods of multiple structure alignment, and the whole SCOP dataset will be taken as testing cases for a comprehensive verification of the proposed refinement algorithms for general multiple structural alignment approaches.
Table 1. Convergence of Human Ribonuclease A-like(RNase A-like) by iterative refinement algorithm.
Avg. aligned residues Avg. RMSD value The sets of common
aligned residues Initial 96.75 1.363832 73 1st 99.25 1.228669 81 2nd 99.50 1.221756 80 The times of refinement 3rd 99.00 1.206968 81 4th 98.75 1.197455 81 5th 98.75 1.197453 81 6th 98.75 1.197453 81 PDB IDs Combinatorial Feature Analysis Hierarchical Clustering
Key Residues analysis
Constrained Multiple Structure Feature Alignment (CMSFA)
Biological Applications
Subgroup Sequences
Clustering and Combinatorial Feature Finding
Consensus Motif Searching
CMFSA Alignment
Table 2. Convergence of Serine Proteases (Subtilisin-like) by iterative refinement algorithm.
Table 3. Convergence of Parvalbumin (EF-hand) by iterative refinement algorithm.
Table 4.Convergence of Subtilases (Subtilisin-like) by iterative refinement algorithm.
References
[1] H.M. Berman, J. Westbrook , Z. Feng , G. Gilliland, T.N. Bhat, H. Weissig, I.N. Shindyalov, and P.E. Bourne, “The Protein Data Bank”, Nucleic Acids Res, vol. 28, no. 1, pp.235-242, 2000.
[2] M. Shatsky, R. Nussinov, and H..J. Wolfson, “A method for simultaneous alignment of multiple protein structures”, Proteins, vol. 56, no. 1, pp.143-156, 2004.
[3] C. Guda, S. Lu, E.D. Scheeff, P.E. Bourne, and I.N. Shindyalov, “CE-MC: a multiple protein structure alignment server”, Nucleic Acids Res, vol. 32, pp.w100-w103, July, 2004.
[4] K. Sumathi1, P. Ananthalakshmi1, M.N.A.Md. Roshan1, and K. Sekar, “3dSS: 3D structural superposition”, Nucleic Acids Res, vol. 34,
[5] B. Su, T. Pai, W. Chou, D. Chang, H. Chang, and W. Chou, ”constrained multiple structure feature alignment”, National Computer Symposium, 2005.
[6] S. Umeyama, “Least-Squares Estimation of
Transformation Parameters Between Two Point Patterns”, IEEE Transactions on Pattern Analysis
and Machine Intelligence, vol. 13, no. 4, April, 1991.
[7] A.M. Lesk, “A toolkit for Computational Molecular Biology. II. On the Optimal Superposition of Two Sets of Coordinates”, Acta Cryst, A42, pp.110-113, 1986.
Avg. aligned residues Avg. RMSD value The sets of common
aligned residues Initial 247.50 1.081330 217 1st 248.25 0.989636 220 2nd 248.25 0.988898 220 The times of refinement 3rd 248.25 0.988897 220 4th 248.25 0.988897 220
Avg. aligned residues Avg. RMSD value The sets of common
aligned residues Initial 106.00 0.927511 102 1st 106.50 0.871092 103 2nd 106.50 0.870260 103 The times of refinement 3rd 106.75 0.873507 104 4th 106.75 0.873579 104 5th 106.75 0.873579 104
Avg. aligned residues Avg. RMSD value The sets of common
aligned residues Initial 244.67 1.184417 201 1st 246.33 1.068764 207 2nd 246.17 1.058753 208 The times of refinement 3rd 246.33 1.059537 209 4th 246.33 1.059521 209 5th 246.33 1.059521 209