Chapter 1
α-
L-Aarabinofuranosidase andβ-
D-xylosidase (Abf/Xyl )Plant cell walls, the major reservoir of fixed carbon in nature, have three major polymeric constituents: cellulose, hemicellulose and lignin [1]. The amount of xylan, which is the predominant hemicellulose, varies in different plants, from as much as 35% of the dry weight of birchwood to as little as 7% in some gymnosperm [2]. Besides terrestrial plants, in which xylans are based
on a β-1,4-linked D-xylosyl backbone, marine algae synthesize xylans of
different chemical structure, based on a β-1,3-linked D-xylosyl backbone [3].
Those containing a mixture of β-1,3 and β-1,4 linkages are found in seaweeds such as Rhodymenia palmata [4, 5]. In some species of the Chlorophecae and the Rhodophceae, where cellulose is absent, xylans form a highly crystalline fibrillar material [6].
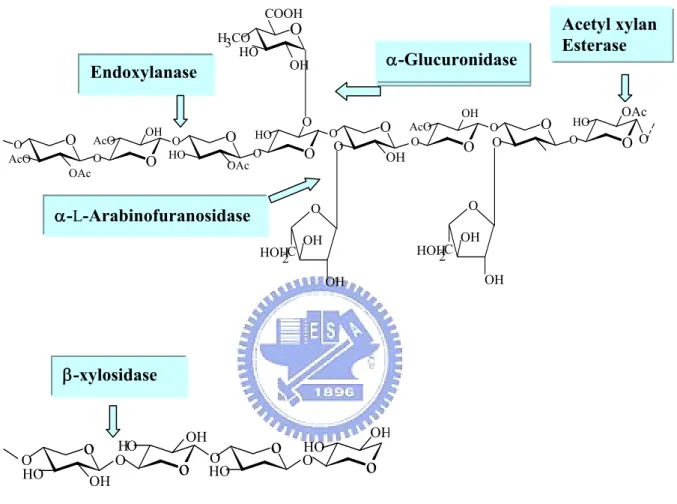
Xylan is the most common backbone structure found in hemicellulose. However, the hemicellulose structure also has branched hetero-polysaccharides, which require a more complex battery of enzymes to achieve hydrolysis. A concerted action of enzymes which randomly cleave inter-monomeric bonds (endo-enzymes), enzymes which remove monomers from the end of the chain (exo-enzymes), and enzymes which hydrolyse dimers is needed. These enzymes include endo-1,4-β-xylanase (1,4-β-D-xylan xylanohydrolase; EC 3.2.1.8), β-xylosidase (1,4-β-D-xylan
xylohydrolase; EC 3.2.1.37), L-glucuronidase, α-L-arabinofuranosidase (EC
3.2.1.55) and acetyl xylan esterase (Figure 1-1). A concerted action of hemicellulolytic enzymes is needed in order to achieve a complete degradation of branched acetyl xylan to carbohydrate monomers [7-9].
α-L-Arabinofuranosidases are exo-type enzymes, which hydrolyze
terminal nonreducing residues from arabinose-containing polysaccharides. These enzymes catalyze the hydrolysis of α-1,2 , α-1,3 and α-1,5
L-arabinofuranosidic bonds in hemicelluloses such as arabinoxylan,
L-arabinan, and other L-arabinose-containing polysaccharides [10-13]. There
is another group of enzymes which degrade arabinan by endo-fashion and are
called endo-1,5-α-L-arabinanases (1,5-α-L-arabinan
1,5-α-L-arabinanohydrolase, EC 3.2.1.99). The α-L-arabinofuranosidases
have been classified into five families of glycanases (families 3, 43, 51, 54, and 62) on the basis of amino acid sequence similarities [14]. In general, the enzymes in GH43 and GH62 hydrolyse unsubstituted α-1,5-arabinan/α-1,5-arabino-oligosaccharides [15, 16] and arabinoxylans, respectively [17], whereas the GH51 and GH54 arabinofuranosidases are believed to exhibit more relaxed substrate specifcity [18]. The two families (51 and 54) of α-L-arabinofuranosidases also differ in substrate specificity for
arabinose-containing polysaccharides. Beldman et al. [19] have classified AFases into three types depending on their mode of action and substrate
specificity. Type-A α-L-arabinofuranosidases preferentially degrade
α-1,5-L-arabinofurano-oligosaccharides to monomeric arabinose and are
inactive towards arabinosyl linkages present in polysaccharides [20,21]. In
contrast, type-B α-L-arabinofuranosidases show activity that debranches
L-arabinose residues from side chains of arabinan or arabinoxylan [22, 23].
arabinofuranohydrolase (axhA) [27] in Aspergillus niger are classified into GH51, GH54, and GH62, respectively. The N-terminal sequence of the first 50 amino acids of α-L-arabinofuranosidase from Bacillus stearothermophilus
T-6 showed high homology with the N-terminal region of
α-L-arabinofuranosidase from Streptomyces lividans 66 [28, 29]. Another
type of arabinan-degrading enzyme has been reported from Erwinia
carotovora [30]. It released arabinose trisaccharides from the nonreducing
end of 1,5-α-L-arabinan, and did not show activity on synthetic
α-L-arabinosides.
Physico-chemical characteristics of
α-
L-arabinofuranosidases
Badal C. Saha summarized the properties of some
α-L-arabinofuranosidases (Appendix 1). Multiple forms of
α-L-arabinofuranosidase have been detected in the culture broth of A.
awamori [31], A. nidulans [32], A. niger [23], A. terreus [33], and P. capsulatum [34]. The α-L-arabinofuranosidase I and
α-L-arabinofuranosidase II purified from the culture filtrate of A. awamori had
MWs of 81,000 and 62,000 and pIs of 3.3 and 3.6, respectively [31]. The
α-L-arabinofuranosidase from A. pullulans is a homodimer with an apparent
native MW of 210,000 and a subunit MW of 105,000 [35]. One
α-L-arabinofuranosidase from P. purpurogenun is a monomer of 58 kDa, with
a pI of 6.5 [36]. The α-L-arabinofuranosidase (MW 110,000) from B.
stearothermophilus L1 consists of two subunits (MWs 52,500 and 57,500),
that (MW 256,000) from B. stearothermophilus T-6 consists of four identical subunits (MW 64,000) [37, 28].
α-L-Arabinofuranosidases have a broad range of pH and temperature
dependence, with optimal activities occurring between pH 3.0–6.9 and 40–75 °C [30, 34, 35, 37]. The purified enzyme from Rhodotorula flava is highly
acid stable, retaining 82% of its activeity after being maintained for 24 h at pH 1.5 and at 30 °C [38]. Optimum activity of this enzyme is at pH 2.0. The α-L-arabinofuranosidase from Corticium rolfsii had an optimum activity at pH
2.5 toward beet arabinan [39]. The α-L-arabinofuranosidase from
Talaromyces emersonii is a dimer (MW 210 000), with a pH and temperature
optima of 3.2 and 70 °C, respectively [40]. In general, the optimal activity of Abf is at the acidic condition.
Application in biotechnology
Hemicellulases have attracted much attention in recent years because of their potential industrial use in biobleaching of paper pulp, bioconversion of lignocellulose material to fermentative products and for the improvement of animal feedstock digestibility [12, 37, 46, 47]. α-L-Arabinofuranosidases are
also capable of hydrolyzing the glycosides of monoterpenes, sesquiterpenes and other alcohols which constitute the aromatic potential of wine, and their exploitation in favor improvement and wine aromatization has been studied in the last several years [48-50].
Molecular biology of
α-
L-arabinofuranosidases
The gene encoding α-L-arabinofuranosidase and β-D-xylosidase from T. reesei RutC-30 was cloned and expressed in S. cerevisiae [41]. The deduced
amino acid sequence of α-L-arabinofuranosidase displays high-level similarity
fusion protein. The deduced amino acid sequence of the catalytic domain of the mature enzyme exhibits extensive identity with the catalytic domains of S.
coelicolor (74%), A. niger (75%), S. lividans (74%), and A. tubingensis (75%),
which are enzymes that belong to family 62 of the glycosyl hydrolases.
Debeche et al. [43] cloned and expressed in Escherichia coil an
α-L-arabinofuranosidase (abfD3) from Thermobacillus xylanilyticus. The
recombinant α-L-arabinofuranosidase (56,071-Da) could be assigned to family
51 of the glycosyl hydrolase classification system [14]. The enzyme is localized within a distinct phylogenic cluster that contains three other
α-L-arabinofuranosidases from taxonomically related bacterial sources [B.
subtilis [44], C. stercorarium [45], and B. stearothermophilus [28]]. The
α-L-arabinofuranosidase I from Streptomyces chartreusis belongs to family 51,
whereas α-L-arabinofuranosidase II from the same organism belongs to family
43 of the glycoside hydrolase family [15]. Interestingly, the Abf of family 54 are much less reported.
Catalytic mechanism of the Glycohydrolases
Glycosidases have been assigned to families on the basis of sequence similarities, there are now 106 such families defined containing over 2500 different enzymes [14]. This classification has been best described and demonstrated on the Web page (http://afmb.cnrs-mrs.fr/ _cazy/CAZY/index.html). These groups of enzymes cleave the glycosidic bond of the substrate in two different manners: retention and inversion of the anomeric configuration. Both mechanisms involve general acid-base catalysis and require two essential residues, which in most glycosyl hydrolases are aspartate and/or glutamate residues (Figure 1-2). Inverting glycosidases catalyse hydrolysis of the glycosidic bond via a single displacement reaction. One of the catalytic residues, acting as a general acid, provides protonic assistance to the departing glycosidic oxygen while the
second, acting as a general base, activates a water molecule which effects a direct displacement at the anomeric centre [51]. Retaining glycosidases, perform hydrolysis via a two-step, double-displacement mechanism. Two key active site carboxylic acid residues [52-55], one functioning as the nucleophile and the other as the general acid/base [53], are involved.
α-L-arabinofuranosidases are assigned to five glycoside hydrolase
families (GHs) 3, 43, 51, 54 and 62, based on sequence similarities [14, 56]. The GH3, GH51 and GH54 were shown to cleave the glycosidic bond with a retention of the anomeric configuration [57]. The GH43, which includes α-L-arabinofuranosidases as well as β-D-xylosidases, was shown to work via
the inverting mechanism [57], whereas the stereochemistry of GH62 is not yet characterized. Different α-L-arabinofuranosidases from bacterial, fungal and
plant sources (Appendix 1) can hydrolyze arabinofuranose moieties at O-5, O-2 and/or O-3 as single substituent, as well as from O-2 and O-3 doubly substituted xylans and xylo-oligomers and arabinans [15, 25, 31, 58, 59]. However, very little is known about the exact biochemical mechanism and the specific catalytic residues involved in the reaction mechanism. One of the major reasons, in the case of α-L-arabinofuranosidases, is the difficulty in
obtaining synthetic substrates bearing different leaving groups, which could act as an essential tool in the identification of catalytic residues of glycosidases [60, 61].
Several strategies for the identification of such residues in other glycosyl hydrolases have been described. For retaining glycosidases, fluorinated
of AbfD3 from Thermobacillus xylanilyticus is an arabinoxylan-debranching enzyme which belongs to family 51 of the glycosyl hydrolase classification [65].
Crystal structures of native or mutant enzymes in complex with substrates, products, non-hydrolyzable substrate-analogues and transition-state analogues provide valuable detailed information regarding the specificity, binding mechanism and transition-state stabilization in retaining glycosidases,
as recently reviewed by Vasella et al. [66]. The α-L-arabinofuranosidase
from Geobacillus stearothermophilus T-6 (AbfA) belongs to the retaining GH-51 family, and its catalytic residues were recently identified, Glu175 is the acid/base, and Glu294 is the nucleophile [67, 68]. Klaus Hovel described high-resolution (1.2-2.0 Å) crystal structures of native and catalytic mutant of AbfA in complex with different substrates. The enzyme is a hexamer, and
each monomer is organized into two domains: a (β/α)8-barrel and a
12-stranded β sandwich with jelly-roll topology. These structures include
the Michaelis complexes with natural and synthetic substrates and the transient covalent arabinofuranosyl-enzyme intermediate with a non-fluorinated substrate. The structures allow thorough examination of the catalytic mechanism, including the two stable states of the glycosylation step, the interactions mediating substrate distortion and the features governing substrate specificity [69].
Recently, the potential for mechanistic studies on the family 54 hydrolases has increased greatly, owing to the complete resolution of the
three-dimensional structure of bifunctional β-D-xylopyranosidase and
α-L-arabinofuranosidase from Aspergillus kawachii IFO4308 (Figure 1-3)
[70]. On the basis of this structure and very preliminary mutagenic studies, Glu-221 and Asp-297 were suggested as candidates for the nucleophile and the general acid/base catalytic residues, respectively. Thirteen enzymes with phylogenetic similarity were chosen and compared. The result of the amino
acid sequence alignment is shown in Appendix 2. Twenty seven residues [D25, D60, D80, D95, D120, D160, E163, D170, D181, E186, D191, E198, D221, E223, D238, E289, D299, E310, D320, E323, E407, D410, D429, E436, D437, D482 and D490 (numbering in abf sequence)] are found to be highly conserved. As compared with the sequence of the Aspergillus kawachii
IFO4308 α-L-arabinofuranosidase and the inspected X-ray structure, D219,
E221 and D297 in Aspergillus kawachii IFO4308 enzyme, corresponding to D221, E223 and D299 in Trichoderma koningii G-39 enzyme are present in the active site. The function of Glu-223 as a nucleophile, which is conserved in the `DXE' (Glu-X-Asp) sequences of family 54 enzymes, has not yet been confirmed by means of site-directed mutagenesis. In order to gain a better understanding of the detailed mechanism and of the active-site topology of the family 54 hydrolases, an expressed and purified enzyme is essential. The
bifunctional β-D-xylopyranosidase and α-L-arabinofuranosidase from
Trichoderma koningii G-39 is one such suitable candidate. Here we report
the development of a recombinant P. pastoris clone for the expression of recombinant abf gene and combine physical and chemical studies to investigate the catalytic mechanism of the enzyme, and propose Glu-223 and Asp-299 as the essential nucleophile and general acid/base, respectively, as determined by site-directed mutagenesis and kinetic study.
O O O O AcO OAc AcO OH OAc O O O O HO HO O O OHO O OH AcO O O O O O COOH H3CO HO OH O O OH OH HOH2C O C 2 HOH OH OH O O OH OAc O Endoxylanase α-Glucuronidase α-L-Arabinofuranosidase Acetyl xylan Esterase β-xylosidase
o
o
O Oo
Oo
O HO OH HO OH HO HO OHFigure 1-1. The basic structural components of xylan, and the hemicellulases responsible for its degradation.
O HO HO O R OH C O O -C O HO O H H O HO HO HO C O O C O -O -O H+ O HO HO OH C O HO C O -O +O R OH Inverting Mechanism O HO HO O R OH C O O -C O HO O HO HO OH C O O C O -O O HO HO OH C O C O HO Retaining Mechanism O H H OH -O H H
N-acetyl-D-glucosamine
Alpha-L-arabinofuranose
Figure 1-3. The three-dimensional structure of bifunctional α-L-arabinofuranosidase and β-D-xylopyranosidase from Aspergillus kawachii
Chapter 2
Expression, purification and characterization of a
bifunctional α-L-arabinofuranosidase/β-D-xylosidase
from Trichoderma koningii G-39
Abstract
A gene of α-L-Arabinofuranosidase (Abf) from Trichoderma koningii
G-39 was successfully expressed in Pichia pastoris. The recombinant enzyme was purified to > 90% homogeneity by a cation-exchanged chromatography. The purified enzyme exhibits both
α-L-arabinofuranosidase and β-D-xylosidase (Xyl) activities with
p-nitrophenyl-α-L-arabinofuranoside (pNPAF) and
2,4-dinitrophenyl-β-D-xylopyanoside (2,4-DNXP) as substrate, respectively.
The stability and the catalytic feature of the bifunctional enzyme were characterized. The enzyme was stable for at least 2 h at pH values between 2 and 8.3 at room temperature when assayed for Abf and Xyl activities. Enzyme activity decreased dramatically when the pH exceeded 9.5 or dropped below 1.5. The enzyme lost 35% of Abf activity after incubation at 55 °C for 2 h, but retained 95% of Xyl activity, with 2,4-DNXP as substrate, under the same conditions. Further investigation of the active site topology of both enzymatic functions was performed with the inhibition study of enzyme
transarabinofuranosyl and transxylopyranosyl activities, indicating both enzymatic reactions proceed through a two-step, double displacement mechanism.
2-1. Introduction
The plant cell wall, the major source of fixed carbon in nature, is a composite structure containing mainly three structural polysaccharides, cellulose, hemicellulose, and lignin. L-Arabinose is a common component in
some hemicelluloses, such as arabinan, arabinoxylan, and arabinogalactan
[71]. Many fungi and bacteria survive in nature by producing a group of
hydrolytic enzymes that synergistically catalyze the degradation of plan
polysaccharides to gain nutrients. α-L-Arabinofuranosidase (Abf, EC
3.2.1.55), one of the key enzymes of the hemicellulase system, is an exo-type enzyme. It involves in degrading the arabinose-containing polysaccharides from the terminal of nonreducing end [72-74]. The release of arabinose side chains by α-L-arabinofuranosidase from hemicellulose is a critical step in the
microbial degradation of natural polysaccharides [3].
α-L-Arabinofuranosidase is tremendously useful in biobleaching of paper pulp
[46], bioconversion of lignocellulose material to fermentative products [37],
and improvement of animal feedstock digestibility [12, 47]. It also
hydrolyzes the glycosides of monoterpenes, sesquiterpenes, and other alcohols that constitute the aromatic potential of wine. The application of
α-L-arabinofuranosidase in flavor improvement and wine aromatization has
been studied [48-50].
Over 2500 different glycosyl hydrolases (GH) have been assigned to 106 families on the basis of amino acid sequence similarities
α-L-arabinofuranosidase families, a detailed mechanistic study of family
GH51 was recently reported [68] but mechanistic details for the other family members remain unknown. This lack of information reflects difficulties in obtaining synthetic substrates bearing different leaving groups [60, 61], as well as problems with effective recombinant protein expression. We
reported herein that an α-L-arabinofuranosidase, a member of GH54, from
Trichoderma knonigii was successfully expressed, purified and characterized
for the first time. The accomplishment will make further investigation of the
mechanism and essential residues of GH54 α-L-arabinofuranosidase become
2-2. Experimental Section
Materials and Chemicals
2,4-Dinitrophenyl-β-D-xylopyranoside (2,4-DNXP) was prepared in one
step using Sharma’s method [75]. Buffers,
p-nitrophenyl-α-L-arabinofuranoside (pNPAF), and chemicals for synthesis
were purchased from Aldrich-Sigma Chemical Co. (St. Louis, MO, USA).
The antibiotic zeocin was obtained from Invitrogen (Carlsbad, CA, USA).
Vent polymerase from New England Biolabs (Ipswich, MA, USA) was used in
polymerase chain reactions (PCR). Restriction endonucleases and T4 DNA ligase were obtained from Roche Applied Science (Mannheim, Germany). Plasmid pPICZαB and Pichia pastoris strain GS115 were purchased from Invitrogen (Carlsbad, CA, USA). Escherichia coli strain JM109 (ECOS, Taipei, Taiwan) served as the host for recombinant plasmids. Oligonucleotides were synthesized by Integrated DNA Technologies (Mission Biotechnology, Taipei, Taiwan). Low molecular-weight protein marker standards for electrophoresis were purchased from Amersham Biosciences (Piscataway, NJ, USA).
Media and buffers
The media BMGY, BMMY, YPD, YPD plus zeocin, LB, and low-salt LB plus zeocin are described in the EasySelect™ Pichia Expression Kit manual (version F) and the pPICZα A, B, C manual (Invitrogen, Appendix 3).
glucose. Zeocin was added to YPD to a final concentration of 100 mg/mL. LB medium contained 1% tryptone, 0.5% yeast extract, and 1% NaCl. Low-salt LB plus zeocin medium contained 0.5% NaCl and 25 mg/mL zeocin.
Construction of expression vector
A vector containing the abf gene from Trichoderma koningii G-39 was the generous gift of Prof. T.-H. Hseu from the National Tsing Hua University,
Hsinchu, Taiwan. To insert the abf gene in an expression vector, a
N-terminal primer (5'-TTAAGAAGGAGCTGCAGCAATGGGGCCTTG-3')
and a C-terminal primer (5'-GCTCGAATTCGGTACCTTAAGCAAAACC-3'), containing PstI and
KpnI restriction enzyme sites, respectively, were used for PCR amplification
of the abf gene. The amplified DNA, containing the mature abf gene, was purified using the Viogen kit (Viogen, Mountain View, CA, USA), digested with PstΙ and KpnΙ, again purified using the Viogen kit, and ligated, using T4 DNA ligase, into PstΙ/KpnΙ-digested pPICZαB (an E. coli/P. pastoris shuttle vector). E. coli JM109 cells were transformed with the ligation mixture and zeocin-resistant colonies were selected. The presence of the expected DNA insert in a zeocin-resistant clone was confirmed by analysis of PstI/KpnI restriction fragments of the recombinant plasmid, and by sequencing of recombinant DNA using commercial 5’AOX, 3’AOX, and α-factor primers (Invitrogen). The recombinant plasmid, pPICZαB-abf, was linearized and introduced into P. pastoris strain GS115 by electroporation as outlined in the
Pichia expression kit EasySelect manual (version F, Invitrogen). The
electroporation mixture was plated onto YPD-zeocin agar medium. After 72 h at 28 °C, colonies were picked for direct colony PCR using the 5'- and 3'-AOX1 primers, followed by DNA sequence analysis of the resulting PCR products. The colony PCR reaction mixture (23.5 µL) consisted of 2.5 µL of resuspended bacterial colony, 10 pmol of each primer (each in 1 µL), 0.5 µL
of a mixture in which each dNTP was present at 25 mM, 15 µL sterile water,
2.5 µL 10 × PCR buffer with 20 mM MgSO4 (50 mM KCl, 10 mM Tris HCl,
pH 8.3), and 1 µL of Taq polymerase (5 U/µL). DNA was amplified in 25 cycles (1 min at 94 °C, 1 min at 55 °C, and 2 min at 72 °C) with a final extension of 7 min at 72 °C. An Applied Biosystems GeneAmp PCR system 9700 machine was used (Appendix 4). Clones positive in this test were then screened for Abf protein expression.
Protein production and purification
Recombinant cultures of P. pastoris (pPICZαB-abf) were incubated overnight at 28 °C, shaking at 180 rpm, in 250 mL BMGY medium contained in 1 L Erlenmeyer flasks, until OD600 values of 7–8 were attained. The cells
were then transferred to BMMY medium to induce the expression of Abf. After 120 h of incubation, the cells were pelleted by centrifugation at 4200 xg for 30 min, and supernatant containing secreted enzyme was clarified by filtering through 0.5 µm filters. The proteins in the supernatant were precipitated by the addition of ammonium sulfate to 80–85% saturation. The precipitate was resuspended in 3 mL of sodium acetate buffer (20 mM, pH 4.5). The solution was then desalted using a 5 mL HiTrap desalting column (Pharmacia, Uppsala, Sweden). The filtrate (4 mL) was loaded onto three 5 mL cation-exchange HiTrap SP columns (Pharmacia) pre-equilibrated with 20 mM sodium acetate buffer, pH 4.5. The columns were eluted with a 150 mL linear gradient of NaCl (0–300 mM) at a flow rate of 1 mL/min.
SDS-PAGE according to Laemmli [76] in comparison to the molecular weights of standard proteins (14–97 kDa).
Enzyme assays and kinetics
Abf activity was assayed with pNPAF as substrate by determining the amount of p-nitrophenol (or p-nitrophenolate) released. In the activity assay, a suitable amount of purified recombinant Abf was added in 0.5 mL of acetate buffer (100 mM, pH 4.1) containing 1 mM pNPAF. The reaction was monitored at 348 nm (the isosbestic point of p-nitrophenol/p-nitrophenolate)
with Δε value of 2426 M−1cm−1. For determining the activity of Xyl,
2,4-dinitrophenyl β-D-xylopyranoside (2,4-DNXP) was used as substrate.
The absorption coefficient of 2,4-nitrophenolate/2,4-nitrophenol was determined to be 6000 M−1cm−1 at pH 4.1. Kinetic studies were performed by monitoring the production of phenolates on a Hewlett–Packard model 8452A Diode Array spectrophotometer with a circulating-water bath set at 25 °C.
pH stability and Thermostability
For thermostability experiments, 300 µL portions of purified α-L-arabinofuranosidase were heated in microcentrifuge tubes at 25 °C, 35 °C,
45 °C, 55 °C, 60 °C and 65 °C. The concentration of purified enzyme was 0.2 µg/mL in 50 mM acetate buffer, pH 5.0. After being heated for the appropriate time interval, 30 µL of samples were then removed to assay the residual activity in pH 4.1 (100 mM NaOAc), 25 °C. For pH stability experiments, enzyme samples (same as above) were incubated in a series of buffers with pH 1.5, 2.5, 3.5, 4.3, 5.6, 7.2, 8.3 and 9.5 at 25 °C. Samples were removed for assay at different time intervals. The reactions were either
monitored at the 348 nm for p-nitrophenol or at the 400 nm for 2,4-dinitrophenolate. The activity measured at pH 4.1 (100 mM NaOAc) and 25 °C served as a control.
Transglycosylation activity
Enzyme (~2 units) was added in 1 mL ammonium acetate buffer, pH 4.5, containing 20% alcohol and 10 mM 2,4-DNXP (or pNPAF for transarabinosylation). The reaction proceeded at 40 °C, overnight. The reaction mixture was concentrated to dryness. The crude solid was then resuspended in 0.5 mL of water to form a yellowish solution, which was then subjected to extraction by chloroform (0.5 mL, once) and ethyl acetate (0.5 mL, twice). The resulting aqueous solutions were lyophilized and exchanged
with 0.5 mL of [2H] water for NMR analysis. The NMR analyses were
shown as follows: Methyl-α-L-arabinofuranoside, 4.81 (d, 1H, C1-H, J = 1.5 Hz), 3.92 (m, 2H, C2-H, C4-H), 3.83 (m, 1H, C3-H), 3.70 (dd, 1H, J = 3.36, 8.9 Hz), 3.58 (dd, 1H, J = 5.79, 6.5 Hz), 3.30 (s, 3H). Methyl-β-D-xylopyranoside, 4.15 (d, 1H, C1-H, J = 7.8 Hz), 3.80 (q, 1H, C5-Heq, J = 5.4 Hz), 3.45 (m, 1H, C4-H), 3.38 (s, 3H, CH3), 3.27 (t, 1H, C3-H, J = 9.0 Hz), 3.17 (t, 1H, C5-Hax, J = 11.4 Hz), 3.08 (dd, 1H, C2-H, J = 9.3 Hz).
2-3. Results and Discussion
Protein purification
The abf gene from T. koningii, encoding α-L-arabinofuranosidase, was
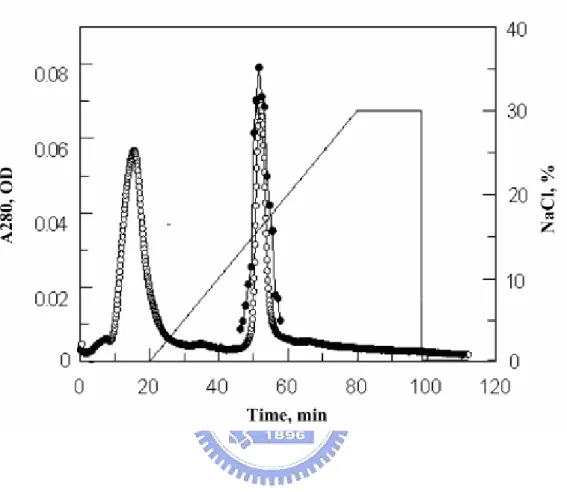
first inserted into plasmid pET22. The recombinant protein was highly expressed but formed an inclusion body. Although much effort was expended in attempts to refold this recombinant Abf, the endeavors were unsuccessful. The abf gene was next cloned into the pPICZαB vector and expressed in P. pastoris GS115. The resulting recombinant protein was fused with three extra amino acids (Ala-Ala-Met) at the N-terminus of the mature Abf. The recombinant Abf was expressed and secreted into the culture medium. Three consecutive steps involving ammonium sulfate precipitation, desalting, and HiTrap SP (cation exchange) chromatography were employed for enzyme purification. The chromatogram of Abf purification was shown in Figure 2-1. The fractions with Abf activity was eluted at 45-60 min, corresponding to the gradient region of 150-180 mM NaCl. The purified Abf and the secreted proteins in the medium with different induction time were analyzed by SDS-PAGE with the molecular weight of Abf at ~ 50 kDa, as shown in Figure 2-2. The purified Abf, with a purity >90%, was obtained and used for further study.
Characteristics of recombinant Abf
The substrate specificity of the recombinant enzyme was tested with a
variety of glycosides, including 2,4-dinitrophenyl-β-D-xylopyranoside
(2,4-DNXP), p-nitrophenyl-α-L-arabinopyranoside,
p-nitrophenyl-β-D-glucopyranoside, p-nitrophenyl-β-D-galactopyranoside, p-nitrophenyl-β-D-N-acetylglucosamine, p-nitrophenyl-β-D-mannopyranoside,
investigated, only 2,4-DNXP, and pNPAF were hydrolyzed effectively. The activities with other non-arabinofuranosides as substrates were less than 0.5% of that with pNPAF. The enzyme showed a preference for substrates with α-L-arabinofuranose and xylopyranose at the glycone moiety. The catalytic
activity (kcat/Km) of Abf (with pNPAF as substrate) and Xyl (with 2,4-DNXP
as substrate) were determined, at pH 4.1, to be 65625 M–1s–1 and 17900 M–1s–1, respectively. These results suggested that the protein be a bifunctional
enzyme with both α-L-arabinofuranosidase and β-D-xylosidase activity.
Although many α-L-arabinofuranosidases were found to exhibit β-D-xyloside
activity [77-79], the bifunctional activity of
α-L-arabinofuranosidase/β-D-xylosidase (Abf/Xyl) was less discussed. This
bifunctional feature is probably due to the consequence of the spatial similarity of the hydroxyl groups and glycosidic bond orientations in β-D-xylopyranosides and α-L-arabinofuranosides, which can accommodate in
a same active site. Alternatively, the enzyme may possess two different catalytic sites for each reaction. Further kinetic investigation or x-ray structure analysis may be required for clarifying the catalytic feature of this bifuctional enzyme.
The pH stability of the enzyme was investigated. In general, the enzyme was stable for at least 2 h at pH values between 2 and 8.3, at room temperature. However, its activity decreased dramatically when the pH
exceeded 9.5 or dropped below 1.5 (data not shown) for both Abf and Xyl
To further investigate the active site topology of both enzymatic functions, we used reversible inhibition of enzyme activities. Both α-L-arabinofuranosidase activity and β-D-xylosidase activity was monitored in
the presence of inhibitor. The effects of substrate concentrations on the hydrolysis rates of 2,4-DNXP and pNPAF, using a fixed concentration of the enzyme, and different concentrations of methyl-α-L-arabinofuranoside (MAF),
were investigated. The results revealed that MAF inhibition is noncompetitive towards 2,4-DNXP as substrate but competitive towards
pNPAF. Estimated Ki values were > 16 mM in both cases. Since the
competitive inhibition occurs when the inhibitor competes with the substrate to bind to the enzyme, whereas, the noncompetitive inhibition is caused by the binding of the inhibitor to the enzyme at another site; a noncompetitive inhibitor can bind to either the free enzyme or the enzyme/substrate complex, we conclude that the Abf and the Xyl catalytic reactions are performed at distinct enzyme sites. This argument is, in fact, supported by the study where a protein crystal was soaked either in 125 mM xylose or a 5% (w/v) solution of a xylooligosaccharide mixture, after which no concentration of electron density corresponding to sugar moiety binding was detected [70]. This suggested that the Abf active site can accommodate neither xylose nor xylosides.
Transglycosylation activity
The recombinant enzyme was found to be stable in the presence of methanol in NaOAc buffer (pH 4.5). When substrate (pNPAF or 2,4-DNXP) was catalyzed by enzyme in the presence of 20% methanol, both enzymatic products derived from hydrolysis and transglycosylation were obtained (as
shown in Scheme 2-I). The yields of methyl-α-L-arabinofuranoside
(MAF) and methyl-β-D-xylopyranoside (MX), estimated from NMR spectra
2,4-DNXP were used as substrate for Abf and Xyl catalysis correspondingly. The NMR assignments of MAF and MX were confirmed according to literatures [80, 81].
Proposed the catalytic mechanisms of the bifunctional Abf/Xyl
In general, glycosyl hydrolases cleave the glycosidic bond of the substrate by two different mechanisms, the retention and the inversion of the anomeric configuration. Both mechanisms require two essential residues, which in most glycosyl hydrolases are residues with carboxylic acid side-chain such as aspartate or glutamate. Inverting glycosidases catalyze the hydrolysis of the glycosidic bond via a single displacement reaction. One of the catalytic residues, acting as a general acid, provides protonic assistance to the departing glycosidic oxygen, while the second, acting as a general base, activates a water molecule which that effects a direct displacement at the anomeric centre and to form the product with the inversion of the anomeric configuration [51]. Retaining glycosidases catalyze the hydrolysis via a two-step, double-displacement mechanism, as shown in
figure 2-4. Two key active-site carboxylic acid residues [52-55] are involved. One functions as the nucleophile and the other functions as the general acid/base [53]. In the first step (the glycosylation step), the nucleophile attacks the anomeric carbon of the glycoside, while the acid/base catalyst protonates the glycosidic oxygen, thereby assisting the leaving of the aglycon moiety. This leads to the formation of a covalent glycosyl-enzyme
methyl-α-L-arabinofuranoside and methyl-β-D-xylopyranoside, we concluded
that the catalytic reactions of the recombinant enzyme for both Abf and Xyl activity proceed via a two-step, double displacement mechanism.
MeOH H2O O OH HO HO O OH HO HO OCH3 OH O OH HO O HO NO2 Abf/Xyl Intermediate MAF pNPAF MeOH H2O Intermediate Abf/Xyl O OH HO HO O NO2 NO2 O OH HO HO OCH3 O OH HO HO OH 2, 4-DNPX MX OH O2N OH NO2 O2N
Figure 2-1. The chromatography of the purification of the recombinant of Abf. The aborbance (-○-) was monitored at 280 nm. The fractions with Abf activity (-●-) was eluted within 150-180 mM NaCl (—).
97 66 45 33 20 kDa M 1 2 3 4
Figure 2-2. SDS-PAGE analysis of Abf obtained from different induction time and the purified protein. Lanes: M, molecular mass markers; 1, recombinant enzyme from day 0 supernatants; 2, recombinant enzyme from day 1 supernatants; 3, recombinant enzyme from day 4 supernatants; 4, pool of active fractions from HiTrap-SP column.
(b) (a)
Figure 2-3. Thermal stability of the recombinant enzyme assay as Abf (a) and Xyl (b). Enzyme was incubated in various temperature: 25 °C (○), 35 °C (●), 45 °C (□), 55 °C (■), 60 °C (△), and 65 °C (▲).
C O O H C O O O HO O NO2 C O O C O O HO O O H H C O O H C O O O HO OH
Possible glycosyl-enzyme intermediate Step 1: Formation
of intermediate
Step 2: Breakdown of intermediate
Figure 2-4. The proposed two-step, double displacement mechanism of Abf/Xyl.
Chapter 3
Mutagenesis and mechanistic study of a family 54
α-
L-arabinofuranosidase from Trichoderma
koningii
Abstract
A GH54 α-L-arabinofuranosidase (Abf) originally from Trichoderma
koningii G-39 was successfully expressed in Pichia pastoris and purified to
nearly homogeneity by cation-exchange chromatography. Extensive mutagenesis of 24 conserved Glu and Asp residues of family 54 was
performed. The kcat values of the D221N and D299N were 7000- and
1300-fold lower than that of the wild-type Abf, respectively, while E223Q was nearly inactive. These results are consistent with implications from the
Aspergillus kawachii α-L-arabinofuranosidase three-dimensional structure.
This structure indicates that E223 of T. koningii Abf function as a nucleophile and D299 as a general acid/base catalyst for the enzymatic reaction, whereas D221 is significant for substrate binding. The catalytic mechanism of wild-type Abf was further investigated by NMR spectroscopy and kinetic analysis. The results showed that Abf is a retaining enzyme. It catalyzes various substrates via the formation of a common intermediate that is probably an arabinosyl-enzyme intermediate. A two-step, double-displacement mechanism involving first the formation, and then the breakdown, of an arabinosyl-enzyme intermediate was proposed. Based on the kcat values of a
series of aryl-α-L-arabinofuranosides catalyzed by wild-type Abf, a relatively
small Brønsted constant, βlg = – 0.18, was obtained, suggesting that the
rate-limiting step of the enzymatic reactions for substrates (where the leaving
kinetic studies with D299G mutant revealed that the catalytic activity of this mutant depended largely on the pKa values (> 6) of leaving phenols, with
βlg = –1.3. This indicated that the rate-limiting step became arabinosylation
step when D299G was employed. This kinetic outcome supports the idea that D299 is the general acid/base residue. The pH activity profile of D299N provided further evidence strengthening this suggestion.
3-1. Introduction
α-L-Arabinofuranosidases (EC 3.2.1.55) are among key enzymes of the
hemicellulase system, which is tremendously useful in biobleaching of paper pulp [46], bioconversion of lignocellulose material to fermentative products
[37], and improvement of animal feedstock digestibility [12, 47].
α-L-Arabinofuranosidases catalyze the hydrolysis of α-1,2- and α-1,3-
α-1,5-L-arabinofuranosidic bonds in hemicelluloses such as arabinoxylan,
L-arabinan, and other L-arabinose-containing polysaccharides [12, 31].
These enzymes also hydrolyze the glycosides of monoterpenes, sesquiterpenes, and other alcohols, releasing the aromatic potential of wine. The enzymes have general applications in flavor improvement [48-50].
Owing to their industrial importance, a variety of
α-L-arabinofuranosidases have been purified from various sources such as
bacteria [59], fungi [41], and plants [77]. Many genes encoding Abf
enzymes have been cloned and sequenced. On the basis of amino acid sequence similarities [56, 82], α-L-arabinofuranosidases are grouped into five
our glycoside hydrolase (GH) families, termed GH3, GH43, GH51, GH54, and GH62. Although many studies on the industrial applications of α-L-arabinofuranosidases from the above families have been reported, only a
few papers have been published describing detailed catalytic mechanisms, with reference to the specific enzyme residues involved. This lack of information reflects difficulties in obtaining synthetic substrates bearing
different leaving groups [60, 61], as well as problems with effective
recombinant protein expression.
There are two broad mechanisms of glycoside hydrolase cleavage of glycosidic bonds. The original anomeric configuration is either inverted or retained. Both mechanisms require amino acids containing side-chain carboxylic residues as essential group(s). Inverting glycosidases catalyze the
hydrolysis of the glycosidic bond via a single displacement reaction. One of the catalytic residues, acting as a general acid, provides protonic assistance to the departing glycosidic oxygen, while the second, acting as a general base, activates a water molecule that effects a direct displacement at the anomeric center [51]. For retaining glycohydrolases, though a catalytic mechanism, involving the formation of an oxocarbenium ion intermediate, was once
proposed by Phillips [83], further studies with this type of glycosidases
provide strong evidence pointing to a common mechanism that involves the
formation of covalent glycosyl-enzyme intermediates [54, 55, 84, 85], as
previously postulated by Koshland [86]. In general, two key active-site carboxylic acid residues are involved. One serves as the nucleophile and the other serves as a general acid/base. The proposed mechanism was shown in
Figure 3-1. In the first step (the glycosylation step), the nucleophile attacks
the anomeric carbon of the glycoside, while the acid/base catalyst protonates the glycosidic oxygen, thereby assisting the leaving of the aglycon moiety. This leads to the formation of a covalent glycosyl-enzyme intermediate. In the second step (the deglycosylation step), breakdown of the glycosyl-enzyme intermediate proceeds through a general base-catalyzed attack of water at the anomeric center to release the glycose. For kinetic analysis of a catalytic mechanism, a series of artificial substrates, each with a glycose moiety attached to different leaving phenols, is commonly prepared. Typically, the extended Brønsted relationship is to be measured. As enzymes in the same family presumably possess a similar catalytic mechanism, solving the mechanistic action of a particular enzyme may help in the understanding of
three-dimensional structure of an α-L-arabinofuranosidase from Aspergillus kawachii IFO4308 [70]. On the basis of protein structure and very
preliminary mutagenic studies, E221 and D297 were suggested as candidates for the nucleophile and the general acid/base catalytic residues, respectively. In this study, we confirm the catalytic functions of essential Abf residues by site-directed mutagenesis and kinetic analysis of mutant enzyme properties. Further, the enzymatic mechanism of a GH54 family member is described and discussed, for the first time, in this thesis.
3-2.
Experimental procedures
MaterialsThe materials are described in the 2-2 experimental section. BMGY,
BMMY, and YPD media were prepared according to the manufacturer’s instructions and 2-2 experimental section. The medium used for 1 L fermentations was that described in the Pichia fermentation guidelines.
Substrates synthesis
The synthesis of phenol-linked arabinofuranosides differs slightly from methods followed in the preparation of glycosides where the glycose has a six-membered ring. To obtain the arabinofuranoside as the major final
product, methyl-α-L-arabinofuranoside was synthesized and used as the
starting material for further reactions. The complete procedure has been
published [87]. All substrates, including phenyl-α-L-arabinofuranoside
(PAF), p-cyanophenyl-α-L-arabinofuranoside (pCPAF),
m-nitrophenyl-α-L-arabinofuranoside (mNPAF),
p-nitrophenyl-α-L-arabinofuranoside (pNPAF),
4-chloro-2-nitrophenyl-α-L-arabinofuranoside (CNPAF), and
2,5-dinitrophenyl-α-L-arabinofuranoside (2,5-DNPAF) were synthesized
according to this published protocol. All substrates were purified by column chromatography and structures confirmed by NMR, which yielded the
following data: methyl-α-L-arabinofuranoside, 4.81 (d, 1H, C1-H, J = 1.53
Hz), 7.12 (d, 2H, J = 8.89 Hz), 5.71 (s, 1H, C1-H), 4.30 (dd, 1H, C2-H, J = 1.53, 1.55 Hz), 4.07 (dd, 1H, C4-H, J = 2.00, 3.38 Hz), 4.00 (dd, 1H, C3-H, J = 2.53, 3.26 Hz), 3.73 (dd, 1H, J = 3.42, 9.05 Hz), 3.64 (dd, 1H, J = 5.38, 6.94 Hz); m-nitrophenyl-α-L-arabinofuranoside, 7.8 (d, 2H, J = 8.78 Hz), 7.48 - 7.37 (m, 2H), 5.68 (s, 1H, C1-H), 4.29 (dd, 1H, C2-H, J = 1.54, 1.67 Hz), 4.07 (m, 1H, C4-H), 3.99 (m, 1H, C3-H), 3.72 (m, 1H), 3.63 (m, 1H); p-nitrophenyl-α-L-arabinofuranoside, 8.28 (d, 2H, J = 9.34 Hz), 7.26 (d, 2H, J = 9.06 Hz), 5.88 (d, 1H, C1-H, J = 1.1 Hz), 4.46 (dd, 1H, C2-H, J = 1.37, 1.67 Hz), 4.20 (dd, 1H, C4-H, J = 2.74, 3.33 Hz), 4.14 (dd, 1H, C3-H, J = 3.57, 9.06 Hz), 3.77 (dd, 1H, J = 5.67, 7.14 Hz), 3.66 (dd, 1H, J = 5.38, 5.45 Hz); 4-chloro-2-nitrophenyl-α-L-arabinofuranoside, 7.92 (d, 1H, J = 2.74 Hz), 7.58 (dd, 1H, J = 2.74, 6.32 Hz), 7.32 (d, 1H, J = 9.06 Hz), 5.66 (d, 1H, C1-H, J = 1.37 Hz), 4.34 (dd, 1H, C2-H, J = 1.65, 1.92 Hz), 4.12 (m, 1H, C4-H), 3.98 (dd, 1H, C3-H, J = 2.47, 3.57 Hz), 3.73 (dd, 1H, J = 3.57, 9.05 Hz), 3.63 (dd, 1H, J = 5.49, 6.86 Hz); 2,5-dinitrophenyl-α-L-arabinofuranoside, 8.27 (s, 1H), 8.01 (d, 2H, J = 1.1 Hz), 5.78 (d, 1H, C1-H, J = 1.37 Hz), 4.32 (dd, 1H, C2-H, J = 1.37, 2.47 Hz), 4.09 (m, C4-H, 1H), 4.02 (dd, 1H, C3-H, J = 2.74, 3.84 Hz), 3.79 (dd, 1H, J = 3.02, 9.05 Hz), 3.68 (dd, 1H, J = 4.66, 7.41 Hz) (Appendix 5).
Vector construction for Pichia expression system
Vector construction for Pichia expression system were described in the 2-2 experimental section. Clones positive in this test were then screened for Abf protein or Abf proteins expression by initial growth, with shaking at 180 rpm, in 50 mL BMGY medium contained in 250 mL Erlenmeyer flasks.
Recombinant DNA expression in cultures at OD600 values of 7-8 was induced
by transferring the cells to BMMY medium. At 24, 48, 72, 96, and 120 h postinduction, methanol, to a final concentration of 0.5% (v/v), was added to cultures. After 160 h of incubation, cultures were centrifuged and proteins in
the supernatants analyzed by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) using a 12% (w/v) polyacrylamide separating gel with a 5% (w/v) stacking gel. Clones producing the protein of interest were identified by the presence of an appropriately sized (50 kDa) supernatant protein band.
In vitro site-directed mutagenesis
The abf gene was mutagenized using the QuickChange site-directed mutagenesis kit (Stratagene, San Diego, CA, USA). The primers used to generate the mutations were as follows (mutations are underlined): D170N(+), 5'-CATGTATGCGGTTCTCAATGGTACACAC-3' and D170N(–),
5'-GTGTGTACCATTGAGAACCGCATACATG-3'; D221N(+), 5'-GATCATGGCTAATCTCGAGAACGGCTTG-3' and E223Q(–),
5'-CAAGCCGTTCTGGAGATCAGCCATGATC-3'; E223G(+), 5'-GATCATGGCTGATCTTCGGCAAGGCTTG-3'; and E223G(–),
5'-CAAGCCGTTGCCGAGATCAGCCATGAT C-3'; D299N(+), 5'-CTCGGCATTGGCGGCAACAACAGC-3' and D299N(–),
5'-GCTGTTGTTGCCGCCAATGCCGAG-3'; D299G(+), 5'-CATTGGCGGCGGCAACAGCAACGGC-3' and D299G(–),
5'-GCCGTTGCTGTTGCCGCCGCCAATG-3'; E310Q(+), 5'-TCTATCAGGGCGTCATGACCTCTGGCTATC-3' and E310Q(–),
5'-GATAGCCAGAGGTCATGACGCCCTGATAGA-3'. Note that D221N (+) and E223Q(–) primers were used to performed both mutations. Primers creating other mutations that did not cause significant loss in enzyme activity
The Abf and Abf mutant proteins production and purification steps were
described in the 2-2 experimental section. To minimize enzyme
cross-contamination during purification, fresh HiTrap SP columns were used in the purification of each mutant protein.
Kinetic studies
Enzyme activity was determined at pH 4.1 by monitoring the hydrolysis of pNPAF to release p-nitrophenol (or p-nitrophenolate). A Hewlett-Packard model 8452A spectrophotometer equipped with a circulating water bath at 25 °C was used. The pH-dependence of enzyme activity in both wild-type and mutant enzymes was assessed using pNPAF as substrate with assay pH values in the range 1.9-6.5. Reactions were monitored at the isosbestic point (348 nm) of p-nitrophenol and p-nitrophenolate and kinetic constants calculated on
the basis that the molar absorption coefficient (Δε) was 2426 M−1cm−1.
Buffers used in this study were glycine (pH 1.8 to 3.5), sodium acetate (pH 4.0 to 5.5), morpholinoethanesulfonic acid (pH 5.5 to 6.5), phosphate (pH 6.5 to 7.5), and Bicine (pH 7.5 to 9.5). The Michaelis constant was determined for each synthetic substrate from the Michaelis–Menten equation, using a double reciprocal plot. Reaction rates were determined, in triplicate, at pH 6.5 (in morpholinoethanesulfonic acid buffer, 100 mM) at each of seven different substrate concentrations. The wavelengths employed and the Δε (M−1cm−1) values obtained at those wavelengths for each arabinofuranoside substrate were as follows: 2,5-DNPAF, 440 nm, 3616; CNPAF, 400 nm, 2603; pNPAF, 400 nm, 3077; mNPAF, 380 nm, 330; pCPAF, 290 nm, 672; PAF, 278 nm, 754.
Mass spectroscopy
Mass spectra were recorded using a Micromass Q-TOF mass spectrometer. Protein samples were injected into the mass spectrometer via
an HPLC system equipped with a PLRP-S column (5 μm, 300 Å, 1-50 mm) using a 5-90% (v/v) acetonitrile gradient containing 0.1% (v/v) formic acid as the eluant. Some 5-10 μg amounts of proteins were injected.
3-3. Results and discussion
Enzyme expression and purificationThe resulting recombinant enzyme was expressed and described in the 2-3 results and discussion. The purified enzyme (> 90% homogeneity) was
analyzed by SDS-PAGE (Figure 3-2a) and mass spectrometry (Figure 3-2b),
and gave mol wt values of ca. 50 kDa and 50045 (major peak), respectively. The theoretical mol wt is 49445 Da. The discrepancy between the measured and calculated mol wt values is likely because of protein glycosylation in
Pichia. This argument is somewhat supported by the presence of a series of
mass peaks (50045, 50208, 50369; Figure 3-2b) with differences of 161-163
Da, which may indicate the glycosylation of protein.
pH profiles of the wild-type and the E223G mutant
It is common that the catalytic reactivity of many retaining glycohydrolases is mainly mediated by two active site carboxylates (aspartate and/or glutamate). A bell-shaped activity profile reflected two apparent pKa is
often observed. The pH-dependent activity assay showed that the purified
enzyme had optimum Abf activity at pH 2.8–3.2 (Figure 3-3). The
pH-dependent activity curve of recombinant Abf exhibited a bell shape, with two apparent pKa values of 1.8 ± 0.1 and 4.2 ± 0.1. It is of interest that both
pKa values were about 2 pKa units lower than those found for other
glycohydrolases.
Mutagenic study
The first protein structure of a family 54 α-L-arabinofuranosidase, from A. kawachii, was recently resolved [70]. This structure is valuable for structural
simulation of enzymes in the same family. Based on sequence comparisons, our enzyme has 73% identity and > 85% homology to the A. kawachii
α-L-arabinofuranosidase. By inspection of the protein structure (Figure 3-4),
residues appearing in the active site of A. kawachii α-L-arabinofuranosidase
and T. koningii (shown in parentheses) may be listed: C176 (C178), D219 (D221), E221 (E223), D297 (D299), and S299 (S301). All these residues are
highly conserved in 12 enzymes of the GH54 family (Figure 3-5). The
carboxyl groups of E221 (E223 in T. koningii Abf) and D297 (D299 in Abf) are located on either side of the anomeric C-1 carbon of enzyme-bound arabinofuranose, strongly suggesting that these residues serve as nucleophile and general acid/base residue, respectively. To confirm the catalytic roles of these residues, an extensive mutagenesis study was performed on E223, D299, and 22 other Asp or Glu residues (D95, D120, D160, E163, D170, D181, E186, D191, E198, D221, D238, E289, E310, D320, E323, E407, D410, D429, E436, D437, D482, and D490), which are highly conserved in GH54 family members. We changed the putative catalytic residues, aspartate (D) and glutamate (E), to asparagine (N), glutamine (Q) or glycine (G) by site-directed mutagenesis. All crude mutant enzymes with mutations in D170, D221, E223, D299 or E310 largely lost enzyme activity with pNPAF as substrates
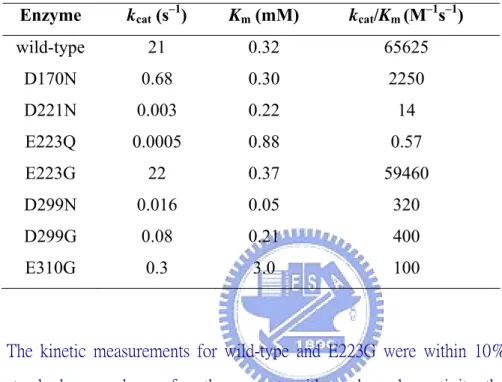
(Appendix 7). Some of these mutant enzymes were further purified and studied. Kinetic parameters are summarized (Table 3-1). The Km values of
mutant enzymes, ranging from 0.22–0.32 mM, are quite similar to that of
wild-type Abf, except for the mutants D299N, and E310G, where the Km
values were 0.05 mM, and 3.0 mM, respectively. The kcat values of the
mutant enzymes D170N, D221N, D299N, D299G, and E310G decreased (with respect to the wild-type Abf) by factors of 31, 7000, 1300, 262, and 70,
hydrolase family 54 members, has been postulated to act as a nucleophile, this study uses site-directed mutagenesis for direct confirmation of this function. Residue D221 (D219 in the A. kawachii enzyme) engages in strong hydrogen
bonding with the C3 and C5 sugar hydroxyl groups [70]. The large
decreases in activity observed when these residues are mutated presumably occurs because the substrate is now incorrectly oriented in the active site. The residues D170 and E310 are more than 20 Ǻ distant from the active site. The activity loss of the E310G mutant results in part from a 10-fold increase in Km value, compared to the unmutated enzyme. A perturbation of general
protein structure by this mutation should perhaps be considered.
One unexpected finding is that the E223G mutant still possesses extremely high activity. Its kcat (22 s–1) and kcat/Km (59460 M–1s–1) values are
nearly identical to those of wild type Abf. After careful inspection of the active-site structure of A. kawachii enzyme, the Asp-189 (Asp-191 in our Abf) was found to locate closely to the nucleophile, E221 (E223 in our Abf). The orientation and distance (4–5 Ǻ) between Asp-189 and the C-1 position of the substrate is perfect for an inverting-type of catalysis to take place. It is very likely that when the site-chain of the nucleophile is replaced by proton (ie, E223G), more space will be gained and, therefore, allow water molecule to diffuse deeply into the active site, while it is no room around the nucleophile if the Glu is mutated into Gln. This hypothesis has been tested by analyzing the activity of double mutant (D191N/E223G) and the stereochemistry of E223G catalysis. The results from this study will be published elsewhere.
In many glycohydrolases, exogenous nucleophiles such as azide, formate, and other anions have been shown to enhance the catalytic activity of enzymes mutant in residues that serve as nucleophiles or provide general acid/base
functions [88-91]. Activity enhancement of a mutant by addition of a
nucleophile (for example, azide) and formation of a stereospecific product (α- or β-glycosyl azide) offers a useful technique for identifying essential residues
of glycosidases. In this study, however, addition of high concentrations of azide (up to 2 M) did not rescue the activities of the D299G, D299N, E223Q or E223G mutants, presumably because the active site was located relatively deep in Abf that the azide ion could not diffuse to the site. Similar observations were reported previously in a study on the A. kawachii IFO4308 enzyme [70]. Nevertheless, comparing the pH activity profiles (kcat vs pH) of
the wild-type Abf and the D299N mutant (Figure 3-3) may provide an insight into the essential function of D299. The activity of the D299N mutant was almost constant in the range of pH 3–6.5 and with a trend of decrease at pH <2.4, indicating the absence of the second pKa point. Also, the low Km value
(0.05 mM) of the D299N mutant suggests accumulation of the glycosyl-enzyme intermediate, whose hydrolysis is then accelerated by the general acid/base catalytic action of the enzyme. Unfortunately, owing to the various degree of post-glycosylation on the recombinant Abf, an unequivocal result of mass spectrometric analysis in attempt to show the presence of the glycosyl-enzyme intermediate was unsuccessful. In sum, the results of the site-directed mutagenesis and kinetic studies, in combination with structure analysis, point to E223 and D299 as the essential nucleophile and general acid/base residues of Abf, respectively. The function of D299 will be further discussed below.
Transglycosylation and product partition
arabinofuranosidases, an arabinofuranoside product may be expected to undergo fast mutarotation to form four arabinose tautomers. These are α- and β-L-arabinofuranosides and α- and β-L-arabinopyranosides [57]. To
overcome this limitation, a method involving transglycosylation of the Abf using methanol as glycosyl acceptor was employed. The advantage of this strategy is the formation of a methyl glycoside that cannot mutarotate. For most retaining enzymes, although the formation of a covalent enzyme intermediate is expected, it is difficult to detect as the lifetime is short. In the past, useful 2-fluoroglycoside products have been obtained using a specific
glycosyl-enzyme trapping technique [62, 92-94]. However, perhaps because
2-fluoroarabinofuranoside is difficult to synthesize, this strategy has not yet been used in any study of α-L-arabinofuranosidase. We used an alternative,
indirect, method. If a constant chemical bias may be noted in different enzyme products, the formation of a common intermediate in an enzymatic reaction may be inferred. Our study showed that the Abf exhibited strong transglycosylation activity when methanol was used as an arabinofuranosyl acceptor. Here, a suitable amount of pNPAF, pCPAF and CNPAF were enzymatically hydrolyzed in acetate buffer (pH 4.1) containing 12% (v/v)
methanol. The solutions were then dried and exchanged with D2O several
times before 1H-NMR analysis. For all three reactions, the 1H-NMR spectra (measured at 25 ºC) of the sugar moieties (in the range of 3-6 ppm) were nearly identical. In principle, five different end products with arabinosyl ring structures should be observed (Figure 3-6a). According to the literature
[80] and our study, the C1 protons of each sugar ring were assigned as follows: methyl-α-arabinofuranoside (4.86 ppm, J = 1.03 Hz), α-arabinofuranose (5.17 ppm), β-arabinofuranose (5.22 ppm, J = 3.8 Hz), α-arabinopyranose (4.43
ppm, J = 5.9 Hz), and β-arabinopyranose (5.16 ppm, J = 3.24 Hz) (Figure
3-6b). Based on peak assignment and the integration of the C1 proton on the sugar ring, the ratio of methyl-α-arabinofuranoside/arabinose was calculated
from each spectrum. Regardless of the substrates, these ratios were nearly constant, and averaged 1.04 ± 0.02 (1.04 for pCPAF, 1.06 for pNPAF, and 1.03 for CNPAF). This suggests that a common intermediate, most likely an arabinosyl-enzyme structure, occurs in the reaction pathways. As the product of transglycosylation in this experiment is methyl-α-arabinofuranoside, we can confirm that the Abf is indeed a retaining enzyme.
Substrate reactivity and Brønsted plot
Kinetic assessment of substrates bearing different leaving phenols is a
common strategy in studying mechanistic actions of glycohydrolases [95].
For glycohydrolases with two-step mechanisms (formation of a glycosyl-enzyme intermediate in a glycosylation step followed by breakdown of the intermediate in a deglycosylation step), the aglycon moiety is cleaved in the glycosylation step of the reaction (Figure 3-1). Therefore, the reaction rate of the first step dictates the ease by which the leaving group may be released from the substrate. A strong correlation between the pKa of the
phenol leaving group and the activity of the enzyme should be observed if the first step is rate-limiting. To determine the rate-limiting step of hydrolysis catalyzed by the recombinant Abf, we worked to prepare
aryl-α-L-arabinofuranosides bearing different leaving phenols (pKa values 5.15 to
9.99) and to perform steady-state kinetic analysis using these substrates. The reaction temperature was held at 25 ºC to reduce spontaneous substrate hydrolysis. Although Abf is specific with regard to the glycon moiety of the
relationship, which has been shown to be useful in understanding the
mechanism of enzyme action [51, 96-97]. Based on the kcat values, an
extended Brønsted plot was constructed by plotting the logarithm of kcat
against the pKa of the leaving phenol (Figure 3-7a). A plot with a slightly
downward trend was obtained, with a slope (βlg value) of –0.18. That the
detailed structures of the leaving phenols do not affect the kinetic parameters of the reaction may indicate that the dearabinosylation step (breakdown of the arabinosyl-enzyme intermediate) is the rate-limiting step. Alternatively, although it may be considered unlikely, the low Brønsted constant could indicate that the arabinosylation step is the slow step and that an early transition state is attained. However, the very weak reaction inhibition shown by MAF (Ki > 16 mM) minimized this possibility. If the transition
state were substrate-like, strong inhibition would be expected. We further studied substrate reactivity using the D299G mutant enzyme. A new plot with βlg = –1.3 was obtained (Figure 3-7a). For substrates with pKa > 6, the
absence of general acid/base catalysis resulted in a strong correlation between the pKa of the phenol leaving group and enzyme activity. Clearly, formation
of the arabinosyl-enzyme intermediate is now the rate-limiting step. However, with 2,5-DNPAF (a good substrate, with pKa = 5.15), the log kcat
value is clearly lower than expected if the enzyme reaction were to proceed as outlined above. This indicates that the dearabinosylation step becomes at least partially rate-limiting when 2,5-DNPAF breakdown is catalyzed by the D299G enzyme. In addition to alteration of catalytic properties, the D299G mutation also affects enzyme Km values with different substrates (Table 2).
The Km values decrease as the pKa values of the leaving phenols change in the
substrates. For the wild-type enzyme, such changes in Km values are not
obvious. These data showed that as the ability of the aglycon moiety to depart the enzyme increased, the more glycosyl-enzyme intermediate
represented as [E][S]/Σ[ES]. Thus, the more the glycosyl-enzyme
intermediate accumulates, the lower the Km value. Such behavior may be
expected in mutants affected in the general acid/base residue, as a good leaving group elevates the rate of the first catalytic step. The rate of the second step remains slow as the basic residue, which (in unmutated enzymes) activates the water molecule, is missing.
The kcat/Km values are informative with respect to the first irreversible
step. For retaining glycoside hydrolases, the Brønsted relationship obtained by plotting kcat/Km values of substrates against the pKa values of the leaving
phenols provides information about the glycosylation step [54, 61]. As may be seen (Figure 3-7b), the Brønsted constants (βlg) are –0.19 and –1.2 for
wild-type Abf and the D299G mutant, respectively. This suggests that the arabinosylation step catalyzed by the wild-type Abf enzyme is not sensitive to the leaving abilities of different phenols, as the general acid/base residue protonates the oxygen of the glycosidic bond, thus imparting constant leaving abilities to different phenols. When the general acid/base residue is absent, however, the catalytic efficiency (kcat/Km) for the tested substrate became
highly sensitive to the leaving ability of phenol groups (βlg = –1.2). A
typical kinetic consequence of the general acid/base mutation is that for substrates requiring strong acid assistance (such as pCPAF and mNPAF), the first reaction step is much slower (12000–14000-fold decreases were noted in this study) than is the case with substrates that need less acid assistance (such as 2,5-DNPAF). The kinetic behavior displayed by the D299G mutant and the pH activity profile of D299N, when compared with data from the
Table 3-1. Michaelis–Menten parameters for the hydrolysis of
pNPAF by wild-type and mutant enzymes at pH 4.1 and 25 °C.
Enzyme kcat (s–1) Km (mM) kcat/Km (M–1s–1)
wild-type 21 0.32 65625 D170N 0.68 0.30 2250 D221N 0.003 0.22 14 E223Q 0.0005 0.88 0.57 E223G 22 0.37 59460 D299N 0.016 0.05 320 D299G 0.08 0.21 400 E310G 0.3 3.0 100
The kinetic measurements for wild-type and E223G were within 10% standard error, whereas for other mutants with much weaker activity, the errors were within 20%.
Table 3-2. Km and kcat values of wild-type Abf using various
aryl-α-L-arabinofuranosides at pH 6.5 Unit: kcat / Km (M–1s–1), kcat (s–1)
Km, mM kcat kcat / Km log kcat log (kcat / Km)
Substrate pKa WT D299G WT D299G WT D299G WT D299G WT D299G 2,5-DNPAF 5.15 0.28 0.10 8.25 0.69 29542 6761 0.92 –0.16 4.47 3.83 CNPAF 6.45 0.42 0.20 6.52 0.12 15633 572 0.81 –0.94 4.19 2.76 pNPAF 7.18 0.31 0.21 2.84 0.014 9215 64 0.45 –1.86 3.96 1.81 mNPAF 8.39 0.54 0.65 2.02 0.0003 7369 0.46 0.31 –3.52 3.87 –0.33 pCPAF 8.49 0.30 0.69 1.70 0.0004 5592 0.58 0.23 –3.41 3.75 –0.25 PAF 9.99 0.28 – 1.23 – 4334 – 0.09 – 3.64 –
O HO OH O HO NO2 A arabinosylation BH O HO OH O HO NO2 A BH O HO OH HO A B arabino sylation OH H arabinosyl-enzyme intermediate dearabin osylation O HO OH HO A B OH H dearabinosylation O HO OH HO A BH OH α-L-arabinofuranoside
Figure 3-1. Proposed reaction mechanism of a retaining
kDa M 1 97.0 66.0 45.0 (a) (b) 30.0 20
Figure 3-2. SDS-PAGE (a) and mass spectrometry (b) analysis of the
recombinant α-L-arabinofuranosidase. Lanes: M, markers; 1, recombinant
8 6 4 2 0 0.030 0.025 0.020 0.015 0.010 k of D 2 99N , s cat -1 8 6 4 2 0 40 30 20 10 0 pH k of W T A bf, s cat -1
Figure 3-3. pH activity profiles of wild-type Abf (○) and the D299N mutant
enzyme (●). The kcat values of wild-type and D299N mutant were
Figure 3-4. The active site of the GH54-family enzyme from Aspergillus
kawachii IFO4308 (1WD4) with arabinofuranose in place. The
corresponding amino acids in the Trichoderma koningii Abf are labeled in parentheses.
170 181 186 191 221 223 289 299 310
U38661 166: YAVLDGTHYNGACCFDYGNAETNSRDTGN 194 215:GPWIMADLENGL 226 286:MSKEGAIILGIGGDNSNGGQGTFYEGV AB085904 164: YAVLDGTHYNDACCFDYGNAETSSTDTGA 192 213:GPWIMVDMENNL 224 284:MSKEGAIILGIGGDNSNGAQGTFYEGV Z69252 166: YAVLDGTHYNGACCFDYGNAETNSRDTGN 194 215:GPWIMADLENGL 226 286:MSKEGAIILGIGGDNSNGAQGTFYEGV AF367026 172: YAVLDGTHYNDACCFDYGNAETSSTDTGN 200 221:GPWVMADLENGL 232 292:MSKEGAIILGIGGDNSNGAQGTFYEGV AB073861 172: YAVLDGTHYNSACCFDYGNAEVSNTDTGN 200 221:GPWIMADLENGL 232 292:MSKEGAIILGIGGDNSNGAQGTFYEGV AB073860 172: YAVLDGTHYNSACCFDYGNAEVSNTDTGN 200 221:GPWIMADLENGL 232 292:MSKEGAIILGIGGDNSNGAQGTFYEGV L23502 164: YAVLDGTHYNDACCFDYGNAETSSTDTGA 192 213:GPWIMVDMENNL 224 284:MSKEGAIILGIGGDNSNGAQGTFYEGV U39942 164: YAVLDGTHYNDACCFDYGNWQTSSTDTGA 192 213:GPWLMVDMENNL 224 284:MSKEGAIILGIGGDNSNGAQGTFYEGV Y13759 170: YAVLDGTHYNDGCCFDYGNAETSSLDTGN 198 219:GPWIMADLENGL 230 291:MSKEGAIILGIGGDNSNGAQGTFYEGA AY495375 166: YAVLDGTHYNDACCFDYGNAEISNTDTGN 194 215:GPWLMADLENGL 226 286:MSLEGAIILGIGGDNSNGAQGTFYEGV AJ310126 166: YAVLDGTHYNGACCFDYGNAETSSTDTGN 194 215:GPWIMADLENGL 226 285:MKKEGAIILGIGGDNSNGAQGTFYEGV AF306764 167: YAVLDGTHYNGGCCFDYGNAETNNLDTGN 195 216:GPWVMADLENGL 227 287:MSKEGAIILGIGGDNSNGAQGTFTEGA
Figure 3-5. Data from a multialignment exercise, using partial sequences,
of family GH54 α-L-arabinofuranosidases. Biology WorkBench 3.2
CLUSTALW (San Diego Supercomputer Center, CA, USA) software was used. All enzyme sequences were derived from published gene sequences. GenBank accession details are: U38661 from Hypocrea koningii G-39, AB085904 from A. kawachii IFO 4308, Z69252 from Hypocrea jecorina
RutC-30, AF367026 from Penicillium purpurogenum, AB073861 from Aspergillus oryzae RIB40, AB073860 from Aspergillus oryzae HL15,
L23502 from Aspergillus niger, U39942 from Aspergillus niger, Y13759 from Emericella nidulans argB2, AY495375 from Aureobasidium pullulans, AJ310126 from Fusarium oxysporum f. sp. Dianthi, and AF306764 from
O OH HO O HO CN O OH HO O HO NO2 O OH HO O HO Cl NO2 Common intermediate O OH HO HO Enzyme MeOH H2O O OH HO HO O OH HO HO OCH3 OH O OH OH OH HO + O OH HO HO OH Enzyme Enzyme Enzym e (a) (b)
Figure 3-6. Stereochemical properties and common intermediates of Abf catalysis. (a) Enzymatic reactions, using various substrates, in the presence of methanol. (b) A partial NMR spectrum (chemical shift 3.4–5.4 ppm) of the end-products. Peak assignment is given in the text.
10 8 6 4 2 0 -2 -4 pKa of phenols log k cat (a) 10 8 6 4 6 4 2 0 -2 pKa of phenols log k /Kcat m (b)