Correspondence and requests for reprints:Dr. V. Courtney Broaddus
Address:1001 Potrero Avenue, Bldg. 1, Rm 150, San Francisco, CA, 94110 USA
Bortezomib-induced Apoptosis in the Treatment of Non-small Cell Lung Cancer
Tsung-Ming Yang
1,2,3, Dario Barbone
2,3, and V. Courtney Broaddus
2,31
Division of Pulmonary and Critical Care Medicine, Chang Gung Memorial Hospital, Chiayi, Taiwan;
2
Lung Biology Center, San Francisco General Hospital;
3
Comprehensive Cancer Center, University of California San Francisco, CA 94143
Introduction
Lung cancer, which can be classified into non- small cell lung cancer (NSCLC) and small cell lung cancer (SCLC), is the leading cause of cancer- related death in the world
1. NSCLC, which accounts for 80 to 85% of all lung cancers, is distinguished from SCLC by its clinical course and its response to chemotherapy and radiotherapy
2. More than 70% of
NSCLC patients present with unresectable locally advanced or metastatic disease
3, so chemotherapy with or without radiotherapy remains the mainstay of lung cancer treatment. Despite advances in the past few decades, current chemotherapy regimens have only limited efficacy with modest survival benefit and significant toxicity in NSCLC patients.
Novel therapies, especially those targeting cell
Abstract
Despite advances in the past few decades, current chemotherapy regimens have had only limited efficacy with modest survival benefit and significant toxicity in non-small cell lung cancer (NSCLC) patients.
Novel therapies, especially those targeting cell signaling pathways, are now under extensive investigation against many tumor types. Proteasome inhibition, one of the novel cancer therapies, has been shown to have widespread ability to induce apoptosis in vitro in a broad spectrum of tumor cell lines. Bortezomib (Velcade) is the first proteasome inhibitor to enter clinical trials. It has demonstrated encouraging efficacy against multiple myeloma, where it is now an approved therapy. In in vitro studies of lung cancer, bortezomib has been found to induce marked apoptosis by itself or when used together with other novel therapy agents or chemotherapy agents. Despite these promising results, bortezomib has not shown great efficacy against lung cancer in clinical trials. In this review, we describe the role of the proteasome in cell homeostasis and apoptosis, the molecular mechanisms of bortezomib-induced apoptosis in preclinical studies of NSCLC cells, and its efficacy to date in the clinical trials. We consider possible reasons why proteasome inhibition may not be as effective in lung cancer and ways in which the efficacy might be improved in the future.( J Intern Med Taiwan 2009; 20: 106-119 )
Key Words: TRAIL, Proteasome inhibitor, Acquired resistance, Multicellular resistance, Three- dimensional spheroid
signaling pathways, are now under extensive investigation against many tumor types. Proteasome inhibition, one of the novel cancer therapies, has been shown to have widespread ability to disrupt tumor cell homeostasis without inducing toxicity in normal cells and to induce apoptosis in vitro in a broad spectrum of tumor cell lines
4. Indeed, when either used alone or combined with other approaches, proteasome inhibition has generated intense interest as a therapeutic approach targeting cancer.
Bortezomib (Velcade) is the first proteasome inhibitor to enter clinical trials. It has demonstrated encouraging efficacy against multiple myeloma, where it is now an approved therapy
5. In in vitro studies of lung cancer, bortezomib also has shown promise; bortezomib has been found to regulate many molecules that are associated with tumor progression and treatment resistance, such as NFκB
6, pro-apoptotic and anti-apoptotic Bcl-2 family proteins
7, and p53
8. In addition, bortezomib often induces apoptosis by itself or when used together with other novel therapy agents or chemotherapy agents
9. Because of the promising results in these preclinical studies, bortezomib has now been tested in many clinical trials of NSCLC
10-19. In this review, we describe the role of the proteasome in cell homeostasis and apoptosis, the molecular mechanisms of bortezomib-induced apoptosis in preclinical studies of NSCLC cells, and its efficacy to date in the clinical trials. We consider possible reasons why proteasome inhibition has not be as effective in lung cancer as in multiple myeloma and strategies by which the efficacy could potentially be improved in the future.
The role of apoptosis in cancer treatment
Defects in apoptosis contribute to the development and treatment resistance of cancer
Although cancer was initially thought to
result from uncontrolled proliferation driven by on- cogenes, there is increasing evidence that the ability to evade apoptosis is also important for cancer to develop
20. Whereas inhibition of apoptosis is not sufficient to cause cancer by itself, we have learned from animal models that, when inhibition of apoptosis is combined with activation of growth stimulatory oncogenes, cancers can then develop
21. As cancer cells acquire the ability to evade apoptosis, they also acquire resistance to the apoptosis that is induced by chemotherapeutic agents
22. Although chemotherapeutic agents can induce cell cycle arrest, senescence, and autophagy
23, emerging evidence suggests that many chemotherapeutic agents induce cancer cell death through apoptosis
22. Furthermore, resistance to chemotherapy has been linked directly to defects in apoptosis
24. The hope for improved cancer therapy depends on a detailed understanding of apoptotic pathways so that strategies to induce apoptosis or to lower the apoptotic resistance against standard therapies can be developed
25.
Molecular machinery of apoptosis
Apoptosis is a programmed cell death that involves a series of biochemical events leading to a variety of morphological changes, including blebbing, cell shrinkage, nuclear fragmentation, chromatin condensation, and chromosomal DNA fragmentation
26. The mechanisms of apoptosis are highly complicated and involve a cascade of activation of caspases, a family of calcium- dependent cysteine proteases. Caspases can be divided into initiator and effector caspases. Initiator caspases, such as caspase 8 and caspase 9, are activated following several initial apoptotic stimuli and then activate the effector caspases. Activation of the effector caspases, such as caspase 3, caspase 6, and caspase 7, results in the enzymatic cleavage of cellular proteins leading to apoptosis (Figure 1).
As one would expect, there is extensive regulation and control over the activation of these caspases.
107 Bortezomib in the Treatment of Non-small Cell Lung Cancer
There are at least 2 major pathways (the extrinsic and intrinsic pathway) that act on the mitochondria to initiate apoptosis and multiple signals that modulate their activity (e.g. p53 and other damage pathways and survival pathways).
The extrinsic pathway
The extrinsic pathway initiates apoptosis via the interaction of transmembrane receptors and death ligands, such as Fas, TNF-α, and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)
27. After the death ligands bind to their corresponding transmembrane receptors, a death- inducing signaling complex (DISC) is formed that then recruits and cleaves the initiator caspase, caspase 8
28. This death receptor-mediated apoptosis can be inhibited by FLIP, a protein that competes with caspase 8 thereby interfering with the initiation of apoptosis
29. In addition, FLIP may have effects on the intrinsic pathway (see below) where, in overexpression studies, FLIP can interfere with Bax
30. Despite this intriguing information, further studies will be needed to confirm whether FLIP acts beyond its well recognized functions in inhibiting death receptor signaling via caspase 8.
The intrinsic pathway
The intrinsic pathway initiates apoptosis via Fig.1. A simplified schematic representation of the two major apoptotic pathways. Figure modified from Shivapurkar et al
22.
the loss of mitochondrial membrane potential and release of multiple pro-apoptotic proteins, including cytochrome c and Smac/DIABLO, from the mitochondria into the cytosol
31. Following release into the cytosol, cytochrome c forms a multiprotein complex with Apaf-1 and procaspase 9, called an apoptosome. After forming the apoptosome, procaspase 9 is activated and then activates the effector caspases. Smac facilitates apoptosis by inhibiting the IAPs (inhibitor of apoptosis proteins), which can bind and inhibit active caspase 9 and the effector caspases.
The intrinsic and extrinsic pathway are linked together, so that proteins in one pathway can affect those in the other pathway
32. Ultimately, the intrinsic and extrinsic pathways converge at the mitochondria, a major integrator of apoptotic signals and effector of apoptosis.
Mitochondrial machinery
Mitochondria regulate apoptosis by integrating
diverse death signals via a family of related
molecules called Bcl-2 family proteins. The Bcl-2
family proteins can be divided into anti-apoptotic
and pro-apoptotic groups. The best known anti-
apoptotic Bcl-2 family proteins include Bcl-2, Bcl-
xL, Bcl-w, A1 and Mcl-1. The major pro-apoptotic
Bcl-2 family proteins are Bax and Bak, which are
required to mediate apoptosis
33,34. There is also a
subset of BH3-only molecules that reside normally
in the cytoplasm, including Bid, Bad, Bim, Bik,
PUMA, and Noxa, that can mediate various pro-
apoptotic signals, whether from the loss of
adherence, disruption of cytoskeleton or DNA
damage, and translocate to the mitochondria where
they can then trigger apoptosis by interacting with
the anti- and the pro-apoptotic Bcl-2 proteins. One
persuasive theory to explain the interaction of these
proteins holds that the anti-apoptotic Bcl-2 proteins
function by binding and buffering the BH3-only
proteins thereby preventing them from binding to
and activating the pro-apoptotic proteins, Bax and
Bak
35. With sufficient activation of the BH3-only molecules or a sufficient reduction in the anti- apoptotic buffering, BH3-only molecules can then bind and activate Bax and Bak, leading to mitochondrial outer membrane permeabilization (MOMP) and the release of proapoptotic proteins as described, such as cytochrome c and Smac/
DIABLO
36.
The p53 and other stress pathways
The tumor suppressor p53 plays a critical role in maintaining the integrity of cellular DNA. In the presence of extensive DNA damage, p53 functions as a regulatory molecule either to facilitate DNA repair or to initiate apoptotic cell death
37. The p53 protein, as a damage sensor, may facilitate or induce apoptosis by actions upon both the intrinsic and extrinsic pathway
38. In the intrinsic pathway, p53 activates pro-apoptotic members of Bcl-2 family proteins as for example by transcriptional activation of Bax
39and by activating the BH3-only molecules, Puma and Noxa
40. In the extrinsic pathway, p53 transactivates receptors for death ligands Fas
41and DR5
42. Of course, many tumors have inactive p53, thereby enabling them to evade apoptosis via this pathway. In such cells without a functioning p53 pathway, the JNK pathway may mediate similar pro-apoptotic signals; for example, our laboratory has shown that the JNK pathway activates the BH3- only molecule, Bim
43.
Survival pathways
In contrast, pro-survival signaling pathways tend to inhibit apoptosis in cells. Such survival pathways may be important anti-apoptotic me- chanisms in tumor cells, in which many such pathways are known to be hyperactive. NFκB is one important survival pathway found to be activated in many cancer cells. NFκB -regulated genes are involved in the regulation of apoptosis, proliferation, and differentiation
44. NFκB is normally inhibited by IκB, and the major mechanism of NFκB activation involves the
proteasomal degradation of the inhibitory IκB following its phosphorylation and ubiquitination
44. After the degradation of IκB, NFκB translocates into the nucleus and promotes the transcription of many target genes. NFκB activation inhibits apoptosis in most cell systems by inducing expression of anti-apoptotic proteins such as Bcl-2, Bcl-XL, A1, cIAPs, as well as pro-survival cytokines such as IL-6
45. Another key survival pathway is the PI3K/Akt pathway which is upregulated in many cancers and also plays a role in the inhibition of apoptosis
46. Activation of the PI3K/Akt pathway inhibits apoptosis in part by the phosphorylation and inactivation of Bad
47and caspase 9
48and the stimulation of the NFκB pathway
49. In addition, activation of the PI3K/Akt pathway stabilizes MDM2
50, which blocks p53 activation and thus prevents p53-mediated apoptosis. Other survival signaling pathways such as MAPK are also involved in the regulation of apoptosis
51. Survival pathways tend to be hyperactive in tumors and thus serve as excellent targets for anti-cancer therapies
52.
Many proteins in the apoptosis and survival pathways just described are regulated either directly or indirectly by proteasomal degradation. Inhibition of the proteasome appears to upset the balance between the pro- and anti-apoptotic proteins, often in a pro-apoptotic direction
6-8. Therefore, the proteasome has been investigated in preclinical and clinical studies as a novel target for cancer therapy.
The role of the proteasome in apoptosis
Proteasomes are large barrel-like protein complexes that are primarily localized in the cytosol but also can be found in the nucleus, endoplasmic reticulum, and plasma membrane
53. The main functions of the proteasome are to degrade unneeded or damaged proteins and to regulate the turnover of short-lived proteins that control cell-
109 Bortezomib in the Treatment of Non-small Cell Lung Cancer
cycle progression, signal transduction, the in- flammatory process and ultimately apoptosis
53. More than 80% of all cellular proteins are processed by the proteasome
54.
The proteasome consists of two 19S regulatory particles and a 20S "core" of four stacked rings around a central pore (Figure 2). The two inner β rings contain six active proteolytic sites on the interior surface of the rings, and the two
outer α rings function as a "gate" through which proteins enter the β rings. Proteins are marked for degradation by being tagged with chains of small proteins called ubiquitins. The resulting po- lyubiquitinated proteins are recognized by the regulatory particles and enter the central pore of the proteasome where they are degraded to peptides of seven to eight amino acids long. These peptides will be further degraded into amino acids that can later be used to synthesize new proteins. By control of intracellular protein levels, the proteasomal degradation pathway is essential for many cell processes
53.
The molecular mechanisms of bortezomib-induced apoptosis
By its myriad effects on the cell, the pro- teasome may have multiple ways by which it induces apoptosis (Figure 3). It is possible that different mechanisms predominate in different tumors or that multiple mechanisms interact in each tumor to induce apoptosis. Even if we only consider NSCLC, several mechanisms have been identified by which bortezomib and proteasome inhibition induce apoptosis.
I. Bortezomib inhibits survival pathways.
The NFκB pathway is thought to be an important target of bortezomib
55. By blocking the activation of the NFκB pathway by preventing the proteasomal degradation of IkB, bortezomib may enhance the response to chemotherapy
56. In one study of lung cancer lines, for example, NFκB was upregulated by treating the NSCLC cell lines A549 and H157 with the chemotherapeutic agent, ge- mcitabine, leading to enhanced transcription of all NFκB -regulated genes and a resistance to gemcitabine
57. Bortezomib, which inhibited this NFκB activation, was able to sensitize these cell lines to gemcitabine-induced apoptosis. In this and in many studies, bortezomib is able to enhance the response of Fig.2. The structure of proteasome and the
ubiquitins-proteasome pathway for protein degradation. Proteins are tagged with chain of ubiquitins. The resulting polyubiquitinated proteins are recognized by the 19S regulatory particles, which unfold the protein and remove the ubiquitin from the proteins. The proteins are then degraded to peptides of seven to eight amino acids long.
Fig.3. A simplified schema of the molecular me-
chanism of bortezomib-induced apoptosis.
resistant tumor cells to chemotherapy
58. Despite the consistent evidence that bortezomib inhibits the activation of NFκB pathway, it remains unclear whether this inhibition is the major mechanism of bortezomib-induced apoptosis.
Indeed, bortezomib may inhibit other survival pathways. For example, there is evidence that bortezomib inhibits the PI3K/Akt pathway and the p44/42 MAPK pathway
59,60. By these actions, bortezomib may also reduce tumor cell resistance to apoptosis
51.
II. Bortezomib alters the balance of pro-apoptotic and anti-apoptotic proteins.
There is increasing evidence that an important mechanism of bortezomib-induced apoptosis is the regulation of the balance of pro-apoptotic and anti- apoptotic Bcl-2 family proteins
7. In general, bortezomib up-regulates BH3-only proteins Bim, Noxa and Bik, and down-regulates anti-apoptotic Bcl-2 family proteins Bcl-2 and A1
61-64. Whether bortezomib induces up-regulation of pro-apoptotic proteins Bax and BH3-only protein PUMA is still controversial and may depend on the different cell types used in the experiments
61, 65. Although bortezomib has also been shown to up-regulate the anti-apoptotic Bcl-2 family protein Mcl-1 in NSCLC
62, 66, the net effect of bortezomib appears to be to shift the balance in a pro-apoptotic direc- tion. Indeed, as we discuss later, because the upregulation of Mcl-1 may limit the pro-apoptotic actions of bortezomib, current approaches are underway to inactivate Mcl-1 in hopes of enhancing the pro-apoptotic effects of bortezomib.
III. Bortezomib enhances stress pathways, p53 and JNK.
Because the degradation of p53 can be mediated by the ubiquitin-proteasome pathway
67, bortezomib treatment results in a concentration- and time-dependent accumulation of p5
38. This accumulation of p53 may promote apoptosis through activation of both the extrinsic and intrinsic
apoptosis pathways
38or may induce cell cycle arrest. In one preclinical study of NSCLC, bor- tezomib induced more apoptosis in a cell line with wild-type p53 (H460) than in a cell line with mutant p53 (H322), suggesting that bortezomib induced apoptosis via a p53-dependent pathway
8. Nonetheless, the same study showed evidence of p53-independent pathways, suggesting multiple mechanisms of apoptosis induction in lung cancer cells. JNK, the other major stress pathway, is directly activated by proteasome inhibition and the JNK pro-apoptotic activation of Bim may enhance the effects of proteasome inhibition on Bim protein accumulation
43, 68.
IV. Bortezomib arrests the cell cycle
In general, bortezomib induces apoptosis in proliferating but not in quiescent cells
4, suggesting that the cell cycle may play a role in the apoptotic effect of bortezomib. Controlled transitions between cell cycle stages depend on the timely activation of cyclin/cyclin-dependent kinase (CDK) complexes.
The CDK inhibitors p21 and p27 are short-lived pro- teins that induce cell cycle arrest by inhibiting the formation of CDK-cyclin complex. Because p21 and p27 are degraded by the proteasome
69, bortezomib increases the levels of these two CDK inhibitors, an increase which can result in cell cycle arrest. In NSCLC cell lines, bortezomib treatment was shown to arrest the cell-cycle in the G2-M phase by increasing the levels of the CDK inhibitors and cyclins A and B
8and this arrest was associated with an increased apoptosis. In another study, introduction of antisense p27 oligonucleotides into an squamous carcinoma cell line blocked the proteasome inhibitor-induced apoptosis
70, suggesting that accumulation of p27 was the key to the cell cycle arrest and apoptosis.
V. Bortezomib has multiple additional effects There is some evidence that bortezomib may increase the generation of reactive oxygen species.
In experiments using the NSCLC cell line H460,
111 Bortezomib in the Treatment of Non-small Cell Lung Cancer
bortezomib exposure increased the generation of reactive oxygen species and cytochrome c release
71. In another study, bortezomib combined with a histone deacetylase inhibitor synergistically enhanced the generation of reactive oxygen species in NSCLC and induced marked apoptosis
72. Of interest, the antioxidant agent Tiron which inhibited the bortezomib-induced reactive oxygen species generation, also blocked cytochrome c release and the bortezomib-induced apoptotic cell death
71,73. Thus generation of reactive oxygen species may also have a role in the induction of bortezomib- induced apoptosis perhaps by altering mitochondrial membrane potential and the release of cytochrome c from mitochondria
71.
Multiple other mechanisms may play a role.
For example, in NSCLC cell lines, bortezomib was also found to induce increased surface expression of death receptors DR5 and thus might sensitize cells to TRAIL-induced apoptosis despite up- regulation of the expression of the apoptosis inhibitor, survivin
74.
Summary of Bortezomib Actions Leading to Apoptosis
Thus, many preclinical studies have shown promising results in the induction of apoptosis in NSCLC cell lines and encouraged the use of bortezomib in NSCLC clinical trials. However, for unknown reasons, bortezomib has had its greatest benefit in hematologic malignancies, especially in multiple myeloma.
Bortezomib in cancer clinical trials
Multiple myeloma shows the best response to bortezomib
Bortezomib has been highly effective in patients with multiple myeloma. In relapsed or refractory multiple myeloma, bortezomib alone caused an overall response rate of 41-43%
75,76, and the overall responses were increased to 64-90%
when bortezomib was combined with other
chemotherapy
77, 78. In newly diagnosed multiple myeloma, bortezomib alone caused a overall response rate of more than 40%
79. When bortezomib was combined with other chemotherapy in newly diagnosed multiple myeloma as front-line treatment, the overall response rates were more than 80-90%
80-82. This benefit was associated with only minimal toxicity, mostly thrombocytopenia and electrolyte abnormalities
83. The effect of bortezomib thus was targeted to the tumor cells with little collateral damage to normal tissues.
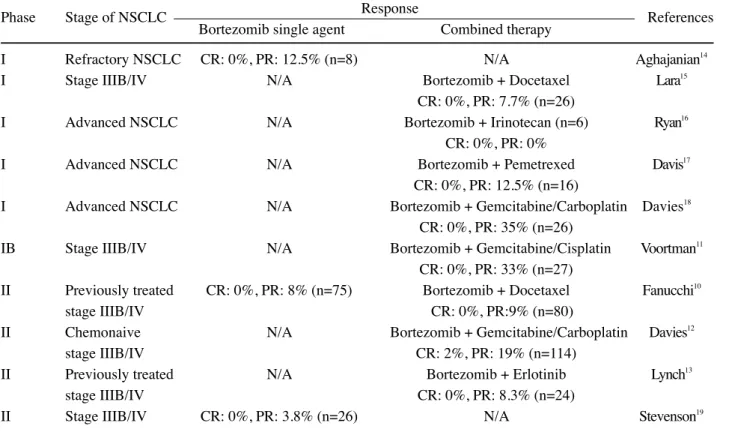
NSCLC: Reality has not yet lived up to its promise
In contrast to the encouraging results of
bortezomib in multiple myeloma, the response to
bortezomib in patients with NSCLC has not been so
promising. A summary of the results of bortezomib
in NSCLC clinical trials is listed in Table 1. One
clinical trial of bortezomib in previously treated
advanced NSCLC showed only modest single-agent
activity with an 8% partial response rate, and the
complete response rate was 0%
10. When bortezomib
was combined with docetaxel, the partial response
was 9%, and again the complete response rate was
0%
10. Besides docetaxel, bortezomib has also been
used in combination with other chemotherapeutic
agents and targeted therapy. When combined with
gemcitabine/carboplatin, bortezomib induced an
overall response rate of 21% with a complete
response rate of 2% in patients with chemotherapy-
naive stage IV and selected stage IIIB (pleural
effusion) NSCLC
12. When combined with EGFR
tyrosine kinase inhibitor erlotinib in a phase II
clinical trial, bortezomib did not show sufficient
activity and the trial was halted
13. Toxicities
included grade 3 or 4 adverse effects such as
fatigue, neutropenia, and dyspnea,
10with a high
incidence of neutropenia when bortezomib was
used in combination with docetaxel (53%). In
general, the clinical result of bortezomib treatment
has been disappointing with only a 4-13% partial
response rate when used as single agent, and 8-35%
Table 1: Results of Bortezomib in NSCLC Clinical Trials
Phase Stage of NSCLC Response References
Bortezomib single agent Combined therapy
I Refractory NSCLC CR: 0%, PR: 12.5% (n=8) N/A Aghajanian
14I Stage IIIB/IV N/A Bortezomib + Docetaxel Lara
15CR: 0%, PR: 7.7% (n=26)
I Advanced NSCLC N/A Bortezomib + Irinotecan (n=6) Ryan
16CR: 0%, PR: 0%
I Advanced NSCLC N/A Bortezomib + Pemetrexed Davis
17CR: 0%, PR: 12.5% (n=16)
I Advanced NSCLC N/A Bortezomib + Gemcitabine/Carboplatin Davies
18CR: 0%, PR: 35% (n=26)
IB Stage IIIB/IV N/A Bortezomib + Gemcitabine/Cisplatin Voortman
11CR: 0%, PR: 33% (n=27)
II Previously treated CR: 0%, PR: 8% (n=75) Bortezomib + Docetaxel Fanucchi
10stage IIIB/IV CR: 0%, PR:9% (n=80)
II Chemonaive N/A Bortezomib + Gemcitabine/Carboplatin Davies
12stage IIIB/IV CR: 2%, PR: 19% (n=114)
II Previously treated N/A Bortezomib + Erlotinib Lynch
13stage IIIB/IV CR: 0%, PR: 8.3% (n=24)
II Stage IIIB/IV CR: 0%, PR: 3.8% (n=26) N/A Stevenson
19CR: complete response rate, PR: partial response rate
when combined with other chemotherapeutic agents
72-81.
Possible explanations for the inefficacy of bortezomib in lung cancer
clinical trials
Despite the promising result of bortezomib treatment in patients with multiple myeloma and in preclinical studies of NSCLC cell lines, the response to bortezomib in NSCLC clinical trials has been unexpectedly disappointing. In considering this, we propose various possible explanations for this different treatment response to bortezomib between multiple myeloma and lung cancer. These possible explanations might provide clues for future study to improve the clinical response of NSCLC to bortezomib or to develop other novel therapies.
Possible specific mechanisms of bortezomib resistance in NSCLC:Molecular differences bet-
ween the two tumors
It is possible that there are differences in the molecular consequences of bortezomib treatment between lung cancer and multiple myeloma. By reviewing the mechanisms just described for the action of bortezomib, we present some interesting differences that have been reported between multiple myeloma and lung cancer studies.
As mentioned, bortezomib is known to alter the balance of pro-apoptotic and anti-apoptotic Bcl-2 family proteins. In general, after bortezomib treatment, there is increased expression of pro- apoptotic BH3-only proteins, such as Bim, PUMA, Noxa, and Bik and decreased expression of anti- apoptotic Bcl-2 family proteins, such as Bcl-2 and A1
7. However, in lung cancer cell lines H460 and SW 1573 following bortezomib treatment, Mcl-1 was found to be up-regulated
62,66, while Mcl-1 was shown to be down-regulated and cleaved in multiple
Bortezomib in the Treatment of Non-small Cell Lung Cancer 113
myeloma cells MM.1S, NCI-H292, and U266
84,85and perhaps also in patients with multiple myeloma
86. Indeed, the Mcl-1 expression appeared to determine the sensitivity to the bortezomib; for example, in one bortezomib-resistant multiple myeloma cell RPMI-8226, down-regulation of Mcl-1 sensitized the cells to bortezomib-induced apoptosis
85. This finding suggests that the different effects on Mcl-1 after bortezomib treatment might explain why bortezomib would be more effective in multiple myeloma but not in NSCLC. It would also suggest that interference with Mcl-1 could enhance the responses of lung cancer to bortezomib.
Anti-apoptotic molecules such as FLIP may be elevated by bortezomib differently in the two malignancies. Indeed, in one study, following bortezomib, FLIP was down-regulated in he- matopoietic malignancies, such as multiple myeloma
87. In contrast, FLIP was up-regulated in NSCLC after bortezomib exposure
74. Indeed, FLIP, best known as an inhibitor of death receptor signaling, may have anti-apoptotic effects at the mitochondria and, if so, FLIP upregulation may inhibit the response to chemotherapy
30. Again, if this were found to be a general response in lung cancer, interference with FLIP might improve the response of lung cancer to bortezomib.
Bortezomib-induced upregulation of p53 may have different effects because of inherent differences in the mutation and functional inactivation of p53 in the two tumors. A mutation of p53 gene is found in less than 20% of multiple myeloma patients
88while, on the other hand, mutations of the p53 gene and the resultant loss of tumor suppressor function are found in about 50%
of NSCLC
89. Mutations of the p53 gene can inac- tivate the protein and remove the key sensor of DNA damage
90or of other stress signals, such as hypoxia and oncogene hyperexpression, that can induce apoptosis through p53
91. Indeed, cell lines with mutant p53 are less sensitive to bortezomib-
induced apoptosis than cell lines with wild-type p53
8. Thus, even if bortezomib successfully upregulates p53, the high incidence of p53 mutation in NSCLC might account for a lower efficacy of bortezomib in NSCLC than in tumors with a lower incidence of p53 mutations.
Possible general mechanisms of bortezomib resistance in NSCLC: Differences between hematopoeitic and solid tumors
Clearly, there are general differences between any hematopoeitic and any solid tumor that should be considered. Indeed, many solid tumors demonstrate an apoptotic resistance that appears to be common among solid tumors, and can be modeled in vitro using 3D models. The mechanisms of this type of resistance, often called acquired or multicellular resistance
92, are less well understood than the specific molecular effects discussed above.
We will consider a few of them.
Solid tumors may have less drug penetrance and thus a lower effective drug concentration than hematopoeitic tumors
93. In at least one dosing study, however, bortezomib was found to have good drug penetration in many tissues, including bone marrow, kidney, liver and lung
94. In fact, the maximal bortezomib concentrations in lung and bone marrow after single or repeat dose administration were similar. Although this study did not test the penetrance into the interior of a tumor itself, this study suggests that penetrance would be adequate.
These dosing studies however do not address
whether the drug that penetrates may function well
in the interior of a solid tumor, where acidosis and
hypoxia may limit the response. In addition, as
mentioned, bortezomib works best against
proliferating cells; if so, the less active core of a
solid tumor may not respond well to bortezomib,
even if the drug successfully reaches the tumor
center. However, despite the many possible
differences we can envision between the two tumor
types, there are currently no data to point to a key
difference that explains the lack of effect of bortezomib in lung cancer.
When compared to non-solid hematopoeitic malignancies, solid tumors display a broad resistance that may be a consequence of their 3D shape. Such broad resistance to treatment may be mimicked in the laboratory when monolayers are allowed to grow into small 3D aggregates of cells called spheroids
92,95. Although the reason why bortezomib induces significant apoptosis in preclinical studies using NSCLC cell lines grown as monolayers but has poor response rate in clinical trials of lung cancer patients remains uncertain, one possibility is that the cells in monolayer culture do not exhibit the apoptotic resistance of the actual tumors. Indeed, the 3D multicellular spheroid model is thought to retain many of the characte- ristics of the tumor and to display resistance to chemotherapy and radiotherapy more similar to the actual tumor
96. Of interest, we have recently found that NSCLC cells are sensitized to apoptosis by bortezomib when they are grown as monolayers, but acquire a high level of resistance to bortezomib after they form multicellular spheroids. We are now investigating the features in the 3D spheroids that account for this apoptotic resistance. Our hope is that manipulations in vitro that restore sensitivity to bortezomib may be translated ultimately into the clinic.
Conclusion
Bortezomib is one of the novel therapeutic agents considered for lung cancer. In preclinical studies of NSCLC, bortezomib induces apoptosis through alteration of many signaling pathways. In addition, when combined with other chemothe- rapy agents or novel molecular targeted agents, bortezomib shows additive or synergistic activities.
These promising results of preclinical studies have promoted the use of bortezomib in NSCLC clinical trials. In contrast to the significant response of
bortezomib in multiple myeloma, the response of bortezomib as a single agent or in combination with other chemotherapy agents or targeted therapies in NSCLC clinical trials has been disappointing.
Some clinical trials of bortezomib in NSCLC are still ongoing, and the results of these trials might determine whether bortezomib has a role in the treatment of NSCLC. Further investigations to identify differences between bortezomib-sensitive tumors, such as multiple myeloma, and lung cancer may help to elucidate the essential molecular mechanism of bortezomib-induced apoptosis and the means to enhance the response to bortezomib in lung cancer.
Acknowledgments
The authors would like to acknowledge the support of NIH RO1 CA095671 and the Chang Gung Memorial Hospital of Taiwan.
References
1.Ettinger DS. Overview and state of the art in the management of lung cancer. Oncology (Williston Park) 2004; 18: 3-9.
2.Lally BE, Urbanic JJ, Blackstock AW, Miller AA, Perry MC.
Small cell lung cancer: have we made any progress over the last 25 years? Oncologist 2007; 12: 1096-104.
3.Ettinger DS. Is there a preferred combination chemotherapy regimen for metastastic non-small cell lung cancer?
Oncologist 2002; 7: 226-33.
4.Almond JB, Cohen GM. The proteasome: a novel target for cancer chemotherapy. Leukemia 2002; 16: 433-43.
5.Richardson PG, Barlogie B, Berenson J, et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med 2003; 348: 2609-17.
6.Milligan SA, Nopajaroonsri C. Inhibition of NF-kappa B with proteasome inhibitors enhances apoptosis in human lung adenocarcinoma cells in vitro. Anticancer Res 2001; 21:
39-44.
7. Fennell DA, Chacko A, Mutti L. BCL-2 family regulation by the 20S proteasome inhibitor bortezomib. Oncogene 2008;
27: 1189-97.
8. Ling YH, Liebes L, Jiang JD, et al. Mechanisms of proteasome inhibitor PS-341-induced G(2)-M-phase arrest and apoptosis in human non-small cell lung cancer cell lines.
Clin Cancer Res 2003; 9: 1145-54.
9. Schenkein DP. Preclinical data with bortezomib in lung cancer. Clin Lung Cancer 2005; 7 Suppl 2: S49-55.
10.Fanucchi MP, Fossella FV, Belt R, et al. Randomized phase 115 Bortezomib in the Treatment of Non-small Cell Lung Cancer
II study of bortezomib alone and bortezomib in combination with docetaxel in previously treated advanced non-small-cell lung cancer. J Clin Oncol 2006; 24: 5025-33.
11.Voortman J, Smit EF, Honeywell R, et al. A parallel dose- escalation study of weekly and twice-weekly bortezomib in combination with gemcitabine and cisplatin in the first- line treatment of patients with advanced solid tumors. Clin Cancer Res 2007; 13: 3642-51.
12.Davies AM, McCoy J, Lara PN, et al. Bortezomib
+gemcitabine (Gem)/carboplatin (Carbo) results in encouraging survival in advanced non-small cell lung cancer (NSCLC): Results of a phase II Southwest Oncology Group (SWOG) trial (S0339). Proc Am Soc Clin Oncol 2006; 24:
7017.
13.Lynch TJ, Fenton DW, Hirsh V, et al. Randomized phase II study of erlotinib alone and in combination with bortezomib in previously treated advanced non-small cell lung cancer (NSCLC). Proc Am Soc Clin Oncol 2007; 25: 7680.
14.Aghajanian C, Soignet S, Dizon DS, et al. A phase I trial of the novel proteasome inhibitor PS341 in advanced solid tumor malignancies. Clin Cancer Res 2002; 8: 2505-11.
15.Lara PN, Jr., Koczywas M, Quinn DI, et al. Bortezomib plus docetaxel in advanced non-small cell lung cancer and other solid tumors: a phase I California Cancer Consortium trial. J Thorac Oncol 2006; 1: 126-34.
16.Ryan DP, O'Neil BH, Supko JG, et al. A Phase I study of bortezomib plus irinotecan in patients with advanced solid tumors. Cancer 2006; 107: 2688-97.
17.Davies AM, Ho C, Metzger AS, et al. Phase I study of two different schedules of bortezomib and pemetrexed in advanced solid tumors with emphasis on non-small cell lung cancer. J Thorac Oncol 2007; 2: 1112-6.
18.Davies AM, Ruel C, Lara PN, et al. The proteasome inhibitor bortezomib in combination with gemcitabine and carboplatin in advanced non-small cell lung cancer: a California Cancer Consortium Phase I study. J Thorac Oncol 2008; 3: 68-74.
19.Stevenson JP, Nho CW, Johnson SW, et al. Effects of bortezomib (PS-341) on NF-κB activation in peripheral blood mononuclear cells (PBMCs) of advanced non-small lung cancer (NSCLC) patients: A phase II/pharmacodynamic trial. Proc Am Soc Clin Oncol 2004; 22: 7145.
20.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100: 57-70.
21.Gerl R, Vaux DL. Apoptosis in the development and treatment of cancer. Carcinogenesis 2005; 26: 263-70.
22.Shivapurkar N, Reddy J, Chaudhary PM, Gazdar AF.
Apoptosis and lung cancer: a review. J Cell Biochem 2003;
88: 885-98.
23.Okada H, Mak TW. Pathways of apoptotic and non-apoptotic death in tumour cells. Nat Rev Cancer 2004; 4: 592-603.
24.Pommier Y, Sordet O, Antony S, Hayward RL, Kohn KW.
Apoptosis defects and chemotherapy resistance: molecular interaction maps and networks. Oncogene 2004; 23: 2934-49.
25.Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression.
Nature 2004; 432: 307-15.
26.Elmore S. Apoptosis: a review of programmed cell death.
Toxicol Pathol 2007; 35: 495-516.
27.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science 1998; 281: 1305-8.
28.Kischkel FC, Hellbardt S, Behrmann I, et al. Cytotoxicity- dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor.
Embo J 1995; 14: 5579-88.
29.Scaffidi C, Schmitz I, Krammer PH, Peter ME. The role of c-FLIP in modulation of CD95-induced apoptosis. J Biol Chem 1999; 274: 1541-8.
30.Wang X, Wang Y, Zhang J, et al. FLIP protects against hypoxia/reoxygenation-induced endothelial cell apoptosis by inhibiting Bax activation. Mol Cell Biol 2005; 25: 4742-51.
31.Saelens X, Festjens N, Vande Walle L, et al. Toxic proteins released from mitochondria in cell death. Oncogene 2004;
23: 2861-74.
32.Igney FH, Krammer PH. Death and anti-death: tumour resistance to apoptosis. Nat Rev Cancer 2002; 2: 277-88.
33.Pohland T, Wagner S, Mahyar-Roemer M, Roemer K. Bax and Bak are the critical complementary effectors of colorectal cancer cell apoptosis by chemopreventive resveratrol.
Anticancer Drugs 2006; 17: 471-8.
34.Ruiz-Vela A, Opferman JT, Cheng EH, Korsmeyer SJ.
Proapoptotic BAX and BAK control multiple initiator caspases. EMBO Rep 2005; 6: 379-85.
35.Certo M, Del Gaizo Moore V, Nishino M, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 2006; 9:
351-65.
36.Wei MC, Zong WX, Cheng EH, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 2001; 292: 727-30.
37.Lane DP, Midgley CA, Hupp TR, et al. On the regulation of the p53 tumour suppressor, and its role in the cellular response to DNA damage. Philos Trans R Soc Lond B Biol Sci 1995; 347: 83-7.
38.Vousden KH, Lu X. Live or let die: the cell's response to p53.
Nat Rev Cancer 2002; 2: 594-604.
39.Thornborrow EC, Patel S, Mastropietro AE, Schwartzfarb EM, Manfredi JJ. A conserved intronic response element mediates direct p53-dependent transcriptional activation of both the human and murine bax genes. Oncogene 2002; 21:
990-9.
40.Li J, Lee B, Lee AS. Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-up- regulated modulator of apoptosis (PUMA) and NOXA by p53. J Biol Chem 2006; 281: 7260-70.
41.Bennett M, Macdonald K, Chan SW, et al. Cell surface trafficking of Fas: a rapid mechanism of p53-mediated apoptosis. Science 1998; 282: 290-3.
42.Wu GS, Burns TF, McDonald ER, 3rd, et al. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene.
Nat Genet 1997; 17: 141-3.
43.Abayasiriwardana KS, Barbone D, Kim KU, et al. Malignant
mesothelioma cells are rapidly sensitized to TRAIL-induced apoptosis by low-dose anisomycin via Bim. Mol Cancer Ther 2007; 6: 2766-76.
44.Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol 2002; 3: 221-7.
45.Baldwin AS, Jr. Series introduction: the transcription factor NF-kappaB and human disease. J Clin Invest 2001; 107: 3-6.
46.Stokoe D. The phosphoinositide 3-kinase pathway and cancer.
Expert Rev Mol Med 2005; 7: 1-22.
47.Pastorino JG, Tafani M, Farber JL. Tumor necrosis factor induces phosphorylation and translocation of BAD through a phosphatidylinositide-3-OH kinase-dependent pathway. J Biol Chem 1999; 274: 19411-6.
48.Cardone MH, Roy N, Stennicke HR, et al. Regulation of cell death protease caspase-9 by phosphorylation. Science 1998;
282: 1318-21.
49.Madrid LV, Wang CY, Guttridge DC, et al. Akt suppresses apoptosis by stimulating the transactivation potential of the RelA/p65 subunit of NF-kappaB. Mol Cell Biol 2000; 20:
1626-38.
50.Feng J, Tamaskovic R, Yang Z, et al. Stabilization of Mdm2 via decreased ubiquitination is mediated by protein kinase B/Akt-dependent phosphorylation. J Biol Chem 2004; 279:
35510-7.
51.Dent P, Jarvis WD, Birrer MJ, et al. The roles of signaling by the p42/p44 mitogen-activated protein (MAP) kinase pathway; a potential route to radio- and chemo-sensitization of tumor cells resulting in the induction of apoptosis and loss of clonogenicity. Leukemia 1998; 12: 1843-50.
52.Ghobrial IM, Witzig TE, Adjei AA. Targeting apoptosis pathways in cancer therapy. CA Cancer J Clin 2005; 55:
178-94.
53.Nandi D, Tahiliani P, Kumar A, Chandu D. The ubiquitin- proteasome system. J Biosci 2006; 31: 137-55.
54.Dalton WS. The proteasome. Semin Oncol 2004; 31: 3-9.
55.Panwalkar A, Verstovsek S, Giles F. Nuclear factor-kappaB modulation as a therapeutic approach in hematologic malignancies. Cancer 2004; 100: 1578-89.
56.Cusack JC, Jr., Liu R, Houston M, et al. Enhanced chemosensitivity to CPT-11 with proteasome inhibitor PS-341: implications for systemic nuclear factor-kappaB inhibition. Cancer Res 2001; 61: 3535-40.
57.Denlinger CE, Rundall BK, Keller MD, Jones DR.
Proteasome inhibition sensitizes non-small-cell lung cancer to gemcitabine-induced apoptosis. Ann Thorac Surg 2004;
78: 1207-14.
58.Richardson PG, Mitsiades C, Hideshima T, Anderson KC.
Proteasome inhibition in the treatment of cancer. Cell Cycle 2005; 4: 290-6.
59.Orlowski RZ, Small GW, Shi YY. Evidence that inhibition of p44/42 mitogen-activated protein kinase signaling is a factor in proteasome inhibitor-mediated apoptosis. J Biol Chem 2002; 277: 27864-71.
60.Fujita T, Doihara H, Washio K, et al. Antitumor effects and drug interactions of the proteasome inhibitor bortezomib
(PS341) in gastric cancer cells. Anticancer Drugs 2007; 18:
677-86.
61.Nikrad M, Johnson T, Puthalalath H, et al. The proteasome inhibitor bortezomib sensitizes cells to killing by death receptor ligand TRAIL via BH3-only proteins Bik and Bim.
Mol Cancer Ther 2005; 4: 443-9.
62.Voortman J, Checinska A, Giaccone G, Rodriguez JA, Kruyt FA. Bortezomib, but not cisplatin, induces mitochondria- dependent apoptosis accompanied by up-regulation of noxa in the non-small cell lung cancer cell line NCI-H460. Mol Cancer Ther 2007; 6: 1046-53.
63.Fahy BN, Schlieman MG, Mortenson MM, Virudachalam S, Bold RJ. Targeting BCL-2 overexpression in various human malignancies through NF-kappaB inhibition by the proteasome inhibitor bortezomib. Cancer Chemother Pharmacol 2005; 56: 46-54.
64.Pham LV, Tamayo AT, Yoshimura LC, Lo P, Ford RJ.
Inhibition of constitutive NF-kappa B activation in mantle cell lymphoma B cells leads to induction of cell cycle arrest and apoptosis. J Immunol 2003; 171: 88-95.
65.Yu J, Tiwari S, Steiner P, Zhang L. Differential apoptotic response to the proteasome inhibitor Bortezomib [VELCADE, PS-341] in Bax-deficient and p21-deficient colon cancer cells. Cancer Biol Ther 2003; 2: 694-9.
66.Voortman J, Checinska A, Giaccone G. The proteasomal and apoptotic phenotype determine bortezomib sensitivity of non-small cell lung cancer cells. Mol Cancer 2007; 6: 73.
67.Oren M. Regulation of the p53 tumor suppressor protein. J Biol Chem 1999; 274: 36031-4.
68.Roccaro AM, Hideshima T, Richardson PG, et al. Bortezomib as an antitumor agent. Curr Pharm Biotechnol 2006; 7:
441-8.
69.An B, Goldfarb RH, Siman R, Dou QP. Novel dipeptidyl proteasome inhibitors overcome Bcl-2 protective function and selectively accumulate the cyclin-dependent kinase inhibitor p27 and induce apoptosis in transformed, but not normal, human fibroblasts. Cell Death Differ 1998; 5:
1062-75.
70.Kudo Y, Takata T, Ogawa I, et al. p27Kip1 accumulation by inhibition of proteasome function induces apoptosis in oral squamous cell carcinoma cells. Clin Cancer Res 2000; 6:
916-23.
71.Ling YH, Liebes L, Zou Y, Perez-Soler R. Reactive oxygen species generation and mitochondrial dysfunction in the apoptotic response to Bortezomib, a novel proteasome inhibitor, in human H460 non-small cell lung cancer cells. J Biol Chem 2003; 278: 33714-23.
72.Denlinger CE, Rundall BK, Jones DR. Proteasome inhibition sensitizes non-small cell lung cancer to histone deacetylase inhibitor-induced apoptosis through the generation of reactive oxygen species. J Thorac Cardiovasc Surg 2004; 128: 740-8.
73.Fernandez Y, Miller TP, Denoyelle C, et al. Chemical blockage of the proteasome inhibitory function of borte- zomib: impact on tumor cell death. J Biol Chem 2006; 281:
1107-18.
117 Bortezomib in the Treatment of Non-small Cell Lung Cancer
74.Liu X, Yue P, Chen S, et al. The proteasome inhibitor PS-341 (bortezomib) up-regulates DR5 expression leading to induction of apoptosis and enhancement of TRAIL-induced apoptosis despite up-regulation of c-FLIP and survivin expression in human NSCLC cells. Cancer Res 2007; 67:
4981-8.
75.Richardson PG, Sonneveld P, Schuster M, et al. Extended follow-up of a phase 3 trial in relapsed multiple myeloma:
final time-to-event results of the APEX trial. Blood 2007;
110: 3557-60.
76.Orlowski RZ, Nagler A, Sonneveld P, et al. Randomized phase III study of pegylated liposomal doxorubicin plus bortezomib compared with bortezomib alone in relapsed or refractory multiple myeloma: combination therapy improves time to progression. J Clin Oncol 2007; 25: 3892-901.
77.Ozaki S, Tanaka O, Fujii S, et al. Therapy with bortezomib plus dexamethasone induces osteoblast activation in responsive patients with multiple myeloma. Int J Hematol 2007; 86: 180-5.
78.Kropff M, Bisping G, Schuck E, et al. Bortezomib in combination with intermediate-dose dexamethasone and continuous low-dose oral cyclophosphamide for relapsed multiple myeloma. Br J Haematol 2007; 138: 330-7.
79.Richardson PG C-KA, Schlossman RL, et al. Phase II trial of single agent bortezomib (VELCADE) in patients with previously untreated multiple myeloma (MM). Blood (ASH Annual Meeting Abstracts) 2004; 104: 336.
80.Jagannath S, Durie BG, Wolf J, et al. Bortezomib therapy alone and in combination with dexamethasone for previously untreated symptomatic multiple myeloma. Br J Haematol 2005; 129: 776-83.
81.Wang M, Giralt S, Delasalle K, Handy B, Alexanian R.
Bortezomib in combination with thalidomide-dexamethasone for previously untreated multiple myeloma. Hematology 2007; 12: 235-9.
82.Oakervee HE, Popat R, Curry N, et al. PAD combination therapy (PS-341/bortezomib, doxorubicin and dexame- thasone) for previously untreated patients with multiple
myeloma. Br J Haematol 2005; 129: 755-62.
83.Ludwig H, Khayat D, Giaccone G, Facon T. Proteasome inhibition and its clinical prospects in the treatment of hematologic and solid malignancies. Cancer 2005; 104:
1794-807.
84.Podar K, Gouill SL, Zhang J, et al. A pivotal role for Mcl-1 in Bortezomib-induced apoptosis. Oncogene 2008; 27: 721-31.
85.Gomez-Bougie P, Wuilleme-Toumi S, Menoret E, et al. Noxa up-regulation and Mcl-1 cleavage are associated to apoptosis induction by bortezomib in multiple myeloma. Cancer Res 2007; 67: 5418-24.
86.Navas TA, Nguyen AN, Hideshima T, et al. Inhibition of p38alpha MAPK enhances proteasome inhibitor-induced apoptosis of myeloma cells by modulating Hsp27, Bcl-X(L), Mcl-1 and p53 levels in vitro and inhibits tumor growth in vivo. Leukemia 2006; 20: 1017-27.
87.Mitsiades N, Mitsiades CS, Poulaki V, et al. Molecular
sequelae of proteasome inhibition in human multiple myeloma cells. Proc Natl Acad Sci U S A 2002; 99: 14374-9.
88.Neri A, Baldini L, Trecca D, et al. p53 gene mutations in multiple myeloma are associated with advanced forms of malignancy. Blood 1993; 81: 128-35.
89.Zochbauer-Muller S, Gazdar AF, Minna JD. Molecular pathogenesis of lung cancer. Annu Rev Physiol 2002; 64:
681-708.
90.Harris CC. p53 tumor suppressor gene: from the basic research laboratory to the clinic--an abridged historical perspective. Carcinogenesis 1996; 17: 1187-98.
91.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell 1997; 88: 323-31.
92.Desoize B, Jardillier J. Multicellular resistance: a paradigm for clinical resistance? Crit Rev Oncol Hematol 2000; 36:
193-207.
93.Tannock IF, Lee CM, Tunggal JK, Cowan DS, Egorin MJ.
Limited penetration of anticancer drugs through tumor tissue: a potential cause of resistance of solid tumors to chemotherapy. Clin Cancer Res 2002; 8: 878-84.
94.Hemeryck A, Geerts R, Monbaliu J, et al. Tissue distribution and depletion kinetics of bortezomib and bortezomib- related radioactivity in male rats after single and repeated intravenous injection of 14 C-bortezomib. Cancer Chemother Pharmacol 2007; 60: 777-87.
95.Desoize B, Gimonet D, Jardiller JC. Cell culture as spheroids:
an approach to multicellular resistance. Anticancer Res 1998;
18: 4147-58.
96.Olive PL, Durand RE. Drug and radiation resistance in spheroids: cell contact and kinetics. Cancer Metastasis Rev 1994; 13: 121-38.
Bortezomib引發細胞凋亡在非小細胞肺癌治療之應用
楊聰明1,2,3
Dario Barbone
2,3V. Courtney Broaddus
2,31嘉義長庚紀念醫院 呼吸胸腔科
2
Lung Biology Center, San Francisco General Hospital;
3
Comprehensive Cancer Center, University of California San Francisco, San Francisco, CA 94143, USA
摘 要
雖然癌症化學治療藥物在過去十數年間有顯著進步,非小細胞肺癌的化學治療卻仍沒有 令人滿意的療效,而且常伴隨著明顯的藥物副作用。目前有許多標靶治療藥物被研究於治療 癌症,其中蛋白解體抑制劑 (proteasome inhibitor)被發現可以導致許多腫瘤細胞株發生細胞凋 亡(apoptosis)。Bortezomib (Velcade)是第一個進入臨床試驗的蛋白解體抑制劑,由於對多發性 骨髓瘤療效顯著,因此已被核准用於治療多發性骨髓瘤。實驗也發現單獨使用 Bortezomib或 同時合併其他標靶或化學治療藥物可引發肺癌細胞株發生顯著的細胞凋亡,故Bortezomib也 進入臨床試驗治療非小細胞肺癌,但這些臨床試驗結果卻不如預期中理想。本文中我們描述 蛋白解體在細胞恆定及細胞凋亡機轉中的角色、Bortezomib導致癌細胞株發生細胞凋亡的機 轉、以及在非小細胞肺癌臨床試驗之療效,並進一步探討 Bortezomib在非小細胞肺癌臨床試 驗療效不如預期的可能原因、以及未來如何改善其療效之可能研究方向。
Bortezomib in the Treatment of Non-small Cell Lung Cancer 119