國立臺灣大學生命科學院基因體與系統生物學學位學程 博士論文
Genome and Systems Biology Degree Program College of Life Science
National Taiwan University Doctoral Dissertation
紅豆杉科與傘松科植物色質體基因組分析:洞悉其核質 體 DNA 與重組基因群
Study on the Plastomic Organizations of Taxaceae and Sciadopityaceae:
Insights into Their Nuclear Plastid DNAs and Chimeric Gene Clusters
許智堯 Chih-Yao Hsu
指導教授:趙淑妙 博士 Advisor: Shu-Miaw Chaw, Ph.D.
中華民國 105 年 7 月
July 2016
紅豆杉科與傘松科植物色質體基因組分析:洞悉其核 質體 DNA 與重組基因群
Study on the Plastomic Organizations of Taxaceae and Sciadopityaceae: Insights into Their Nuclear Plastid
DNAs and Chimeric Gene Clusters
致謝
一轉眼間五年的時間過去了!在此特別感謝指導教授趙淑妙 博 士在這段時間的指導與提攜;以及吳宗賢 博士於研究上的指導與經 驗分享,使得我的研究可以順利進行;感謝 Edi Sudianto 不厭其煩地幫 我修改論文英文;感謝科技部、中研院對於本研究的經費支持及台灣 大學提供的研究生獎學金。感謝論文口試期間王亞男 教授、可文亞 教授、林崇熙 教授、莊樹諄 教授、陳豐奇 教授及蔡怡陞 教授的批 評與指正,使得本論文可以修改的更加完備。
此外亦要感謝王婷湞學妹當我口試的記錄,以及試驗室裡趙秀鳳 小姐與許許多多的學弟妹,陪我度過了這幾年的實驗室生活,有他們 的幫助與關心使得實驗室的生活充滿了樂趣與希望,在此致上最深的 感謝與祝福。
當然也要感謝學程裡的同學們,有他們的陪伴讓我在學校時不會 孤單一個人,真的很幸運可以與大家一起修課、討論、學習成長,我 想這也是博士生涯中最值得懷念的部分了。祝福你們研究順利。
最後,本論文謹獻給我最親愛的家人,感謝你們的支持與鼓勵,
讓我無後顧之憂的完成學業。
祝福各位身體健康!再次謝謝你們。
TABLE OF CONTENTS
中文摘要 ... I ABSTRACT ... III
CHAPTER 1. Background and Significance ... 1
1.1 General Characters of Plastids and Plastomes ... 1
1.2 Plastomic Organization ... 2
1.3 Plastomic Rearrangements ... 3
1.4 Transcription of Plastid Genes ... 5
1.5 Regulation of Plastid Gene Transcription ... 6
1.6 Applications of Plastomic sequences for Addressing Plant Evolution ... 8
1.7 Plastid Inheritance ... 9
1.8 What are Gymnosperms? ... 10
1.9 Why Cupressophyta? ... 11
1.10 Research Purposes ... 12
CHAPTER 2. Ancient Nuclear Plastid DNA in the Yew Family ... 14
2.1 Introduction ... 14
2.2 Materials and Methods ... 16
2.2.1 DNA Extraction, Sequencing, and Genome Assembly... 16
2.2.2 Genome Annotation and Sequence Alignment ... 16
2.2.3 Exploration of Single-Nucleotide Polymorphisms (SNPs), Indels, and Simple Sequence Repeat (SSR) Sequences ... 17
2.2.4 Construction of Ancestral Plastomic Organization ... 17
2.2.5 PCR Amplification, Cloning, and Sequencing ... 17
2.2.6 Phylogenetic Tree Analysis ... 18
2.2.7 Estimation of Mutations in Nuclear Plastid DNAs and Their Plastomic Counterparts ... 18
2.2.8 Plastome Map and Statistical Analyses ... 19
2.3 Results ... 19
2.3.1 Reduction and Compaction of the Plastome of T. mairei ... 19
2.3.2 Intra-species Variations in the Plastomes of T. mairei ... 20
2.3.3 Retrieval of Ancestral Plastome Sequences in Taxaceae ... 21
2.3.4 Characteristics of Potential Nupt Amplicons ... 22
2.3.5 Evolution of Nupt Sequences in Taxaceae ... 23
2.3.6 Ages of Nupts in Taxaceae ... 24
2.4 Discussion ... 24
2.4.1 Labile Plastomes of Yew Family and Their Impact on Phylogenetic Studies ... 24
2.4.2 PCR-Based Approach in Investigating Nupts: Pros and Cons ... 26
2.4.3 Nupts Are Molecular Footprints for Studying Plastomic Evolution ... 27
CHAPTER 3. Birth of Four Chimeric Plastid Gene Clusters in Sciadopitys verticillata ... 29
3.1 Introduction ... 29
3.2 Materials and Methods ... 30
3.2.1 DNA Extraction ... 30
3.2.2 Sequencing, Plastome Assembly, and Genome Annotation ... 31
3.2.3 Estimates of Dispersed Repeats and Plastomic Inversions ... 31
3.2.4 Detection of Isomeric Plastomes ... 32
3.2.5 Detection of RNA Transcripts in Chimeric Gene Clusters ... 32
3.3 Results and Discussion ... 32
3.3.1 Loss of IRA from S. verticillata Plastome ... 32
3.3.2 Pseudogenization of Four tRNA Genes after Tandem Duplications ... 34
3.3.3 Evolution of Plastid trnI-CAU Genes in S. verticillata ... 34
3.3.4 Presence of Two Isomeric Plastomes in S. verticillata ... 35
3.3.5 Birth of Four Chimeric Gene Clusters ... 36
3.3.6 Evolutionary Effects of Novel Chimeric Gene Clusters ... 38
CHAPTER 4. Conclusions ... 40
CHAPTER 5. Future Prospectives ... 42
FIGURES ... 43
TABLES ... 62
REFERENCES ... 69 PUBLICATIONS ... 84
LIST OF FIGURES
Figure 1. The phylogenetic tree of endosymbiotic evolution ... 43
Figure 2. Fate of cyanobacterial genes and the intracellular targeting of their products in the flowering plant Arabidopsis thaliana... 44
Figure 3. A schematic explanation for the amplification of ancestral plastomic DNAs
transferred from plastids to the nucleus. ... 45 Figure 4. Hypothetical evolutionary scenarios for plastomic rearrangements in Taxaceae. .... 46
Figure 5. An alignment revealing two premature stop codons in the chlB sequence of Cep-2…
... 47
Figure 6. A dot-plot comparison of the plastomes of Amentotaxus formosana and Taxus mairei ... 48
Figure 7. A neighbor-joining tree inferred from a whole-plastome alignment ... 49
Figure 8. Stacked histogram for single-nucleotide polymorphisms (SNPs), indels, and indel lengths of the T. mairei plastome ... 50
Figure 9. Distribution of single-nucleotide polymorphisms (SNPs), indels, and simple
sequence repeats (SSRs) in the plastomes of Taxus mairei ... 51
Figure 10. An unrooted tree inferred from the locally collinear block matrix generated from comparative plastomes ... 52
Figure 11. Origin of the obtained PCR amplicons examined by maximum-likelihood
phylogenetic analyses ... 53 Figure 12. Alignment of seven rps8 sequences. ... 54
Figure 13. Percentage of nucleotide mutation classes in nupts and their plastomic counterparts.
... 55
Figure 14. Plastome map of Sciadopitys verticillata. ... 56
Figure 15. Comparison between the two copies of trnI-CAU genes in the Sciadopitys plastome. ... 57
Figure 16. Co-existence of two isomeric plastomes in Sciadopitys. ... 58
Figure 17. Postulated scenarios for the plastomic inversions in Sciadopitys ... 59
Figure 18. Birth of chimeric gene clusters in the Sciadopitys plastome ... 60
LIST OF TABLES
Table 1. Plastid and mitochondria RNA polymerases in higher plants. ... 62
Table 2. PCR primers used for the nupt study. ... 63
Table 3. Characteristics of obtained PCR amplicons in the nupt study. ... 64
Table 4. Mutations in nupts and their plastomic counterparts. ... 65
Table 5. Primers used in Sciadopitys project. ... 66
Table 6. Genes predicted in the plastome of Sciadopitys. ... 67
Table 7. Presence of trnI-CAU copies in the plastomes of cupressophytes. ... 68
摘 要
針葉樹植物 (conifers)分為松科 (Pinaceae) 與柏門 (Cupressophyta) 兩大群,其中以柏門植物最具多樣性及高經濟價值。現生的柏門植物又分為五個 科,共約有 400 多種。種子植物 (seed plants) 色質體基因組 (plastomes)結 構相當保守,但柏門植物的色質體基因組結構卻有高度的變異性。為了更深入的 探討色質體基因組重組在演化上的意義與影響,本論文利用比較色質體基因體學 的方法研究以下兩個題目。
第一,提出新的方法篩選及研究紅豆杉科植物核質體 DNA (nuclear plastid DNA or nupt)。核質體 DNA 是一群由色質體基因組轉移到核基因組中的 DNA 片段。
核質體 DNA 的移轉為核基因組提供了豐富的遺傳資源,也提高了核基因組的遺傳 多樣性。但目前為止,沒有針葉樹植物核質體 DNA 的相關研究,因此本研究定序 了台灣穗花杉 (Amentotaxus formosana) 及台灣紅豆杉 (Taxus mairei) 的完 整色質體基因組,並利用比較基因體學的方法分析穗花杉、紅豆杉及粗榧屬的色 質體基因組排列方式,進而推測出紅豆杉科植物祖先色質體基因組的組成與排列 方式。由此,我們設計專一性的引子來增幅祖先型及現生紅豆杉科植物色質體基 因組的非共線型區域 (non-syntenic region)。利用這方法我們一共篩選出 12.6 kb 的核質體 DNA,這些核質體 DNA 明顯地累積較多 GC 變成 AT 的突變。此外,藉
由比較祖先型核質體 DNA 與現生核質體 DNA 的rps8 基因,我們發現現生核質體
DNA 的rps8 基因轉譯起始碼子包含了一個 C 變成 U 的 RNA 修飾。我們的研究進
一步的指出紅豆杉科植物的色質體基因組大約在白堊紀就已經轉移到核基因組 中,這些核質體 DNA 不僅保留了祖先型色質體基因組排列方式與核酸組成,也提 供了線索,讓我們可以瞭解與研究針葉樹色質體基因組的演化過程。
第二,利用實驗的方法驗證與探討色質體基因組序列重組對於傘松科植物演 化上的意義與影響。種子植物的色質體基因轉錄時大多是以操縱組 (operon) 的 模式進行,也就是多個基因同時轉錄 (polycistronic transcription); 種子植
物的色質體基因組操縱組相當保守,即使像針葉樹的色質體基因組排列方式經過 多次的重組,但它們的操縱組也很少遭到破壞。本研究定序了完整的傘松 (Sciadopitys verticillata)色質體基因組。傘松科植物目前僅存一種,基因體 比較分析後發現傘松的色質體基因組有三項特點: 色質體基因組排列經過多次 的 重 組 、 包 含 了 三 組 連 續 並 退 化 的 tRNA 基 因 (trnV-GAC, trnQ-UUG 及
trnP-GGG )、有一個特殊的反向重複序列 (inverted repeat),而這個反向重
複序列可形成不同構型 (isomeric) 的色質體基因組。此外,傘松的色質體基因 組因重組打斷了多個原本的操縱組,而這些斷裂的操縱組卻重新組合而形成四個 重組基因群 (chimeric gene clusters)。我們的數據顯示這些新的重組基因群 保留有原本的啟動子(promoter) ,且可以順利地轉錄出其相對應的 RNA 序列。本研究結果使我們能更深入的了解為何針葉樹植物色質體基因組具有多樣性及 高複雜度的特性。
關鍵詞: 色質體基因組、基因組重組、演化、核質體 DNA、基因群、紅豆杉科、
傘松科、針葉樹
ABSTRACT
Cupressophytes (Cupressophyta or conifers II) is the largest of the two conifer groups, the other being Pinaceae. Cupressophytes comprise about 400 species in five families. They are the most diversified and economically valuable group in conifers. Our previous studies showed that gene organizations of their plastid genomes (plastomes) are highly variable among the few elucidated plastomes of cupressophytes. To further decipher the evolution of plastomic re-organization, I carried out comparative plastome studies of two cupressophytes families; Taxaceae and Sciadopityaceae.
Here we reported two major findings. First, I proposed a new strategy for identification and evolutionary studies of nuclear plastid DNAs (nupts) in Taxaceae.
Plastid-to-nucleus DNA transfer provides a rich genetic resource to the complexity of plant nuclear genome architecture. To date, the evolutionary fates of nupt remain unknown in conifers. We have sequenced the complete plastomes of two yews,
Amentotaxus formosana and Taxus mairei. Comparative plastomic analyses
revealed possible evolutionary scenarios for plastomic reorganization from ancestral to extant plastomes in the three sampled Taxaceae genera, Amentotaxus,Cephalotaxus, and Taxus. Specific primers were designed to amplify non-syntenic
regions between ancestral and extant plastomes, and 12.6 kb of nupts were identified based on phylogenetic analyses. These nupts have significantly accumulated GC-to-AT mutations, suggesting a nuclear mutational environment shaped by spontaneous deamination of 5-methylcytosin. The ancestral initial codon of rps8 is retained in the Taxus nupts, but its corresponding extant codon is mutated and requires C-to-U RNA-editing. These findings suggest that nupts can helprecover scenarios of the nucleotide mutation process. We also demonstrated that the Taxaceae nupts we retrieved may have been retained since the Cretaceous period of Mesozoic Era and they carry the information of both ancestral genomic organization and nucleotide composition, which offer clues for understanding the plastome evolution in conifers.
Second, we used experimental data to show the evolutionary impact of plastomic rearrangements in Sciadopityaceae. Many genes in the plastid genomes of seed plants are organized in polycistronic transcription units known as operons.
These plastid operons are highly conserved, even among conifers whose plastomes are highly rearranged. We sequenced the complete plastome sequence of
Sciadopitys verticillata (Japanese umbrella pine), the sole member of
Sciadopityaceae. The Sciadopitys plastome is characterized by extensive inversions, pseudogenization of tRNA genes after tandem duplications, and a unique pair of inverted repeats involved in the formation of isomeric plastomes. We showed that plastomic inversions in Sciadopitys have led to the shuffling of remote operons, resulting in the birth of four chimeric gene clusters. Our data also suggested that these chimeric gene clusters have adopted pre-existing promoters for the transcription of genes. This newly deciphered plastome of Sciadopitys advances our current understanding of how the conifer plastomes have evolved toward increased diversity and complexity.Keywords: Plastome, Genomic reorganization, Evolution, Nupt, Gene cluster, Taxaceae, Sciadopityaceae, Conifer.
CHAPTER 1
Background and Significance
1.1 General Characters of Plastids and Plastomes
Plastids are specialized organelles found in land plants and green algae. They usually contain diverse pigments and are responsible for photosynthesis. Chloroplast contains a type of plastids with green pigments, also known as chlorophylls. The chloroplast is enclosed by two envelope membranes that consist of an inner and an outer membrane (Wise et al., 2006). Chloroplasts use light energy and carbon dioxide as resources to produce starch and oxygen, which maintains the atmospheric oxygen level and provides essential energy for all of the lives on earth (Bryant et al., 2006).
Plastids were once free-living cyanobacteria engulfed by a eukaryotic precursor about two billion years ago (Martin et al., 2002; Archibald, 2009). They have their own genomes but the genome sizes are relatively small as compared to those of free-living prokaryotic ancestors (Figure 1). For example, the genomes of two reported cyanobacteria, Nostoc PCC7120 and N. punctiforme, are 6.4 Mb and 9 Mb in sizes with
~5,400 and ~7,200 proteins encoded, respectively (Timmis et al., 2004). In contrast, the plastid genomes (plastomes) of seed plants have an average size of about 145 kb with only 20 to 200 proteins encoded (Timmis et al., 2004; Jansen RK, 2012). This extreme reduction of plastomes was hypothesized as a result of bulk gene transfers from plastomes to the nucleus during early plastome evolution (Martin et al., 1998; Sheppard et al., 2008). Previous studies demonstrated that during the evolutionary history of plastomes, the vast majority of cyanobacterial genes were either lost or transferred to the nucleus and mitochondria of their host cells (Timmis and Scott, 1983; Mochizuki et
al., 2008). For instance, a comparative study of the three genomes of Arabidopsis (nuclear, mitochondrial, and plastid genomes) and the cyanobacterial genomes indicated that Arabidopsis nuclear genome includes about 4,100 genes that are of cyanobacterial origin and approximately 1,300 of these were sent back to plastid (Kleine et al., 2009) (Figure 2). These data suggest that the DNA transfer from plastid to nucleus resulted in massive relocation of organelle genes and shaped the size of nuclear genome during organelle evolution.
Interestingly, the transfer of genes or DNA fragments between plastomes and nucleus is still an on-going process in flowering plants. Michalovova et al. (2013) analyzed the nuclear-encoded plastid DNA (nupt; Richly and Leister, 2004) in six completely sequenced plant species (Arabidopsis thaliana, Vitis vinifera, Sorghum
bicolor, Glycine max, Oryza sativa, and Zea mays). Their results indicated that most nupts are close to centromeres and that longer nupts showed lower divergence from
plastid DNA than shorter nupts in these six species. Taken together, these data suggested that nupts are newly transferred into nuclear genomes. Why do plastids tend to transfer genes to the nucleus? Population genetic study indicated that deleterious mutations can accumulate rapidly in asexual populations but can be minimized through sexual recombination (Muller, 1932, 1964; Moran, 1996; Allen, 2015; de Vries et al., 2016).Therefore, to avoid the deleterious mutations in the asexual plastids, gene transfer from the plastome to nucleus could increase the recombination rates, reduce the genetic load of plastomes (Martin et al., 1998), and contribute to a great diversity of new genetic materials for the generation of new genes (Timmis et al., 2004).
1.2 Plastomic Organization
Plastomes of most land plants are highly conserved in size and structure. In general,
the plastomes of seed plants are circular with conserved genomic organization, including a large single copy (LSC), a small single copy (SSC), and a pair of inverted repeats (IRs) (Palmer 1983, 1991). The size of seed plant plastomes ranges from 120 to 160 kb, depending on different species (Palmer, 1985). The typical IRs of seed plant plastomes are about 20 to 25 kb.
However, a number of seed plant groups have lost (or highly reduced) one of their IRs. The IRs were likely independently lost at least five times in seed plants phylogeny (Jansen and Ruhlman, 2012). Within angiosperms, loss of an IR has been found in the plastomes of Fabaceae (Wojciechowski et al., 2004), Geraniaceae (Downie and Palmer, 1992) and two genera of Orobanchaceae (Palmer et al., 1991). Among gymnosperms, IR loss was only reported in conifers, Pinaceae, and cupressophytes (Raubeson and Jansen, 1992; Lin et al., 2010; Wu, Lin et al., 2011; Hsu et al., 2014). Recent comparative plastomic analyses suggested that Pinaceae lost IRB and that cupressophytes lost IRA, and that plastomes of conifers not only have lost an IR but also exhibited frequent plastome rearrangements (Wu, Wang et al., 2011; Wu et al., 2014).
However, whether these genome organizations are evolutionarily adaptive or associated with the loss of IR remain to be investigated.
1.3 Plastomic Rearrangements
Plastome rearrangements have been considered an evolutionary adaptation because they can create new gene clusters or isomeric forms (Cui et al., 2006). As mentioned earlier, most of the seed plant plastomes are structurally conserved. However, there are some exceptional cases in both angiosperms and gymnosperms.
In angiosperms, Campanulaceae, Fabaceae, and Geraniaceae have experienced bursts of gene order changes (Jansen and Ruhlman, 2012). Comparative analyses among
18 Campanulaceae plastomes indicated that their genome orientation changed at least 42 times, including 18 large insertions (over 5 kb), five IR expansions/contractions, and several small inversions (Cosner, 1993; Cosner et al., 2004). Fabaceae are known to exhibit a high degree of gene order change in their plastomes, including the loss of IR, transfer of plastomic genes to the nucleus, intron losses, gene duplications, and inversions (Milligan et al., 1989; Gantt et al., 1991; Doyle et al., 1995; Doyle et al., 1996; Wojciechowski et al., 2004; Cai et al., 2008; Magee et al., 2010). The plastomes of Geraniaceae are highly rearranged because of the genome inversions and expansion/contraction of IR regions (Chumley et al., 2006; Guisinger et al., 2008;
Blazier et al., 2011; Guisinger et al., 2011; Weng et al., 2013). For example, the plastome of Pelargonium hortorum has undergone at least 12 inversions and eight IR expansion/contraction changes (Chumley et al., 2006). However, reconstruction of evolutionary scenarios for these genome changes is not possible due to insufficient sampling. More genome sequencing data within each genus will be required to construct a reliable evolutionary model (Jansen and Ruhlman, 2012).
In gymnosperms, cupressophytes have been reported as the only group that exhibits a high level of gene order change. Cryptomeria japonica, the first completed plastome in cupressophytes, accumulates a lot of direct and inverted repeats, and at least 15 inversions in its plastome (Hirao et al., 2008). The gene order and genome structure of Cryptomeria plastomes are significantly different compared to previously reported land plant plastomes. In addition, similar to the plastome of Cryptomeria, those of
Agathis dammara, Nageia nagi, Calocedrus formosana, and four Juniperus species also
have extensive rearrangements (Guo et al., 2014; Wu and Chaw, 2014). However, to date, no experimental data has ever demonstrated that the rearranged plastomes of cupressophytes can create new co-transcriptional gene clusters.1.4 Transcription of Plastid Genes
Plastids are plant organelles that posses their own genomes and transcription systems (Allen, 2015). Because of their cyanobacterial origin, plastids retain a prokaryotic-type transcription system and a eubacterial RNA polymerase (Igloi and Kössel, 1992; Ishihama, 2000). Therefore, plastids and eubacteria have a similar gene expression system. Most of the plastid-encoded genes are arranged in operons. These genes in operons are co-transcribed into polycistronic RNA precursors, some of which may be post-processed and cut into monocistronic units for translation (Barkan, 1988;
Stern et al., 2010; Meierhoff et al., 2003). Moreover, their operon organizations are usually conserved among higher plants (Jansen and Ruhlman, 2012). The plastids of higher plants include at least two types of plastomic transcription systems ─ the plastid-encoded RNA polymerase (PEP) and the nuclear-encoded plastid RNA polymerase (NEP) (Shiina et al., 2005).
PEP, a bacterial-type gene expression system, is a highly efficient transcription component in higher plants and contributes over 80% of all plastid transcripts (Zhelyazkova et al., 2012). Basically, it is composed of two major subunits, the core enzyme and sigma factor subunits. The core enzyme subunit is encoded by plastomes (rpoA, rpoB, rpoC1, and rpoC2) and catalyzes the activity of RNA synthesis (Ohyama et al., 1986; Hudson et al., 1988; Sexton et al., 1990). These four rpo genes are present in the plastomes of photosynthetic plants and algae and share high sequence similarity with their bacterial counterparts (Morden et al., 1991; Sakai et al., 1998). In contrast, the activity of the plant PEP system needs additional nuclear-encoded subunit, called sigma factor. These sigma factor subunits are encoded by nuclear genome (SIG1, SIG2, SIG3,
SIG4, SIG5, and SIG6) and responsible for recognition of promoter regions and
transcription initiations (Shirano et al., 2000; Kanamaru et al., 2001; Hanaoka et al.,2003; Privat et al., 2003; Tsunoyama et al., 2004; Ishizaki et al., 2005) (Table 1). Allison (2000) suggested that multiple sigma factors may lead the PEP to specific promoters during plastid and plant development.
NEP, a phage-type RNA polymerase, has high sequence similarity with both T3/T7 phage and mitochondrial polymerase enzymes (Allison et al., 1996; Kapoor et al., 1997;
Gray and Lang, 1998; Hedtke et al., 2000;). NEP is specifically encoded by three nuclear genes (RpoTp, RpoTm, and RpoTmp) (Table 1) (Hedtke et al., 1997). The rpoT family is present in most plant species (Ortelt and Link, 2014). RpoTp proteins play an important role in a catalytic subunit of NEP and serve as the second transcription activity in higher plant plastids (Young et al., 1998; Kobayashi et al., 2001). RpoTmp proteins might be responsible for the early stage of seedling development and in the greening process of leaves. Experimental evidence from Arabidopsis showed that RpoTmp-deficient mutants had a significant delay of the greening process, altered leaf shape, and a defect in the light-induced accumulation of several plastid mRNAs (Baba et al., 2004). In addition, a biochemical study on spinach showed an unidentified nuclear-encoded RNA polymerase, NEP-2 (Bligny et al., 2000). In contrast to NEP, the NEP-2 containing fractions do not include a phage-like enzyme. NEP-2 can recognize the T7 and rrnPc promoters, and is responsible for the transcription of rRNA operon (Shiina et al., 2005).
1.5 Regulation of Plastid Gene Transcription
Regulation of plastid gene transcription is still unclear. Three major mechanisms might account for their regulation. First, there is a partition of labor in which PEP transcribes genes associated with photosynthesis and NEP transcribes housekeeping genes (Hajdukiewicz et al., 1997; Pfannschmidt, Nilsson, Tullberg et al., 1999;
Pfannschmidt, Nilsson, and Allen, 1999). Liere and Maliga (2001) proposed that NEP plays a major role at early stage of plastid development and transcribes plastome-localized genes involved in PEP core enzyme subunit. Subsequently, it was demonstrated that PEP does transcription during plastid development and is responsible for the transcription of photosynthesis-related genes (Liere and Maliga, 2001).
Second, transcription of plastid genes might be regulated by multiple promoters (Legen et al., 2002; Liere and Börner, 2007; Börner et al., 2015). An alternative promoter has evolved to perform plastomic transcription because many plastid genes are transcribed by both the NEP and PEP promoters. For example, Legen et al. (2002) analyzed the plastid transcription profiles of the entire plastome from a wild-type and PEP-deficient tobacco. Their results indicated that the functional integration of PEP and NEP is feasible and that the plastid genes are completely transcribed well in both wild-type and PEP-deficient tobacco.
Third, the entire plastome can be transcribed via read-through transcriptions which allow the expressions of downstream of 3’ untranslated regions (Quesada-Vargas et al., 2005). Northern-blot analyses confirmed that read-through transcripts were present and took about 30% of total transcripts in the transgenic tobacco plastomes (Quesada-Vargas et al., 2005). Moreover, they constructed a transgenic line that disrupts the 16S rrn operon by insertion of a foreign operon. The transcription of disrupted 16S rrn operon should be affected by the terminator of the foreign operon (Quesada-Vargas et al., 2005).
Then the downstream genes of the foreign operon would not be transcribed and the chloroplast protein synthesis would be changed. However, transcription of the whole 16S rrn operon was detected in this transgenic line and the phenotype of this transgenic lines and wild-type were similar (Quesada-Vargas et al., 2005). These results indicated that the read-through transcripts were sufficient for chloroplast development.
1.6 Applications of Plastomic sequences for Addressing Plant Evolution
Plastomes provide rich information for resolution of phylogenies at different elevels of plants because of their lower nucleotide substitution rates. For example, Lin et al.
(2010) reported a comparative plastomic study to resolve the previously controversial classifications in Pinaceae. They used 49 plastomic protein-coding genes common to 19 gymnosperms, including 15 species from eight Pinaceous genera, to reconstruct the phylogenetic trees of Pinaceous genera. Their phylogenetic tree suggested that Cedrus is clustered with Abies-Keteleeria and that Cathaya is closer to Pinus than to Picea or
Larix-Pseudotsuga. Their molecular dating also suggested that Pinaceae first evolved
during Early Jurassic and diversified during mid-Jurassic and Low Cretaceous.Furthermore, Seong and Offner (2013) used the matK gene to construct a phylogenetic tree of conifers. Their study linked the phylogeny and phenotype dates of conifers, like leaf type, seeds, and cones. They hypothesized that the environmental selection was the major force that drives the evolution of leaf phenotypes in conifers. Their study also supported the hypothesis that North American pines originated from Asian pines.
In addition, structural characters of plastomes, such as gene order inversions, genome rearrangements, expansion/contraction of IR, loss/gain of genes, and disruption of operons, can serve as good resources for phylogenetic inference (Raubeson and Jansen, 2005; Wu and Chaw, 2014; Hsu et al., 2016). A phylogenetic study of 81 plastid genes common to 64 seed plants showed a positive correlation among the numbers of gene order changes, gene losses, and lineage-specific rate accelerations (Jansen et al., 2007). The structural changes of plastomes generate a large amount of variability and some of these variations are accumulated during plastome evolution (Maréchal and Brisson, 2010).Wu and Chaw (2014) inferred phylogenetic trees of gymnosperms based on the plastome organizations. They used the matrices compiled from locally collinear
blocks (LCBs) of plastomic architectures to construct a phylogenetic tree for extant gymnosperms. Their phylogenetic trees provided structural evidence for supporting the gnepines hypothesis and for the loss of different IR copies in Pinaceae and cupressophytes. They also suggested that at least two mechanisms, mutational burden (genome reduction) and rearrangement association (genome expansion), are involved in the variation of plastomic size in cupressophytes.
1.7 Plastid Inheritance
Studies have shown that plastid DNA is mainly inherited from the maternal parent in angiosperms (Corriveau and Coleman, 1988; Mogensen, 1996; Birky, 2003; Zhang et al., 2003). To date, only Actinidia speciose reportedly has the paternal inheritance of plastids in angiosperms (Testolin and Cipriani, 1997). About 80% of angiosperms species inherit plastids from their maternal parents, while the remaining species inherit them from both parents (biparental inheritance) (Jansen and Ruhlman, 2012). Hu et al.
(2008) argued that maternal inheritance is the ancestral feature and that maternal inheritance has been converted to biparental inheritance and evolved independently among derived lineages. Phylogenetic studies also indicated that changes in the mode of inheritance are unidirectional. There have been no report of the occurence of maternal inheritance from biparental or paternal ancestors. However, Hansen et al. (2006) showed that intraspecific crosses of Passifloraceae resulted in primarily maternal-inherited plastids, whereas interspecific crosses had paternal-inherited plastids.
This may be due to a functional incompatibility between interspecific genomes, which fails to exclude paternal DNA.
In contrast to angiosperm plastids, most of the gymnosperm plastids are paternally inherited (Stine et al., 1989). Mogensen (1996) further indicated that cycad, ginkgo, and
gnetophyte plastids are of maternal inheritance, and most of the conifer plastids are of paternal inheritance except Cryptomeria (Ohba et al., 1971) and Larix (Szmidt et al., 1987). It is still a mystery why plastid inheritances are so variable among gymnosperms and angiosperms. To fully understand the variation of the modes of plastid inheritance, a broader examination of every representative genus from gymnosperms and angiosperms will be required.
1.8 What are Gymnosperms?
Gymnosperms are a group of seed plants, characterized by their “naked seeds”
(Conway, 2013). Their ovules are naked prior to fertilization. Most of the gymnosperm seeds are borne on the surface of woody, scales, which form a cone. In contrast, angiosperm seeds are covered with mature ovaries or fruits. There are about 1,000 extant species of gymnosperms in five major groups: Pinaceae (conifers I), cupressophytes (conifers II), cycads, ginkgo, and gnetophytes (Gymnosperms on The Plant List, May 2016). Among living gymnosperms, conifers are the most species-rich group, followed by cycads, gnetophytes, and ginkgo.
Gymnosperms are extremely diversified and difficult to classify based on their morphological characteristics alone. In the past decades, molecular data have been widely used to re-examine the traditional classifications of gymnosperms. Chaw et al.
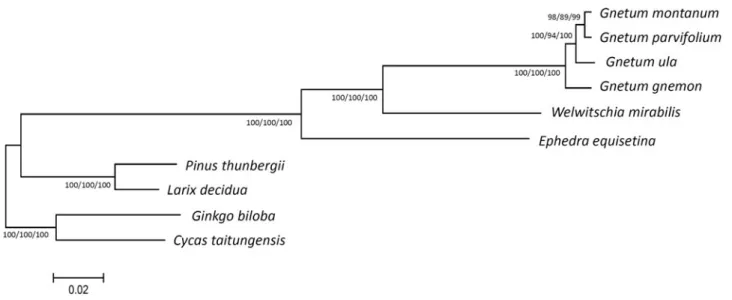
(2000) suggested that the extant gymnosperm orders are monophyletic, and none of them alone is a sister of angiosperms. Later, phylogenetic studies further indicated that cycads are sister to ginkgo (Conway, 2013; Wu et al., 2013). However, the placement of gnetophytes has been controversial. After the studies of Chaw et al. (2000) and Bowe et al. (2000), most studies agreed that gnetophytes are a sister clade to the Pinaceae, widely known as the “gnepines” hypothesis.
Conifers are evolutionarily more recent than the other groups of gymnosperms (Seong and Offner, 2013). They are the most abundant extant group of gymnosperms, containing six families and over 600 species (Conway, 2013). Almost all conifers are trees and evergreens. Although conifers contain much fewer species than the flowering plants, they are ecologically and economically important. They dominate forests in high latitudes of Northern and Southern Hemisphere and high altitudes of tropical and subtropical areas.
1.9 Why Cupressophyta?
Cupressophyta (common name: cupressophytes), the most diversified and valuable group of conifers, include ca. 400 species in five families: Araucariaceae, Cupressaceae, Podocarpaceae, Sciadopityaceae, and Taxaceae. They are of great economic value in terms of wood production, resins, pharmaceutical drugs, and horticulture. Notably, their plastome organization is diverse (Hirao et al., 2008; Wu, Lin et al., 2011), therefore, they are ideal materials for studying the evolution and mechanisms of plastome rearrangements.
Taxaceae (the yew family), a family of cupressophytes, comprises 28 species in six genera: Amentotaxus, Austrotaxus, Cephalotaxus, Pseudotaxus, Taxus, and Torreya.
They are mainly distributed in the Northern Hemisphere. Amentotaxus includes five species restricted to subtropical Southeastern Asia, from West Taiwan across Southern China to Assam in the eastern Himalayas and south of Vietnam (Cheng et al., 2000).
The genus Taxus include seven species, best known for their anti-cancer component taxol. They commonly occur in the understories of moist temperate or tropical mountain forests (de Laubenfels, 1988).
Among the conifer families, Sciadopityaceae is the only one that includes the sole member Sciadopitys verticillata (abbreviated Sciadopitys), which is an evergreen tree that can reach 27 m tall. Its spectacular needle-like leaves are arranged in whorls, like an umbrella. Thus, Sciadopitys verticillata is commonly called the Japanese umbrella pine. Three genome-based (Chaw et al., 2000) and plastome-based (Rai et al., 2008) phylogenetic studies are congruent in placing the genus Sciadopitys as sister to Taxaceae and Cupressaceae. Recent molecular dating suggests that Sciadopitys diverged from other cupressophytes more than 200 million years ago (Crisp and Cook, 2011).
Although Sciadopitys is considered a living fossil endemic to Japan, paleobiogeographic evidence indicates that its ancestors existed in China during the early and middle Jurassic (Jiang et al., 2012).
1.10 Research Purposes
As mentioned above, the plastomes of cupressophytes are highly rearranged.
Therefore, they are ideal materials for studying the following two projects.
First, we aimed to propose a new strategy for the identification and evolutionary study of nupts in yews. Previously, most of the studies on nupts were based on a comparison between nuclear and plastid genomes. This approach, however, is impractical because the nuclear genome sizes of cupressophytes are huge (ranging from 6.3 to 20 Gb/1C; Murray BG., 1998). In this study, we propose an approach to study
nupts based on comparative plastomes. A proof-of-concept study using yew plastomes
is described (in Chapter 2 below).Second, we aimed to understand the evolutionary effect of the plastomic rearrangements. Two questions were asked: (1) Do plastomic rearrangements alter the transcription, translation, or end products of plastomes? And (2) Can these
rearrangements disrupt any functional operons and create new gene-clusters? To date, consequences of genomic rearrangements are still poorly understood in gymnosperms.
Sciadopityaceae provides a unique opportunity to address these questions as its plastome is highly rearranged. Here we used experimental data to approach these two questions (Chapter 3).
CHAPTER 2
Ancient Nuclear Plastid DNA in the Yew Family
2.1 Introduction
Previous comparative genomic studies indicated that on average about 14% of the nuclear-encoded proteins were acquired from the cyanobacterial ancestor of plastids (Deusch et al., 2008). Transgenic experiments also demonstrated a high frequency of plastid-to-nucleus transfers with one event per 11,000 pollen grains or per 273,000 ovules (Sheppard et al., 2008).
Nupts have been discovered in a large number of plant species (Smith et al., 2011).
Nupts can contribute to nuclear exonic sequences (Noutsos et al., 2007) and play an
important role in plant evolution. Nupts may be initially inserted close to centromeres and then fragmented and distributed by transposable elements (Michalovova et al., 2013). The amount of nupts in plants is associated with the nuclear genome size and the number of plastids per cell (Smith et al., 2011; Yoshida et al., 2014).Studies on nupts remains limited to plant species that have complete sequences of both nuclear and plastid genomes. In nuclear genomes, nupt rearrangements may resemble that of plastomes or consisted of mosaic DNA derived from both plastids and mitochondria (Leister, 2005; Noutsos et al., 2005). Notably, a 131-kb nupt of rice was found to harbor a 12.4-kb inversion, which was likely the ancestral characters in the plastome before the transfer (Huang et al., 2005). Recently, Rousseau-Gueutin et al.
(2011) proposed a PCR-based method to amplify nupts containing a specific ancestral sequence that was deleted from the plastomes of viable offspring. Hence, ancestral plastomic characteristics, such as unique indels and gene orders of specific fragments,
may be retained in nupts. Construction of an ancestral plastomic organization should yield valuable clues to retrieve nupts. If a plastomic inversion can distinguish an ancestral plastome from its current counterpart, appropriate primers based on the ancestral plastomic organization should be able to amplify the corresponding nupts that were transferred to the nucleus before the inversion (Figure 3).
Although the first known nupt was identified more than 3 decades ago (Timmis and Scott, 1983), nupts of gymnosperms still remain poorly studied. Conifers, the most diverse gymnosperm group, possess huge nuclear genomes ranging from 8.3 to 64.3 pg (2C) (reviewed in Wang and Ran, 2014) and may have integrated many nupts. The plastomes of conifers are highly rearranged, possibly due to their common loss of a pair of large inverted repeats (Wicke et al., 2011). Numerous plastomic rearrangements have been identified and are useful in reconstructing phylogenetic relationships among taxa and inferring intermediate ancestral plastomes (Wu and Chaw, 2014). Therefore, the conifer plastomes are well suited for evaluating the feasibility of retrieving nupts and surveying their evolution.
In this study, we aim to demonstrate our approach (Figure 3) for mining nupts in yews, and to continue the understanding of the plastome evolution in conifers. To better reconstruct ancestral plastomes of yews, we sequenced two complete plastomes, one from each of the yew genera Amentotaxus and Taxus. The primers based on the recovered ancestral plastomic organization were used to amplify potential nupts. The origins of obtained nupt candidates were then examined by phylogenetic analyses and mutation preferences to ensure that they were indeed transferred plastomic DNA in the nucleus. Here, for the first time, we demonstrate that conifer nupts can be PCR-amplified using our approach and that ancestral plastomic characteristics retained in nupts can be compared with extant ones, providing valuable information for
understanding plastome evolution in conifers.
2.2 Materials and Methods
2.2.1 DNA Extraction, Sequencing, and Genome Assembly
Young leaves of Amentotaxus formosana and Taxus mairei were harvested in the greenhouse of Academia Sinica and Taipei Botanical Garden, respectively. Total DNA was extracted with modified CTAB method with 2% polyvinylpyrrolidone (Stewart and Via, 1993). The DNA was qualified by a threshold of both 260/280 = 1.8–2.0 and 260/230 > 1.7 for next-generation DNA sequencing on an Illumina GAII instrument at Yourgene Bioscience (New Taipei City, Taiwan). For each species, approximately 4 GB of 73-bp paired-end reads were obtained. These short reads were trimmed with a threshold of error probability < 0.05 and then de novo assembled by use of CLC Genomic Workbench 4.9 (CLC Bio, Aarhus, Denmark). Contigs with sequence coverage of depth greater than 50X were blasted against the nr database of the National Center for Biotechnology Information (NCBI). Contigs with hits for plastome sequences with E-value < 10-10 were retained for subsequent analyses. Gaps between contigs were closed by PCR experiments with specific primers. PCR amplicons were sequenced on an ABI 3730xl DNA Sequencer (Life Technologies).
2.2.2 Genome Annotation and Sequence Alignment
Genome annotation involved the use of DOGMA with default option (Wyman et al., 2004). Transfer RNA genes were explored by using tRNA scan-SE 1.21 (Schattner et al., 2005). For each species, we aligned the annotated genes and their orthologous genes of other known conifer plastomes to confirm gene boundaries. Sequences were aligned using MUSCLE (Edgar, 2004) implemented in MEGA 5.0 (Tamura et al., 2011).
2.2.3 Exploration of Single-Nucleotide Polymorphisms (SNPs), Indels, and Simple Sequence Repeat (SSR) Sequences
To estimate the distribution of both SNPs and indels between our newly sequenced plastome of T. mairei and the T. mairei voucher NN014 (NC_020321), the two genomes were aligned by using VISTA (Frazer et al., 2004). The alignment was then manually divided into non-overlapping bins of 200 bp according to the position of our newly sequenced T. mairei plastome. Both SNPs and indels in each bin were estimated by using DnaSP 5.10 (Librado and Rozas, 2009). SSRs of the T. mairei plastome were explored using SSRIT (Temnykh et al., 2001) with a threshold of repeat units > 3.
2.2.4 Construction of Ancestral Plastomic Organization
We performed whole-plastomic alignments between the two yews under study and other conifers, Calocedrus formosana (NC_023121), Cephalotaxus wilsoniana (NC_016063), Cryptomeria japonica (NC_010548), Cunninghamia lanceolata (NC_021437), and Taiwania cryptomerioides (NC_016065), to detect locally collinear blocks (LCBs) using Mauve 2.3.1 (Darling et al., 2010). The yielded matrix of LCBs was used to reconstruct the putative ancestral plastomic organizations on MGR 2.03 (Bourque and Pevzner, 2002), which seeks the minimal genomic rearrangements over all edges of a most parsimonious tree.
2.2.5 PCR Amplification, Cloning, and Sequencing
Ten pairs of specific primers used for amplification of nupt sequences in Taxaceae were manually designed and their sequences and corresponding locations are in Table 2 and Figure 4. PCR amplification involved the use of long-range PCR Tag (TaKaRa LA Taq, Takara Bio Inc.) under the thermo-cycling condition 98℃ for 3 min, followed by
30 cycles of 98℃ for 15 s, 55℃ for 15 s, and 68℃ for 4 min, and a final extension at 72 ℃ for 10 min. Amplicons were checked by electrophoresis. Amplicons with expected lengths were collected and cloned into yT&A vectors (Yeastern Biotech Co., Taipei) that were then proliferated in E. coli. Sequencing the proliferated amplicons involved M13-F and M13-R primers on an ABI 3730xl DNA Sequencer (Life Technologies).
2.2.6 Phylogenetic Tree Analysis
Maximum likelihood trees were inferred from sequences of potential nupts, their plastomic counterparts, and their orthologs in other gymnosperms using MEGA 5.0 (Tamura et al. 2011) under a GTR + G (4 categories) model. Supports for nodes of trees were evaluated by 1,000 bootstrap replications.
2.2.7 Estimation of Mutations in Nuclear Plastid DNAs and Their Plastomic Counterparts
The sequence for each nupt was aligned to the homologous plastome sequences for
A. formosana, C. wilsoniana, T. maire and C. lanceolata using MUSCLE (Edgar, 2004).
To precisely calculate the mutational preference in nupts, all ambiguous sites and gaps were removed from our alignments. Nucleotide divergence between nupts and their plastomic counterparts were derived from mutations in either of these two sequences. A mutation in nupt or its plastomic counterpart was recognized when the corresponding site of the plastomic counterpart or nupt was identical to that of at least two other taxa.
For example, a specific aligned site has “T”, “C”, “C”, “C”, and “C” in Cep-2 nupt, C.
wilsoniana, A. formosana, T. mairei and C. lanceolata, respectively (also see the aligned
position 32 in Figure 5). This site would be recognized as a nonsynonymous mutationfrom “C” to “T” in the Cep-2 nupt as the corresponding amino acid change from Alanine to Valine.
2.2.8 Plastome Mapping and Statistical Analyses
The plastome map of T. mairei was drawn using Circos (http://circos.ca), which is a flexible software for exploring relationships between objects or positions. It was written in Perl language. The physical map will be saved as PNG format in the input folder. In all statistical tests, including Pearson’s correlation test and Student’s t-test, Microsoft Excel 2010 was used.
2.3 Results
2.3.1 Reduction and Compaction of the Plastome of T. mairei
The plastomes of A. formosana (AP014574) and T. mairei (AP014575) are circular molecules with AT contents of 64.17% and 65.32%, respectively. The T. mairei (128,290 bp) plastome has lost five genes (rps16, trnA-UGC, trnG-UCC, trnI-GAU, and
trnS-GGA) compared to that of A. formosana (136,430 bp), which leads to a relatively
smaller plastome size. The coding regions occupy 61.27% of the plastome length in A.formosana and 64.18% in T. mairei. The gene density was estimated to be 0.88 and 0.90
(genes/kb) for the plastome of A. formosana and T. mairei, respectively. In addition, the other two published plastomes for Taxaceae species, C. wilsoniana (NC_016063) and C.oliveri (NC_021110), are 136,196 bp and 134,337 bp, respectively. Altogether, these
data suggest that the plastome of T. mairei has evolved towards reduction and compaction.Dot-plot analysis (Figure 6) reveals three genomic rearrangements between the plastomes of A. formosana and T. mairei, including a relocated fragment of ~18 kb from
psbK to trnC-GCA, a relocated fragment of ~16 kb from trnD-GUC to trnT-UGU, and
an inverse fragment of ~18 kb from 5'rps12 to infA. However, the two plastomes share a unique inverted repeat pair that contains trnQ-UUG in each copy, hereafter termed“trnQ-IR” (Figure 6).
2.3.2 Intra-species Variations in the Plastomes of T. mairei
To date, the plastomes of three T. mairei individuals (T. mairei voucher NN014:
NC_020321, T. mairei voucher SNJ046: JN867590, and T. mairei voucher WC052:
JN867591) have been published. Together with our newly sequenced plastome of T.
mairei, these four plastomes vary slightly in size ranging from 127,665 to 128,290 bp. A
neighbor-joining (NJ) tree inferred from the whole-plastomic alignment between these four individuals and A. formosana is shown in Figure 7. The tree topology indicates that although the plastome size of SNJ046 and WC052 is similar to that of NN014, and that the two plastomes SNJ046 and WC052 form a sister clade to that of our sampled T.mairei.
We also performed a pairwise genome comparison between our T. mairei and voucher NN014 because the latter was designated as the reference sequence (RefSeq) in NCBI GenBank. We detected 858 SNPs and 218 indels between the two plastomes.
Figure 8 shows that the intergenic spacers and coding regions contained nearly equal numbers of SNPs. Most of the indels were found in the intergenic spacers and accounted the difference in plastome size between the two T. mairei individuals. We found 33 indels in the coding regions, but none caused frameshifts. Figure 9 illustrates the distribution of SNPs, indels, and SSRs in the plastome of our sampled T. mairei.
Interestingly, the abundance of SSRs was positively correlated to those of SNPs (Pearson, r = 0.52, p < 0.01). However, no correlation was detected between SSRs and
indels abundance (Pearson, r = 0.02, p = 0.89). In legumes, the region that contains ycf4,
psaI, accD, and rps16 was found to be hypermutable (Magee et al. 2010). In the
plastome of T. mairei, three 200-bp bins that located in the sequence of 5’clpP (position 55,001–55,200), 5’ycf1 (pos. 124,201–124,400), and the intergenic spacer betweenrrn16 and rrn23 (pos. 96,801–97,000) contained the highest sum of SNPs, indels, and
SSRs (Figure 9). Therefore, these loci can be considered intra-species mutational hotspots in T. mairei and can be a potential high-resolution DNA barcodes for population genetics of Taxus.2.3.3 Retrieval of Ancestral Plastome Sequences in Taxaceae
A matrix with 20 locally collinear blocks (LCBs) was generated on the basis of whole plastome alignments between the sampled three Taxaceae and four Cupressaceae species. This matrix of LCBs was then used in reconstructing ancestral plastomic organization. The most parsimonious tree with the corresponding ancestral plastomic organization is shown in Figure 4 and Figure 10, and that the three Taxaceae species form a monophyletic clade while A. formosana is closer to C. wilsoniana than to T.
mairei. This topology is in good agreement with the recent molecular review of the
conifer phylogeny by Leslie et al. (2012). Figure 4 shows the detailed evolutionary scenario of plastomic rearrangements with the intermediate ancestral plastomes in the three examined Taxaceae species. By comparing the ancestral and extant plastomes, we postulated that one, three, and two inversions might have occurred in A. formosana, C.wilsoniana, and T. mairei, respectively, after they had diverged from their common
ancestor. Specific primer pairs were used for amplifying the corresponding ancestral fragments that differ from the extant plastomes in genomic organization (Figure 4). Five (Ame-2, Cep-2, Cep-5, Cep-6, and Tax-4) out of the ten primer pairs were able toproduce amplicons totaling 16.6 kb (see Table 3 for accession numbers).
2.3.4 Characteristics of Potential Nupt Amplicons
The obtained PCR amplicons were sequenced and annotated (Table 3). With the exception of chlB of Cep-2, all putative protein-coding genes contain no premature stop codons. The coding sequence (CDS) of each amplicon was aligned with its plastomic counterparts and orthologs of other cupressophytes, Ginkgo, and Cycas. We used maximum likelihood (ML) trees inferred from concatenated CDSs to examine the origins of these PCR amplicons, with Ginkgo and Cycas as the outgroup (Figure 11). In each tree, the plastomic sequences were divided into three groups (i.e., the Cupressaceae clade, the Taxaceae clade, and the clade comprising Araucariaceae and Podocarpaceae).
Notably, the placements of our PCR amplicons are incongruent among the four trees.
For example, both Ame-2 and Cep-2 were clustered with their plastomic counterparts (Figure 11A). In contrast, Cep-5, Cep-6, and Tax-4 were placed remotely from their plastomic counterparts, indicating that they originated via horizontal transfer (Figure 11B, C, and D).
The ancestral plastomic organization that we used to design primers for amplification of Ame-2 and Cep-2 was rearranged by a 34-kb inversion flanked by trnQ-IRs. These trnQ-IRs were 564 and 549 bp in size for A. formosana and C.
wilsoniana, respectively. IRs of similar sizes can mediate homologous recombination in
the conifer plastomes (Tsumura et al., 2000; Wu et al., 2011; Yi et al., 2013; Guo et al., 2014). As a result, if the trnQ-IR-mediated isomeric plastome is present in our sampled taxa, our PCR approach shall be able to amplify isomeric plastomic fragments. Ame-2 has 100% sequence identity with its plastomic counterpart (Figure 11A) in the CDS, which strongly suggests its origin as an isomeric plastome. Cep-2 differs from itsplastomic counterpart by several mutations, including two premature stop codons in
chlB, of which one of the two cannot be replaced by neither U-to-C nor C-to-U
RNA-editing (Figure 12). Therefore, the origin of Cep-2 is from a horizontal transfer rather than an isomeric plastome.2.3.5 Evolution of Nupt Sequences in Taxaceae
The sequence identity between the four nupts and their plastomic counterparts ranges from 61.71% to 99.08% (Table 4). In fact, differences in aligned sites between
nupts and their plastomic counterparts are derived from two types of mutations. One is
the mutation in nupts and the other is that in plastomes. As shown in Table 4, with the exception of Tax-4, all nupts accumulated more mutations than their plastomic counterparts. The low sequence identity between Tax-4 and its plastome sequences (61.71% in Table 4) may be due to the unusually increased mutations in the latter. In allnupts except Cep-5, at least one potential protein-coding gene had the ratio of
nonsynonymous (dn)/synonymous (ds) mutations > 1, which reflects the effect of relaxed functional constraints in nupts. Figure 13 illustrates nucleotide mutation classes in nupts and their corresponding plastome sequences. We excluded the plastomic counterpart of Cep-2 from the calculation because we observed only one mutation in the sequence. In all nupts, transitional mutations comprise over 50% of the total mutations.The mutation of G to A and its complement C to T (denoted GC-to-AT in Figure 13) had the highest frequency in both nupts and plastome sequences. To examine which of the mutation classes is statistically predominant, we compared the two most abundant classes of mutations. In nupts, the frequency was higher for GC-to-AT than AT-to-GC mutations (t-test, p = 0.018). However, GC-to-AT and AT-to-GC mutations did not differ in plastome sequences (t-test, p = 0.379), suggesting different mutational environments
between nupts and their corresponding plastome sequences.
2.3.6 Ages of Nupts in Taxaceae
Molecular dating of sequences highly depends on mutation rates. Unfortunately, mutation rates in the nuclear genomes of Taxaceae species have not been directly measured. The nupts identified in this study were expected to evolve neutrally. The four-fold degenerated site is a useful indicator in measuring the rate of neutral evolution (Graur and Li, 2000). In nuclear genomes of conifers, the mutation rate at the four-fold degenerate sites was estimated to be 0.64 × 10-9 per site per year (Buschiazzo et al., 2012). In the nupts Cep-2, Cep-5, Cep-6, and Tax-4, we found 29, 117, 100, and 42 mutations among 2,961, 3,380, 2,207, and 1,466 sites, respectively (Table 4). Therefore, the ages of Cep-2, Cep-5, Cep-6, and Tax-4 were estimated to be approximately 15.3, 54.1, 70.8, and 44.8 million years (MY), respectively.
2.4 Discussion
2.4.1 Labile Plastomes of Yew Family and Their Impact on Phylogenetic Studies The phylogenetic relationships among Amentotaxus, Cephalotaxus, and Taxus have not been resolved. Recent molecular studies placed Amentotaxus as sister to Taxus (e.g., Cheng et al. 2000; Mao et al. 2012) or to Cephalotaxus (e.g., Leslie et al. 2012). We found that a 34-kb inversion from trnT to psbK distinguished A. formosana and C.
wilsoniana from T. mairei (Figure 4), which suggests that A. formosana is closer to C.
wilsoniana than to T. mairei. However, the plastome of another Taxus species, T.
chinensis (Zhang et al., 2014), cannot be distinguished from those of A. formosana and
C. wilsoniana by this 34-kb inversion. Of note, this 34-kb inversion is flanked by a pair
of trnQ-IR sequences. We found that the trnQ-IR sequence is commonly present in A.formosana (564 bp), C. wilsoniana (549 bp), T. mairei (248 bp), and T. chinensis (248
bp).The presence of the trnQ-IR pair is able to generate isomeric plastomes in C.
oliveri (Yi et al., 2013) and four Juniperus species (Guo et al., 2014). In Pinaceae,
inverted repeats larger than 0.5 kb could trigger plastomic isomerization, and retention of an isomer was species- or population-specific (Tsumura et al., 2000; Wu et al., 2011).Indeed, Figure 11A revealed that Ame-2 was likely a PCR amplicon derived from the trnQ-IR-mediated isomeric plastome of A. formosana. Therefore, with the presence of an isomeric plastome, the synapomorphic character―the 34-kb inversion―in Figure 4 might be a false positive result caused by insufficient sampling. Nonetheless, our data also suggest that isomeric plastomes be cautiously treated when using genomic rearrangements in phylogenetic estimates.
Disruption of the plastomic operons is rare in seed plants (Jansen and Ruhlman, 2012). We found that the S10 operon of T. mairei was separated into two gene clusters (rpl23-rps8 and infA-rpoA) by an 18-kb inversion (Figure 4). Because the transcriptional direction of the S10 operon is from rpl23 to rpoA (Jansen and Ruhlman, 2012), the gene cluster infA-rpoA in T. mairei likely has to acquire a novel promoter sequence for transcription. Disruption of the S10 operon was previously reported in the plastome of Geraniaceae (Guisinger et al., 2011). However, the evolutionary consequence of plastomic operon disruption has never been studied. In the plastome of
T. mairei, we detected prominently elevated mutations in the two separated gene
clusters of the S10 operon as compared with their relative nupts (Table 4). Interestingly, two (i.e., infA and rps11 in Table 4) of the three protein-coding genes on the plastomic gene-cluster infA-rpoA had dn/ds ratios larger than 1. Whether disruption of the S10 operon results in the positive selection of these two genes requires further investigation.2.4.2 PCR-Based Approach in Investigating Nupts: Pros and Cons
The immense growth of available sequenced nuclear genomes offers great opportunities for investigating nuclear organellar DNA (norgs). The number of norgs could vary depending on the use of different assembly software, versions of released genomes, and search strategies (Hazkani-Covo et al., 2010). A PCR-based approach, such as that of Rousseau-Gueutin et al. (2011) and ours, is free from this problem encountered in genome assembly. The nupts we amplified and reported here are a few examples of nupts. However, considering the huge nuclear genome of conifers which requires high cost and efforts for sequencing and assembly, our PCR-based approach provides a cost-effective way for studying the evolution of nupts.
Using a threshold of >70% sequence identity, Smith et al. (2011) extracted nupts of about 50 kb from the nuclear genome of Arabidopsis. The amount of Arabidopsis nupts decreased to approximately 17.6 kb when the threshold of sequence identity was increased to 90% (Yoshida et al., 2014). It seems that identification of possible nupts is largely influenced by the thresholds. Setting high thresholds might limit the exploration of nupts to only relatively recent transfers (Yoshida et al., 2014). Clearly, the problem of setting thresholds is absent from our PCR-based approach. In this study, sequence identity between nupts and their plastomic counterparts ranged from 61.71 to 99.08%
(Table 4). Thus, one or three of the four presented nupts would not be obtained if we had considered the thresholds of Smith et al. (2011) or Yoshida et al. (2014), respectively.
Only five of our ten primer pairs worked well, and one amplified the DNA fragment of isomeric plastomes rather than nupts. This low success rate may be due to the unsuitable primers used in our PCR experiments. Multiple primer pairs for a specific locus may improve amplification of nupts, as noted by Rousseau-Gueutin et al. (2011).
Plastid-to-mitochondrion DNA transfers are frequent in seed plants (Wang et al., 2007).
Because the mitochondrial genome of Taxaceae is currently unavailable, the possibility that our PCR products were amplicons of mitochondrial plastid DNA could not be ruled out. The phylogenetic tree approach was previously used to examine horizontal DNA transfers (Bergthorsson et al., 2003; Rice et al., 2013), but our tree analyses in Figure 11 could not distinguish the transfer events between plastid-to-nucleus and plastid-to-mitochondrion origins. The mutation rate of nuclear genomes is higher than that of plastomes in plants (Wolf et al., 1987). All of our amplified nupts, except Tax-4, had more mutation sites than their plastomic counterparts (Table 4). Disruption of the S10 operon is likely associated with the elevated mutation in the plastomic counterpart of Tax-4, as mentioned above. Additionally, among our nupts, the AT-to-GC mutation was predominant (Figure 13). These data are similar to the findings for nupts in rice and
Nicotiana (Huang et al., 2005; Rousseau-Gueutin et al., 2011), which reflects a
nuclear-specific circumstance shaped by spontaneous deamination of 5-methylcytosin.2.4.3 Nupts Are Molecular Footprints for Studying Plastomic Evolution
Although mutation rates are relatively low in plant organellar genomes, norgs can serve as “molecular fossils” for genomic rearrangements (Leister, 2005). Similarly, the Taxaceae nupts identified in this study retain the ancestral plastomic organization. In other words, nupts are footprints that are valuable in reconstructing the evolutionary history of plastomic organization and rearrangements.
Dating the age of nupts is critical for elucidating the evolution of nupts. For example, the estimated ages of Cep-2, Cep-5, and Cep-6 nupts are 15.3, 54.1, and 70.8 MY, respectively. Remarkably, these ages conflict with the scenario of plastomic rearrangements because the transfer of Cep-2 predated those of both Cep-5 and Cep-6
(Figure 4). Two plastomic forms derived from trnQ-IR-mediated homologous recombination coexist in an individual of C. oliveri (Yi et al., 2013). This trnQ-IR is also present in the plastome of C. wilsoniana as previously mentioned. We suspect that in C. wilsoniana, the younger Cep-2 nupt might originate from a transferred fragment of the trnQ-IR-mediated isomeric plastome.
Most importantly, nupts can also help in probing RNA-editing sites and improving gene annotations. Figure 12 clearly reveals that the previously annotated rps8 of T.
mairei (vouchers NN014, WC052, and SNJ046) is truncated. Our newly predicted
initial codon, “ACG”, locates 48 bp upstream of the previously predicted site. This“ACG” initial codon was predicted to be corrected to “AUG” via a C-to-U RNA-editing because the corresponding sequence of Tax-4 nupt and other conifers retain a normal initial codon of “ATG” (Figure 12). These data also imply that in T. mairei, the transfer of Tax-4 nupt predates the T-to-C mutation at the second codon position in the initial codon of rps8.
CHAPTER 3
Birth of Four Chimeric Plastid Gene Clusters in Sciadopitys verticillata
3.1 Introduction
Due to the loss of many genes in early endosymbiosis, plastomes are much reduced compared to their cyanobacterial counterparts (Ku et al., 2015). To date, plastomes have invariably retained a small handful of prokaryotic features, including the organization of genes into polycistronic transcription units resembling bacterial operons (Sugiura, 1992;
Wicke et al., 2011). A hallmark of seed plant plastomes is the presence of two 20- to 30-Kb IR (hereafter referred to as “typical IRs,” including IRA and IRB), which typically contain four ribosomal RNAs. However, a few exceptions have been reported.
For example, conifers—the largest gymnosperm group comprising cupressophytes and Pinaceae—have lost a typical IR copy from their plastomes (Raubeson and Jansen, 1992). Recent studies have further suggested that cupressophytes and Pinaceae might have lost different IR copies, with the former losing IRA and the latter losing IRB (Wu, Wang et al., 2011; Wu and Chaw, 2014).
Conifer plastomes are also characterized by extensive genomic rearrangements.
The plastome of Cryptomeria japonica—the first completed plastome of cupressophytes (Hirao et al., 2008)—experienced at least 12 inversions after its split from the basal gymnosperm clade, cycads, whose plastomes have remained virtually unchanged for 280 million years (Wu and Chaw, 2015). The co-existence of four different plastome forms among Pinaceae genera is associated with intra-plastomic recombination mediated by three specific types of short IRs (Wu, Lin et al., 2011). Furthermore,
Cephalotaxus oliveri (Cephalotaxaceae; Yi et al., 2013) and four Juniperus species
(Cupressaceae; Guo et al., 2014) harbor isomeric plastomes that deviate from each other by an inversion possibly triggered by a trnQ-containing IR (“trnQ-IR”). Although conifer plastomes are highly rearranged (Wu and Chaw, 2014), disruptions in their operons are rare. Until recently, only one case was reported in the plastome of Taxusmairei, in which the S10 operon (trnI-rpoA region) was disrupted into two separate
segments by a fragment of approximately 15 Kb (Hsu et al., 2014). However, the impact of such operon disruptions on plastid evolution remains poorly understood.The 25 published cupressophyte plastomes available on GenBank (Dec 2015) represent four of the five cupressophyte families. However, no complete plastome is available for Sciadopityaceae. As part of our continuing efforts to decipher the diversity and evolution of conifer plastomes, we have completed and elucidated the plastome sequence of Sciadopitys. We found that the plastome of Sciadopitys is characterized by several unusual features. For the first time, this study reports the unusual shuffling of operons that results in the re-organization of plastid genes into new chimeric gene clusters.
3.2 Materials and Methods 3.2.1 DNA Extraction
Approximately 2 grams of fresh leaves were collected from an individual of