Organic light-emitting diodes based on charge-neutral Os(
II
) emitters:
generation of saturated red emission with very high external quantum
efficiency
Yung-Liang Tung,
aShin-Wun Lee,
aYun Chi,*
aYu-Tai Tao,*
bChin-Hsiung Chien,
bYi-Ming Cheng,
cPi-Tai Chou,*
cShie-Ming Peng
cand Chao-Shiuan Liu
dReceived 21st September 2004, Accepted 10th November 2004 First published as an Advance Article on the web 1st December 2004 DOI: 10.1039/b414636k
The OLED device using 6% of Os(fptz)2(PPh2Me)2 as the
dopant emitter in a CBP host and BPAPF as hole transporting material shows an external quantum efficiency of 15.3% and luminous efficiency of 21.3 cd A21, power efficiency of 6.3 lm W21at 20 mA cm22. An even higher external quantum efficiency of y20% was achieved at a low current density of y1 mA cm22
.
Since the seminal work of Tang and VanSlyke in electrolumines-cence (EL) devices using organic materials,1the research activities on organic light-emitting diodes (OLEDs) have made significant progress during the past two decades. The OLEDs of this category continuously attract great interest because of their potential in the development of full color flat-panel displays. In this regard, fabrication of OLEDs with energy efficient, saturated red emission becomes essential,2and this has been achieved by using third-row metal Pt(II)- and Ir(III)-containing phosphorescent dopant emit-ters, for which the strong spin–orbit coupling effectively promotes the intersystem crossing from singlet excited states to lower triplet emitting states as well as the enhancement of the T1–S0transition.
3
Theoretically, OLEDs with 100% internal quantum efficiencies may be attained by harnessing both triplet and singlet excitons.4 However, for most of the phosphorescent OLEDs, the device quantum efficiency drops rapidly with increasing current density and thus brightness. This is believed to be due to the fact that triplet excitons relax more slowly and the emission inevitably reaches saturation through a quenching mechanism involving triplet–triplet annihilation.5One way to alleviate the problem is to
use materials with a shorter triplet radiative lifetime. To achieve this goal, a potential category in point may be Os(II) complexes,6
which, in general, possess a shorter triplet-state exciton lifetime (¡a few ms) due to the enhancement of the heavy-metal atom participating in the lowest excited triplet manifolds (either3p–p*,
3
MLCT or the mixed states). More importantly, owing to its divalent state, the oxidation potential at the Os(II) metal center is significantly lower than that of the Ir(III) analogues with +3 oxidation state. The higher oxidation potential of the latter makes it less of an ideal center for carrier direct-trapping and recombination.7
In this communication, we report the syntheses and character-ization of a series of readily sublimable, charge neutral Os(II)
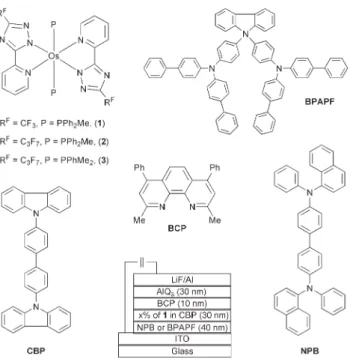
triazolate complexes, Os(fptz)2(PPh2Me)2(1) Os(hptz)2(PPh2Me)2
(2) and Os(hptz)2(PPhMe2)2(3), the molecular structures of which
are shown in Fig. 1. In contrast to the PLED fabricated using the nonvolatile, ionic Os(II) emitters,6,7remarkably high efficiency in red emission has been achieved by fabricating the OLED devices using a co-deposition technique. In particular, with a device configuration of ITO/BPAPF(40 nm)/CBP : 6% of 1(30 nm)/ BCP(10 nm)/Alq(30 nm)/LiF(1 nm)/Al(150 nm), an external quantum efficiency of 15.3% and a luminous efficiency of 21.3 cd A21, power efficiency of 6.3 lm W21 were obtained at 20 mA cm22, yielding CIE coordinates at x 5 0.64 and y 5 0.35. An even higher external quantum efficiency of y20% was achieved at a very low current density ofy1 mA cm22
. This high efficiency is comparable to that obtained for the best green-emitting Ir(III) based OLEDs.8 A maximum brightness of y45 000 cd m22
was recorded at the driving voltage of 15 V. To our knowledge, this result steps up a major advance for red-emitting, small molecule OLEDs fabricated by the co-deposition technique.
*[email protected] (Yun Chi) [email protected] (Yu-Tai Tao) [email protected] (Pi-Tai Chou)
Fig. 1 The molecular structure of the relevant compounds used in this study and configuration of the OLED devices.
These Os(II) emitting materials are designed by bearing the relatively poor electron-donating pyridyl triazolate anion to increase the stability and to neutralize/balance the+2 charge on the Os(II) metal center, the strategy of which is in a way similar to the previously described pyrazolate complexes.9 Synthetic details of the triazole ligands have previously been elaborated.10 The pyridyl triazolate ligands are expected to adopt a trans orientation around the Os(II) center. This is confirmed by the observation of a very downfield ortho-CH signal of the pyridine fragment (d 5 10.24–10.12 ppm), resulting from the notably strong inter-ligand N…H–C hydrogen bond.9Conversely, in order to tune the
phosphorescent emission to the red, phosphine auxiliary ligands are also selected owing to their great electron-donating property (vide infra). Finally, the incorporation of either CF3 or C3F7
fluorinated substituents in 1–3 is essential to reduce the intermolecular interaction, rendering the required volatility to these Os(II) emitting complexes.11
The reaction condition was optimized using a one-pot synthetic strategy, which involved the in situ preparation of the dicarbonyl complexes Os(fptz)2(CO)2 and Os(hptz)2(CO)2 from Os3(CO)12,
followed by conducting phosphine substitution in presence of Me3NO. This modified approach circumvents the tedious isolation
of the above mentioned intermediates,12gives us the desired Os(
II) complexes 1–3 in much improved (. 70%) yields, and hence has a great advantage in scaling up for the industrial application.
The absorption and luminescence spectra of complexes 1–3 in CH2Cl2 are shown in Fig. 2. The strong absorption bands
commonly observed around 400 nm for 1–3 are assigned to the spin-allowed1p–p* transition of the fptz (or hptz) ligands. The
next lower energy absorption band around 450 nm can be ascribed to a spin-allowed metal to ligand charge transfer (1MLCT)
transition, while an equally strong absorption band with peak wavelengths at 543 nm (e 5 1400 M21cm21), 545 nm (e 5 1400 M21cm21) and 550 nm (e 5 1450 M21cm21) for complexes 1, 2 and 3, respectively, can reasonably be assigned to a state mixing of spin–orbit coupling enhanced3p–p* and3MLCT transitions. It is also notable that substitution with stronger donor ligands such as PPhMe2not only causes the spectral red-shift due
to the increase of the metal dp orbital energy but also increases the
entire transition dipole moment, A similar argument has been proposed to account for the systematic spectral variation of the Os(II) polypyridyl phosphine complexes.13
Very intense luminescence was observed for 1–3 with lmax
located at 617 nm, 614 nm and 629 nm, respectively, in CH2Cl2
solution. The significant overlap of the 0–0 onsets between emission and the lowest energy absorption band, in combination with a broad, structureless spectral feature, leads us to conclude that the phosphorescence originates primarily from the 3MLCT
state.14 In comparison to 3 bearing PPhMe2 as coordinating
ligands, complex 2 with the PPh2Me group reveals a y15 nm
hypsochromic shift in lmaxand can qualitatively be rationalized by
a decrease of Os(II) dp energy level due to the stronger electron-withdrawing strength of an additional phenyl substitution. Table 1 lists the corresponding photophysical data for the studied complexes in both solution and solid phases. The observed lifetimes of ca. 0.8–1.0 ms, in combination with the quantum efficiencies of 0.50–0.76, lead us to deduce a radiative lifetime of 1.54, 1.23 and 1.62 ms for 1, 2 and 3, respectively, in degassed CH2Cl2. To our knowledge, the radiative lifetimes for 1–3 are
considerably shorter than those of most reported red emitting Ir(III) complexes.15In the solid state, the emission maximum for these Os(II) phosphors shifts to the red, possibly due to their molecular packing, and the lifetime falls within the range of 0.2– 0.9 ms. The emission quantum efficiencies of 1–3 lie in the range 0.21–0.36 in the solid state. It is notable that the exciton lifetime of 3 is about 4.5 times greater than that of 1 in solid, implying that the OLED device fabricated using 1 should reduce T–T annihilation at the higher driving voltage (vide infra).
Due to its high PL quantum efficiency in the red and excellent redox stability, complex 1 was selected in fabricating a series of multilayer devices of the configuration ITO/HTL(40 nm)/CBP : 1(30 nm)/BCP(10 nm)/AlQ3(30 nm)/LiF(1 nm)/Al(150 nm), where
CBP, BCP and AlQ3stand for
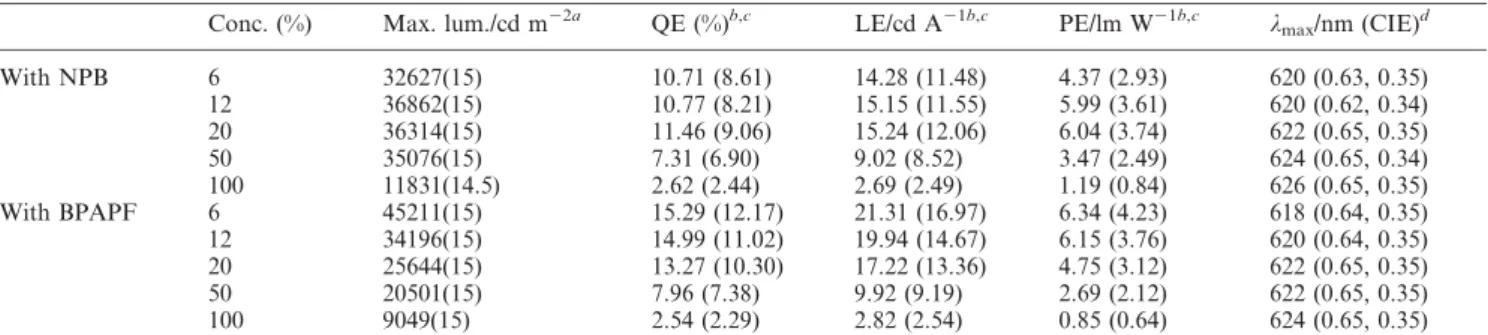
4,49-N,N9-dicarbazolyl-1,19-biphe-nyl, 2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline, and tris(8-hydroxyquinolinato) aluminium(III) respectively. Two distinctive hole transporting materials (HTL) were 4,49-bis[N-(1-naphthyl)-N-phenylamino]biphenyl (NPB) and 9,9-bis{4-[di-(p-biphenyl)ami-nophenyl]}fluorene (BPAPF).16 The doping levels of Os(II) complex 1 varied from 6%, 12%, 20%, 50% to a 100% neat thin film. Device configurations and the molecular structures of the compounds used in these devices are also shown in Fig. 1, while crucial device performance characteristics are collected in Table 2. Bright red emission was observed for all the concentrations applied, even for the one with a pure layer of the Os(II) emitter. With NPB as HTL, the current–voltage–luminance curves, plotted in Fig. 3a, show a rough trend of decreasing current density with increasing concentrations of 1, implying that the phosphorescent dopant sites serve as charge trapping sites.17The EL spectra are given in Fig. 3b. A small amount of emission at y450 nm, identified as originating from NPB, was observed for the low dopant concentration of 6%. This NPB emission diminished upon increasing the doping concentration to 12% and higher. Concurrently, a small red shift of the EL spectra was observed with increasing dopant concentrations, from lmaxy 620 nm for
the 6% device to 625 nm for the neat film device (Fig. 3b), presumably due to the change of the medium polarity.18
Interestingly, upon switching the hole-transport layer from NPB to BPAPF, a significant improvement in both luminescence and
Fig. 2 UV–Vis absorption and normalized emission spectra of 1 (–#–), 2 (–&–) and 3 (–m–) in CH2Cl2at RT; the excitation wavelength: 500 nm.
external quantum efficiency was observed. The EL spectrum is free from BPAPF emission at all dopant concentrations. Such an outcome may be attributed to the higher hole mobility of BPAPF than that of NPB,19so that a shift of the charge recombination area well inside the CBP/Os(II) dopant emitter layer occurs, in view of the similar HOMO energy levels (NPB, 5.2 eV; BPAPF, 5.3 eV) and LUMO energy levels (NPB, 2.2 eV; BPAPF, 2.2 eV) for the two compounds. For comparison, the representative current– voltage–luminance characteristics for the 6% dopant device are depicted in Fig. 3c. A very high initial external quantum efficiency ofy20% and luminous efficiency of 27.8 cd A21were obtained at
1 mA cm22. Considering the coupling out factor, this is reaching nearly 100% internal phosphorescence efficiency.8 Like other phosphorescent emitters, the efficiencies also witnessed a drop with increasing driving voltage (Fig. 3d). At a driving current of 20 mA cm22, the external quantum efficiency is 15.3% and
luminous efficiency is 21.3 cd A21, whereas at 100 mA cm22, the efficiencies remain 12.2% and 17 cd A21respectively. However, it is noted that the decreasing trend in the quantum efficiency/power efficiency versus current density is slower than those reported for the triplet-state emitters.2The key difference is plausibly due to the
remarkably short radiative lifetime (y0.75 ms for 1 in solid), which significantly reduces the triplet–triplet annihilation.
In conclusion, a very efficient synthetic method for the charge neutral Os(II) emitters has been discovered and highly efficient, saturated red color, phosphorescent OLEDs are achieved by co-deposition of the charge neutral Os(II) triazolate complexes with CBP host as the emitting layer. The results demonstrate for the first time the generation of saturated red emission with external quantum efficiency up to 20% among the organometallic emitters composing the third-row Os(II), Ir(III) and Pt(II) elements.3,6 In
addition to the color tuning that should gain considerable interest, other basic photophysical properties, such as the phosphorescence lifetime of the designed Os(II) complexes, are also adjustable through modification of their ancillary coordination ligands to optimize the performance of the triplet-exciton driven OLEDs.
[Os(fptz)2(PPh2Me)2] (1) was prepared as follows. A 50 mL
reaction flask was charged with 3-trifluoromethyl-5-(2-pyridyl)-1,2,4-triazole (fptzH, 298 mg, 1.39 mmol), pulverized Os3(CO)12
(200 mg, 0.22 mmol), and 20 mL of anhydrous diethylene glycol monoethyl ether (DGME). The mixture was heated at 180–190uC for 24 h. After that, the temperature was lowered toy150 uC, freshly sublimed Me3NO (120 mg, 1.59 mmol) dissolved in 12 mL
DGME was added and stirring was continued for 5 min. Finally, PPh2Me (592 mL, 3.18 mmol) was injected into the mixture. In
the meantime, the temperature of solution was raised to 190uC. After 12 h, the reaction was stopped. The solvent was evaporated under vacuum, and the residue was washed with distilled water (20 mL 6 2) to remove the remaining Me3NO. Purification by
silica gel column chromatography (with EA–hexane 5 1 : 1 as eluent), followed by recrystallization from a mixture of EA and hexane at room temperature, yielded a bright red crystalline solid (504 mg, 0.50 mmol) in 75% yield.
Spectral data: MS (FAB,192Os): m/z 1018 (M+), 818 (M+
2 PPh2Me), 618 (M+ 2 2PPh2Me). 1H NMR (400 MHz, d6 -acetone): d 10.26 (d, 2H, JHH56.8 Hz), 7.54 (ddd, 2H, JHH56.8, 7.6, 0.8 Hz), 7.29 (d, 2H, JHH 57.6, 0.8 Hz ), 7.21 (ddd, 2H, JHH 5 7.6, 6.8, 0.8 Hz), 7.24–7.10 (m, 4H), 7.00 (t, 4H, JHH57.6 Hz), 6.92 (t, 4H, JHH57.6 Hz), 6.89–6.84 (m, 4H), 6.69–6.60 (m, 4H), 1.24 (t, 6H, JHP53.4 Hz, CH3). Anal. Calcd.
for C42H34F6N8OsP2: C, 49.60; N, 11.02; H, 3.37. Found: C, 49.61;
N, 10.98; H, 3.50.
Table 1 Photophysical and electrochemical properties for complexes 1–3
1 2 3
UV–Visible absorption e/nma 405 (15500), 457 (2400), 543 (1400) 403 (16000), 457 (2300), 545 (1400) 410 (17000), 465 (2200), 550 (1450)
PL lmaxa 617 (631) nm 614 (618) nm 629 (634) nm Wb 0.62 (0.24) 0.76 (0.36) 0.50 (0.21) tobsb 0.96 (0.18) ms 0.94 (0.58) ms 0.81 (0.91) ms Eox 1=2[DEp] c 0.12 [110] 0.19 [130] 0.11 [130] Ered
1=2or Ecp[DEp]c 22.61 [120] 22.78 [irr] 22.73 [irr]
aein M21 cm21. Samples were recorded in CH
2Cl2 at RT with at least three freeze–pump–thaw cycles. Emission spectra in solution were
excited at 500 nm, while an Ar+laser (514 nm) was used as an excitation source for the solid sample.bData in parentheses are measured in solid state at RT.cAll potentials are measured in a 0.1 M TBAPF6–THF solution and reported in volts using Fc/Fc+as reference, which is
0.18 V anodic of Ag/AgNO3electrode; DEp5 Eap(anodic peak potential) – Ecp(cathodic peak potential) and the data is quoted in mV.
Table 2 Performance characteristics for ITO/HTL/CBP : x% 1/BCP/LiF/Al devices
Conc. (%) Max. lum./cd m22a QE (%)b,c LE/cd A21b,c PE/lm W21b,c lmax/nm (CIE)d
With NPB 6 32627(15) 10.71 (8.61) 14.28 (11.48) 4.37 (2.93) 620 (0.63, 0.35) 12 36862(15) 10.77 (8.21) 15.15 (11.55) 5.99 (3.61) 620 (0.62, 0.34) 20 36314(15) 11.46 (9.06) 15.24 (12.06) 6.04 (3.74) 622 (0.65, 0.35) 50 35076(15) 7.31 (6.90) 9.02 (8.52) 3.47 (2.49) 624 (0.65, 0.34) 100 11831(14.5) 2.62 (2.44) 2.69 (2.49) 1.19 (0.84) 626 (0.65, 0.35) With BPAPF 6 45211(15) 15.29 (12.17) 21.31 (16.97) 6.34 (4.23) 618 (0.64, 0.35) 12 34196(15) 14.99 (11.02) 19.94 (14.67) 6.15 (3.76) 620 (0.64, 0.35) 20 25644(15) 13.27 (10.30) 17.22 (13.36) 4.75 (3.12) 622 (0.65, 0.35) 50 20501(15) 7.96 (7.38) 9.92 (9.19) 2.69 (2.12) 622 (0.65, 0.35) 100 9049(15) 2.54 (2.29) 2.82 (2.54) 0.85 (0.64) 624 (0.65, 0.35)
aValues in the parentheses are the applied driving voltage. bData collected under 20 mA cm22. cValues in the parentheses are the data
[Os(hptz)2(PPh2Me)2] (2) and [Os(hptz)2(PPhMe2)2] (3) were
prepared as follows. The synthetic procedures are essentially identical to those for complex 1, using a similar molecular ratio of 3-heptafluoropropyl-5-(2-pyridyl) 1,2,4-triazole (hppzH), powdery Os3(CO)12, freshly sublimed Me3NO and the phosphine ligands.
The orange–red complex 2 and bright red complex 3 were obtained in 73% and 70% yields, respectively.
Spectral data of 2: MS (FAB,192Os): m/z 1219 (M+), 1019 (M+2 PPh2Me), 818 (M+ 2 2PPh2Me). 1 H NMR (400 MHz, d6-acetone): d 10.24 (d, 2H, JHH 5 6.8 Hz), 7.49 (dd, 2H, JHH56.8, 7.6 Hz), 7.30 (d, 2H, JHH57.6 Hz), 7.18–7.14 (m, 4H), 7.10–7.03 (m, 10H), 6.88 (t, 4H, JHH57.4 Hz), 6.59–6.55 (m, 4H), 1.22 (t, 6H, JHP 5 3.2 Hz, CH3). 19F NMR (470 MHz, d6-acetone): d 2122.6 (s, 4F), 2109.7 (q, 4F, JFF 5 10.0 Hz), 279.8 (t, 6F, JFF510.0 Hz).31P NMR (202 MHz, d6-acetone):
d 218.2 (s). Anal. Calcd for C46H34F14N8OsP2: C, 45.40; N, 9.21;
H, 2.82. Found: C, 45.41; N, 9.27; H, 2.98.
Spectral data of 3: MS (FAB,192Os): m/z 1095 (M+), 957 (M+2 PPhMe2), 8618 (M+ 2 2PPhMe2). 1H NMR (400 MHz, d6-acetone): d 10.12 (d, 2H, JHH 5 6.4 Hz), 7.73 (dd, 2H, JHH56.4, 7.4 Hz), 7.68–7.65 (m, 2H), 7.20 (ddd, 2H, JHH57.4, 6.4, 1.6 Hz), 7.08 (t, 2H, JHH57.6 Hz), 6.90 (t, 4H, JHH57.6 Hz), 6.38–6.33 (m, 4H), 0.86 (t, 6H, JHP53.2 Hz, CH3), 0.61 (t, 6H, JHP53.2 Hz, CH3).19F NMR (470 MHz, d6-acetone): d 2126.1 (s, 4F), 2110.1 (q, 4F, JFF58.3 Hz), 280.0 (t, 6F, JFF58.3 Hz). 31
P NMR (202 MHz, d6-acetone): d 222.1 (s). Anal. Calcd for
C36H30F14N8OsP2: C, 39.57; N, 10.25; H, 2.77. Found: C, 39.43;
N, 10.20; H, 2.90.
Photophysical measurements were taken as follows. Steady-state absorption, emission and phosphorescence lifetime measurements both in solution and solid have been elaborated in our previous reports.9,20 For measuring quantum yields in the solid state, an integrating sphere (Labsphere) was applied, in which the solid sample film was prepared via a vapor deposition method and was excited by a 514 nm Ar+laser line. The resulting luminescence was acquired by an intensified charge-coupled detector for subsequent quantum yield analyses.21
OLED fabrication and data measurement were performed as follows. BCP was purchased from Aldrich and used as received. AlQ3, NPB and BPAPF were synthesized according to literature
procedures, and were sublimed twice prior to use. Patterned ITO substrates with an effective device area of 3.14 mm2were cleaned as described in a previous report.22A 40 nm thick film of BPAPF or NPB was first deposited as the hole transport layer (HTL). The light-emitting layer (30 nm) was then deposited by co-evaporating the CBP host and the phosphorescent dopant from two
Fig. 3 (a) I–V–L characteristics of the devices based on 1 with NPB as HTL, (b) EL spectra of devices with NPB as HTL, as a function of doping concentration, (c) I–V–L characteristics of the devices based on 1 with BPAPF as HTL and (d) external quantum efficiency and luminous efficiency as a function of current density for device ITO/BPAPF/CBP : 6% 1/BCP/LiF/Al.
independent sources, with both deposition rates being controlled with two independent quartz crystal oscillators. A 10 nm thick BCP as a hole and exciton blocking layer (HBL) and 30 nm thick AlQ3 as an electron transport layer were then deposited
sequentially. A thin layer of LiF (1 nm) and a thick layer of Al (150 nm) were followed as the cathode. The current–voltage– luminance of the devices was measured in ambient conditions with a Keithley 2400 Source meter and a Newport 1835C Optical meter equipped with 818ST silicon photodiode.
Acknowledgements
We thank the National Science Council of Taiwan, ROC for the financial supports (NSC 93-2113-M-007-012) and (NSC 93-ET-7-007-003).
Yung-Liang Tung,aShin-Wun Lee,aYun Chi,*aYu-Tai Tao,*b Chin-Hsiung Chien,bYi-Ming Cheng,cPi-Tai Chou,*cShie-Ming Pengc and Chao-Shiuan Liud
aDepartment of Chemistry, National Tsing Hua University, Hsinchu,
300, Taiwan. E-mail: [email protected]
bInstitute of Chemistry, Academia Sinica, Taipei, 115, Taiwan.
E-mail: [email protected]
c
Department of Chemistry and Instrumentation Center, National Taiwan University, Taipei, 106, Taiwan. E-mail: [email protected]
d
Department of Chemistry, SooChow University, Taipei, 111, Taiwan
Notes and references
1 C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913. 2 (a) C. Adachi, M. A. Baldo, S. R. Forrest, S. Lamansky,
M. E. Thompson and P. C. Kwong, Appl. Phys. Lett., 2001, 78, 1622; (b) Y.-J. Su, H.-L. Huang, C.-L. Li, C.-H. Chien, Y.-T. Tao, P.-T. Chou, S. Satta and R.-S. Liu, Adv. Mater., 2003, 15, 884; (c) A. Tsuboyama, H. Iwawaki, M. Furugori, T. Mukaide, J. Kamatani, S. Igawa, T. Moriyama, S. Miura, T. Takiguchi, S. Okada, M. Hoshino and K. Ueno, J. Am. Chem. Soc., 2003, 125, 12971; (d) C. Jiang, W. Yang, J. Peng, S. Xiao and Y. Cao, Adv. Mater., 2004, 16, 537. 3 (a) S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq,
H.-E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304; (b) J. Brooks, Y. Babayan, S. Lamansky, P. I. Djurovich, I. Tsyba, R. Bau and M. E. Thompson, Inorg. Chem., 2002, 41, 3055.
4 M. A. Baldo, D. F. O’Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151. 5 M. A. Baldo, C. Adachi and S. R. Forrest, Phys. Rev. B, 2000, 62,
10967.
6 (a) X. Jiang, A. K.-Y. Jen, B. Carlson and L. R. Dalton, Appl. Phys. Lett., 2002, 80, 713; (b) S. Bernhard, X. Gao, G. G. Malliaras and H. D. Abruna, Adv. Mater., 2002, 14, 433; (c) X. Jiang, A. K. Y. Jen, B. Carlson and L. R. Dalton, Appl. Phys. Lett., 2002, 81, 3125; (d) H.-J. Su, F.-I. Wu, C.-F. Shu, Y.-L. Tung, Y. Chi and G.-H. Lee, J. Polym. Sci., Part A: Polym. Chem., 2005, DOI: 10.1002/pola.2059. 7 (a) B. Carlson, G. D. Phelan, W. Kaminsky, L. Dalton, X. Z. Jiang,
S. Liu and A. K.-Y. Jen, J. Am. Chem. Soc., 2002, 124, 14162; (b) J. H. Kim, M. S. Liu, A. K.-Y. Jen, B. Carlson, L. R. Dalton, C.-F. Shu and R. Dodda, Appl. Phys. Lett., 2003, 83, 776.
8 (a) C. Adachi, M. A. Baldo, M. E. Thompson and S. R. Forrest, J. Appl. Phys., 2001, 90, 5048; (b) M. Ikai, S. Tokito, Y. Sakamoto, T. Suzuki and Y. Taga, Appl. Phys. Lett., 2001, 79, 156.
9 Y.-L. Tung, P.-C. Wu, C.-S. Liu, Y. Chi, J.-K. Yu, Y.-H. Hu, P.-T. Chou, S.-M. Peng, G.-H. Lee, Y. Tao, A. J. Carty, C.-F. Shu and F.-I. Wu, Organometallics, 2004, 23, 3745.
10 (a) P. Passaniti, W. R. Browne, F. C. Lynch, D. Hughes, M. Nieuwenhuyzen, P. James, M. Maestri and J. G. Vos, J. Chem. Soc., Dalton Trans., 2002, 1740; (b) M. H. Klingele and S. Brooker, Coord. Chem. Rev., 2003, 241, 119; (c) J. G. Haasnoot, Coord. Chem. Rev., 2000, 200–202, 131; (d) J.-K. Yu, Y.-H. Hu, Y.-M. Cheng, P.-T. Chou, S.-M. Peng, G.-H. Lee, Y.-L. Tung, S.-W. Lee, Y. Chi and C.-S. Liu, Chem Eur. J., 2004, DOI: 10.1002/chem.200400598. 11 (a) Y. Wang, N. Herron, V. V. Grushin, D. LeCloux and V. Petrov,
Appl. Phys. Lett., 2001, 79, 449; (b) V. V. Grushin, N. Herron, D. D. LeCloux, W. J. Marshall, V. A. Petrov and Y. Wang, Chem. Commun., 2001, 1494.
12 P.-C. Wu, J.-K. Yu, Y.-H. Song, Y. Chi, P.-T. Chou, S.-M. Peng and G.-H. Lee, Organometallics, 2003, 22, 4938.
13 (a) E. M. Kober, B. P. Sullivan, W. J. Dressick, J. V. Caspar and T. J. Meyer, J. Am. Chem. Soc., 1980, 102, 7383; (b) E. M. Kober, J. V. Caspar, R. S. Lumpkin and T. J. Meyer, J. Phys. Chem., 1986, 90, 3722.
14 A. Vlcek, Jr., Coord. Chem. Rev., 1998, 177, 219.
15 Y.-H. Song, S.-J. Yeh, C.-T. Chen, Y. Chi, C.-S. Liu, J.-K. Yu, Y.-H. Hu, P.-T. Chou, S.-M. Peng and G.-H. Lee, Adv. Funct. Mater., 2004, DOI: 10.1002/adfm200400137.
16 C.-W. Ko and Y.-T. Tao, Synth. Met., 2002, 126, 37.
17 X. Gong, J. C. Ostrowski, D. Moses, G. C. Bazan and A. J. Heeger, Adv. Funct. Mater., 2003, 13, 439.
18 V. Bulovic, R. Deshpande, M. E. Thompson and S. R. Forrest, Chem. Phys. Lett., 1999, 308, 317.
19 The hole mobility of BPAPF was estimated to be an order of magnitude higher than that of NPB from time-of-flight measurement, unpublished results.
20 P.-T. Chou, W.-S. Yu, Y.-M. Cheng, S.-C. Pu, Y.-C. Yu, Y.-C. Lin, C.-H. Huang and C.-T. Chen, J. Phys. Chem. A., 2004, 108, 6487. 21 J.-C. de Mello, H.-F. Wittmann and R.-H. Friend, Adv. Mater., 1997, 9,
230.
22 W.-S. Huang, J. T. Lin, C.-H. Chien, Y.-T. Tao, S.-S. Sun and Y.-S. Wen, Chem. Mater., 2004, 16, 2480.