ATRIPPI: An atom-residue preference scoring function for protein-protein
interactions
Kang-Ping Liu
1, Lu-Shian Chang
1and Jinn-Moon Yang
1,2,3*1
Department of Biological Science and Technology, National Chiao Tung University,
Hsinchu, Taiwan
2
Institute of Bioinformatics, National Chiao Tung University, Hsinchu, Taiwan
3Core Facility for Structural Bioinformatics, National Chiao Tung University, Hsinchu,
Taiwan
[email protected]
Abstract
We present an ATRIPPI model for analyzing protein-protein interactions. This model is a 167-atom-type and residue-specific interaction preferences with distance bins derived from 641 co-crystallized protein-protein interfaces. The ATRIPPI model is able to yield physical meanings of hydrogen bonding, disulfide bonding, electrostatic interactions, van der Waals and aromatic-aromatic interactions. We applied this model to identify the native states and near-native complex structures on 17 bound and 17 unbound complexes from thousands of decoy structures. On average, 77.5% structures (155 structures) of top rank 200 structures are closed to the native structure. These results suggest that the ATRIPPI model is able to keep the advantages of both atom-atom and residue-residue interactions and is a potential knowledge-based scoring function for protein-protein docking methods. We believe that our model is robust and provides biological meanings to support protein-protein interactions.
Keywords: protein-protein interaction, atom-atom interacting preference, knowledge-based scoring matrix, residue-residue interaction preference
1. Introduction
Protein-protein interactions are involved in most biological processes. Identifying their associated networks comprehensively is the key to understanding cellular mechanisms [1]. Many experimental and computational methods have been proposed to identify protein-protein interactions. Protein interactions derived from the large-scale experimental methods, such as the two-hybrid system [2] or affinity purifications [3], are often inconsistent and high false-positive rates [4]. Many computational methods have been developed to predict protein-protein interactions by using gene expression profiles [5], known three-dimensional (3D) complexes [6, 7], 3D-domain interologs [8, 9],
and interologs [10]. The development of computational approaches to map interactions seems useful in light of the shortcomings of large-scale experimental methods.
Known 3D structures of interacting proteins provide interacting domains and atomic details for direct physical interactions [11]. The comparative modeling, which a known complex structure comprising homologs of these two sequences is available, has been applied to predict protein-protein interactions [6, 7, 9]. To analyze interacting interfaces from structural complexes is useful to understand the protein-protein mechanism and to develop knowledge-based scoring functions [12-15] for protein-protein interactions. Accurate docking methods often provide substantial structural knowledge of complexes, from which functional information can be studied. Generally, a docking method should have a scoring function which can discriminate correct or near-native docked orientations from incorrect docked ones.
Various approaches have been developed to analyze protein-protein complexes for understand or predicting protein-protein interactions [12, 13, 16-21]. Glaser et al. [13] analyzed residue contact preferences based on a nonredundant set of 621 protein-protein interfaces and derived knowledge-based residue-residue contact preferences. Moont and coworkers [22] studied empirical residue– residue pair potentials. Zhang et al. [21] determined 18 different atom types to estimate effective atomic contact energies. The residue-based methods [13, 22] are limited to reflect hydrogen bonds, disulfide bonds and electrostatic effect; conversely, the limit of atom-based methods [21] is often poor to describe residue propensities (e.g. aromatic-aromatic interactions and amino acid compositions) in protein-protein interfaces. Therefore, combinations of residue and atom properties have been suggested as a possible means to improve performance in measuring protein-protein interactions. [23]
Here, we present a 167-atom-type and residue-specific interaction model for protein-protein interactions (ATRIPPI). This model, which are derived from 641 co-crystallized protein–protein interfaces selected from Protein Data Bank (PDB) [24], is able to consider both residue-residue interactions and the contributions of atom-atom pairs with distance bins. The ATRIPPI model includes group-charged model for electrostatic force, donor-acceptor model for hydrogen bonds and van der Waals contact model for hydrophobic-hydrophobic interactions. ATRIPPI was evaluated on 34 bound and unbound complexes and proved that our ATRIPPI model effectively identified the native and near-native complexes from thousands of decoy structures for these targets.
2. Results and Discussion
We selected a non-redundant data set, which consists of 641 protein-protein interfaces of known high-resolution structures from PDB, to derive both atom-atom and residue-residue interacting preferences. In this data set, 621 protein-protein complexes were proposed by Glaser et al. [13] and 20 antibody-antigen interfaces were collected from PDB. These antibody-antigen complexes are 1fbi, 1fdl, 1iai, 1jhl, 1jrh, 1kip, 1kiq, 1mel, 1nca, and 2jel based on PDB entry. Each antibody-antigen complex consists of two interfaces, which are between the antigen and the light and heavy chains on the antibody, respectively. In this data set, 237 complexes are heterodimers and 404 complexes homodimers and the sequence identity is less than 30% to each other. This set can be divided into some categories, such as oligomeric proteins, enzyme-inhibitor complexes, membrane proteins, and antibody-antigen complexes. The ATRIPPI model was evaluated on 17 bound and unbound complexes with different atom/residue types to discriminate the native state from 2,500 near-native random states. The set consists of 10 complexes selected from 641 dimer complexes and 7 complexes selected form other related works for comparing with other methods. We followed the method [25] to generate 2500 decoys, which are near the native structures,
for each test complex in the data set. Figure 1 shows
the framework of our ATRIPPI model for atom-atom and residue-residue preferences derived from protein-protein interfaces of this data set.
2.1 Atom and residue types
The atoms with different environments, connectivity and chemical nature, would be different in physicochemical properties. Here, we considered all heavy atoms (i.e. non-hydrogen atoms) of 20
amino acids are residue specific, i.e. the atom Cα of
Gly is different from the atom Cα of Ala. Based on
atom name defined in PDB format, the number of
atom types is 167 (Table 1), including 80 and 87
atom types in the backbone and side chain, respectively. We can consider the physicochemical properties of both atom-atom and residue-residue interaction preferences by using this 167-atom types. For example, the hydrogen bonding is able to be identified if the specific pairing atoms are interacting on the respective chains, such as the atom N of Lys interacting to the atom O of Asp ; the atom NH1 of Arg interacting to the atom OD1 of Asp on sidechains. The atom-atom interactions (i.e. the atom CG, CD1, CD2, CE1, CE2 and CZ of Tyr and the atom CG, CD1, CD2, CE2, CE3, CZ2, CZ3 and CH2 of Trp) and the residue-residue interactions (e.g. Tyr and Trp or Leu and Ile) are able to model hydrophobic-hydrophobic interactions. The atom NZ (Lys) and the atom OD1 (Asp) may form the electrostatic interaction if the distance is within 6 Å in this study. An atom pair SG (Cys) and SG (Cys) form the disulfide bond if the distance of this pair is within 2.8 Å. Here, we obtained a residue-residue interaction by summing all of possible atom-atom pairing interactions of these two interacting residues.
SER O THR CA SER C THR N PRO CD PRO CB HIS CE1 Bin 1 : Dis < 3Å Bin 2 : 3Å < Dis < 3.5Å Bin 3 : 3.5Å < Dis < 4Å Bin 4 : 4Å < Dis < 4.5Å
Calculate atom-atom contact frequencies with different distance bins for 167x167 atom-atom interaction types
according to binding areas and contact residues Identify 167 atom types and distance bins of
atom-atom interactions Identify protein-protein binding areas and contact residues for each complex
Calculate 167x167 atom-atom interaction preferences using
Boltzmann distribution for each distance bin matrix
-1.31 1.76 1.07 GLY N -1.68 1.16 -1.31 GLU OE2 1.76 GLY N ... 1.09 GLY CA … 1.16 GLY CA GLU OE2 …
Distance Bin 1 Distance Bin K
… -0.10 0.67 0.21 GLY N -1.45 -1.02 -0.10 GLU OE2 0.67 GLY N ... 0.23 GLY CA … -1.02 GLY CA GLU OE2 Binding area … …
Figure 1. The scheme of the ATRIPPI model.
2.2 Atom-atom interacting types
The ATRIPPI model is able to yield protein-protein interacting properties of hydrogen bonds, electrostatic interactions, and van der Waals contacts
(hydrophobic-hydrophobic) (Figures 2 and 3). These
three preferences are essential interactions to analyze protein-protein interactions. To identify hydrogen bonds, we identify all donor and acceptor atom pairs
(Table 1) that satisfy the distance from 2.5 to 3.5Å.
All carbon atoms separated by 4.0 Å were considered to be interacting through van der Waals
contacts. The salt bridge interaction is inferred for a pair of oppositely charged residues (Arg, Lys or His interacting with Asp or Glu) if they meet the following criteria: (i) The centroids of the side-chain charged groups in oppositely charged residues lie within 4.0 Å of each other [26, 27]; and (ii) at least one pair of Asp or Glu side-chain oxygen atoms and side-chain nitrogen atoms of Arg, Lys or His is within 4.0 Å. To identify disulfide bonds, our program finds SG (Cys) and SG (Cys) atom pairs that satisfy the distance is smaller than 2.2 Å. Based on these conditions, the ATRIPPI model derived 9,705 hydrogen bonds, 965 salt bridges, 41 disulfide bonds and 30,715 van der Waals interactions derived from 641 protein-protein interfaces. In order to observe the interaction preferences of the ATRIPPI model, 167 atom types are divided into 6 classes
(Figures 2 and 3) based on the physicochemical
properties of atom types: (i) atom N in the backbone;
(ii) Atom C and Cα in backbone; (iii) Atom O in
backbone; (iv) atom Cβ and Cγ of side chain; (v) Cδ,
Cε and Cζ of side chain; and (vi) atom N, O and S of
side chain.
Table 1. The 167 atom types, donor and acceptor for
hydrogen bonds, atom formal charge and 20 residue types defined in the ATRIPPI model
Residue types
No. atom
types atom types Gly 4 N a CA C O b O Ala 5 N CA C O C O CB Val 7 N CA C O CB CG1 CG2 Leu 8 N CA C O CB CG CD1 CD2 Ile 8 N CA C O CB CG1 CG2 CD1 Met 8 N CA C O CB CG SD CE
Phe 11 N CA C O CB CG CD1 CD2 CE1 CE2 CZ Tyr 12 N CA C OCB CG CD1 CD2 CE1 CE2 CZ
OH b
Trp 14 NCE3 CZ2 CZ3 CH2 CA C O CB CG CD1 CD2 NE1aCE2 Ser 6 N CA C O CB OG Pro 7 N CA C O CB CG CD Thr 7 N CA C O CB OG1 CG2 Cys 6 N CA C O CB SG Asn 8 N CA C O CB CG OD1 ND2 Gln 9 N CA C O CB CG CD OE1 NE2 Lys 9 N CA C O CB CG CD CE NZ
His 10 N CA C O CB CG ND1 c CD2 CE1 NE2 c
Arg 11 N CA C O CB CG CD NE CZ NH1 c NH2 c
Asp 8 N CA C O CB CG OD1 d OD2 d
Glu 9 N CA C O CB CG CD OE1 d OE2 d
Atom and residue names are taken from the typical PDB format.
a, b the atom types of donor (blue) and acceptor (red) for
hydrogen-bond type, respectively.
c, d the atom types with formal positive and negative charge,
respectively.
2.3 Atom-atom interacting preferences
Figure 2A indicates the atom-atom preferences at
the distance bin, ranging from 3.0 Å to 3.5 Å. In this matrix (167 x 167), the most preferences (red blocks) of pairing atoms are able to form the hydrogen bonds
by the atom pair N and O on the interacting side chains, respectively. This atom pair (N and O) on the sidechain of the charged residues (e.g. Arg, Lys, His, Asp, and Glu) play a central role for hydrogen bonds in the protein interfaces. Among these atom pairs forming hydrogen bonds, the atom pair N of Arg interacting to atom O of Asp and Glu is the most preference because the multiple donor and acceptor atom types of Asn or Gln. In the preference scores at
the distance bin ranging from 3.5Å to 4.0Å (Figure
2B), the preferences of forming hydrogen bonds are
rapidly decreasing and van der Waals interactions
(atom Cδ, Cε, and Cζ in green block) by aromatic and
long side-chain carbon atom are increasing.
O β γCC δ εCζCC C α C (A)3.0Å ~ 3.5Å (B)3.5Å ~ 4.0Å Cγ Cβ O Cζ Cε Cδ Backbone Side chain 3 ~ 4 2 ~ 3 1 ~ 2 0 ~ 1 -1 ~ 0 -2 ~ -1 -3 ~ -2 -4 ~ -3 N O S C Cα N N S O N
Figure 2. The 167x167 atom-atom interaction scoring
matrixes at two distance bins: (A) 3.0 Å to 3.5 Å and (B) 3.5 Å to 4.0 Å. 167 atom types are divided into atom types
on the backbone (atom N, C, Cα and O) and on the side
chain (atom Cβ, Cγ, Cδ, Cε, Cζ, N, O and S) based on atom
names in the typical PDB. The arrangements of atom types are based on the physicochemical properties. The scores reflect the normalized pairing preferences are derived from 641 protein-protein interfaces. The red and blue colors denote the most and least atom-atom interacting preferences, respectively.
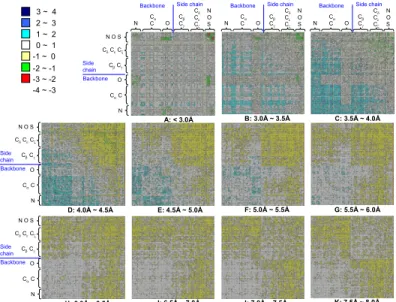
Figure 3 shows the trends of atom-atom
interacting preferences based on 11 protein-protein interacting matrices with 0.5 Å bins ranging from 3 Å to 8.0 Å by considering the contacts between atom types in the 0.0–3.0 Å as a separate bin. These matrices are symmetric and 167 atom types are divided into backbone atom types and sidechain atom types. Here, we analyzed our ATRIPPI model based on sidechain-sidechain, sidechain-backbone, and backbone-backbone interactions by roughly dividing 167 atom types into backbone atom types
(i.e. atom N, C, Cα and O) and sidechain atom types
(i.e. atom Cβ, Cγ, Cδ, Cε, Cζ, N, O and S).
According to Figure 3, we summarized some
observations of atom-atom and residue-residue interacting preferences in the following: (a) Because the large side chains hinder backbone-backbone interactions, the atom-atom interacting preferences on the interacting backbones are small when the
distances of atom pairs are ranging between 3.5 Å
and 5.0 Å (from matrix A to matrix E). The
preferences of backbone-backbone interactions are increasing when the distance of a pairing atom is more than 5.5 Å. (b) On other hand, for the atom-atom preferences of sidechain-sidechain interactions, the preferences are high when the distances of pairing pairs are less than 5.5 Å and the preferences are decreasing when the distance is more than 6.0 Å. (c) The interacting preferences of backbone-sidechain interactions are increasing from 4.5 Å to
8.0 Å (from matrix E to matrix K). (d) The
interacting preferences of sidechain-sidechain interactions are general much larger than ones of backbone-backbone interactions for each distance bin. (e) The hydrogen bonds are formed by a atom pair N and O when their distance is less than 3.5 Å
(e.g. green and red blocks in matrices A and B in
Figure 3). Most of these hydrogen bonds are formed
by atoms N and O on the interacting sidechains, respectively. Some of hydrogen bonds are formed by the atom pair N and O on the interacting backbones.
The matrix A (e.g. <3.0 Å) also shows that the
disulfide interaction is formed by the atom pair S and S of Cys on the respective interacting chains. Except hydrogen bonding and disulfide interactions, other interacting preferences are very low in these two matrices A and B.
2.4 Hydrogen bonds and electrostatic interactions
The effect of the hydrogen bonds is one of the important features in protein-protein interactions. A hydrogen bond is often formed by the donor-acceptor or donor-acceptor-donor atom pairs. The hydrogen-bonding atom types (i.e. donor and
acceptor) of 167 atom types are summarized in Table
1. Figure 4A shows the relationship between
atom-atom preference scores and the distance of pairing donor-acceptor atoms from 3.0 Å to 12.0Å. The preference scores ranging from 2.0 to 4.0 Å are mainly derived from atom pairs N and O of the
interacting side chains (red blocks in Figures 3A and
3B) and partly from interacting backbones on the
protein-protein interfaces. The highest probability of atom pairs forming the hydrogen bonding is between Arg and Glu, reflecting also the tendency for
opposite charge residue pairs (Figure 4B). Compared
with these atom-atom pairs, we found that the number of hydrogen bonds of similar charge residue pairs is quite low because of the electrostatic
repulsion between them (Figure 4B). However, using
18 different protein atom types [14], which were identified by clustering all the heavy atoms of the 20 common amino acids, was unable to reflect the different frequencies of hydrogen bonds for different pairs of residues. 3 ~ 4 2 ~ 3 1 ~ 2 0 ~ 1 -1 ~ 0 -2 ~ -1 -3 ~ -2 -4 ~ -3 A: < 3.0Å B: 3.0Å ~ 3.5Å C: 3.5Å ~ 4.0Å E: 4.5Å ~ 5.0Å D: 4.0Å ~ 4.5Å F: 5.0Å ~ 5.5Å G: 5.5Å ~ 6.0Å I: 6.5Å ~ 7.0Å H: 6.0Å ~ 6.5Å J: 7.0Å ~ 7.5Å K: 7.5Å ~ 8.0Å N O Cβ Cγ Cδ Cε Cζ N O S Cα C N O CβCγ CδCεCζ N O S CαC Side chain Backbone N O CβCγ CδCεCζ N O S CαC Side chain Backbone N O CβCγ CδCεCζ N O S CαC Side chain Backbone
Backbone Side chain
N O Cβ Cγ Cδ Cε Cζ N O S Cα C
Backbone Side chain
N O Cβ Cγ Cδ Cε Cζ N O S Cα C
Backbone Side chain
Figure 3. Eleven 167x167 atom-type scoring matrixes with 0.5 Å bins ranging from 3 Å to 8.0 Å. 167 atom types are
divided into atom types on the backbone (atom N, C, Cα and O) and on the side chain (atom Cβ, Cγ, Cδ, Cε, Cζ, N, O and S)
based on atom names in the typical PDB. The arrangements of atom types are based on the physicochemical properties. The scores reflect the normalized pairing preferences are derived from 641 protein-protein interfaces. The red and blue colors denote the most and least atom-atom interacting preferences, respectively.
0 3 6 9 12 -4 -3 -2 -1 0 1 Edonor-accep to r distance(angstrom) 0 3 6 9 12 -4 -3 -2 -1 0 1
donor-acceptor atom pair for opposite charged residue donor-acceptor atom pair for similar charged residue
Edonor-accep to r distance(angstrom) 0 3 6 9 12 -4 -3 -2 -1 0 1 Eoppo
site charge residue pa
ir distance(angstrom) 0 3 6 9 12 -1.0 -0.5 0.0 0.5 1.0 1.5 2.0
nonpolar atom pair for aromatic-aromatic interaction nonpolar atom pair for backbone-backbone interaction
Enop
olar-nopolar
distance(angstrom) Similar charged atom-atom interactions
opposite charged atom-atom interactions
Non-polar atom-atom interactions (backbone to backbone) Aromatic-aromatic interactions (sidechain to sidechain) A D C B A tom -at om p re fer enc e ( Sij ) A to m -a to m pr efer en ce ( Sij ) A to m -a to m pr ef er en ce ( Sij ) A tom -at om pr ef er e nc e ( Sij ) Distance bins (Å ) Distance bins (Å ) Distance bins (Å ) Distance bins (Å ) opposite charged atom-atom
interactions donor-acceptor atom-atom interactions of hydrogen bonds
Figure 4. The relationship between atom-atom
interacting preferences and distances of the pairing atoms. (A) Hydrogen bonds of the donor-acceptor atom pairings; (B) Hydrogen bonds of donor-acceptor atom pairings with the similar charge and opposite charge; (C) Electrostatic interactions of opposite-charged atom pairings; (D) Van der Waals interactions of carbon atom pairings in interacting aromatic groups and in backbones.
In the ATRIPPI model, the charged groups are atom NE, NH1 and NH2 of Arg; the atom NZ of Lys; the NE2 atom of His; the atom OD1 and OD2 of Asp;
and the atom OE1 and OE2 of Glu. Figure 4C shows
the relations of Sij between opposite charge atoms
and distances of pair atoms. The most preference
scores (Sij) of pair atoms with opposite charges for
electrostatic interactions when their distances are less than 4.5 Å. However, using only a distance cutoff (e.g. <6.0 Å [14]) was often unable to yield electrostatic interactions exactly in protein-protein binding site. The electrostatic interactions often form
salt bridges and hydrogen bonding if their distance is less than 3.0 Å.
2.5 Hydrophobic–hydrophobic Interactions
The percentage of van der Waals interactions, which are mostly used to stabilize a protein-protein interface, is 64.07% of all atom-atom interactions derived from 641 protein-protein interfaces. The most common atom-atom pairs involving van der Waals interactions are from aromatic residues. Owing to the large surface area provided by ring-stacking, the carbon atoms of the residues Phe, Tyr, and Trp are often interacting to the carbon atoms of Phe, Tyr, and Trp. The carbon-carbon interactions in aromatic group exhibit an elevated preference which is agreement with the well-known aromatic-aromatic interactions [28]. In addition, we found that carbon atoms of aliphatic side chains for Val, Leu, Ile and Met have high preferences to interact with carbon atoms of the aromatic side chains for Phe, Tyr, and
Trp (Figures 2 and 3). The favorable atom pairs
between the charged residues (e.g. Lys, His, and Arg) and the hydrophobic residues (e.g. Phe, Tyr, and Trp) suggest that hydrogen bond interactions and van der Waals interactions may exist simultaneously
(Figures 3D, 3E, and 3F). For non-polar contacts
(Figure 4D), the preferences of carbon atom
interactions on the sidechains is much higher than ones of the carbon atoms in backbones. If we used only one distance cutoff < 6.0 Å, the aromatic-aromatic interactions were unable to reflect exactly in protein-protein binding interfaces.
Table 2. The ATRIPPI model results on 17 bound and 17 unbound complexes with different atom and residue types
Unbound structures Bound structures No. hits in top 200 for bound structures Complex name Receptor a Ligand a Complex a RMSD b 167 atom type e 18 atom type f 20 residue type
A. Enzyme-inhibitor complexes
Torpedo Acetylcholinesterase/Fasciculin II 2ace 1fsc 1fssd 0.76 109 136 0
Mouse Acetylcholinesterase/Fasciculin II 1maa 1fsc 1mah 0.6 86 119 0
Subtilisin Novo/Eglin C 1sup 1sbnc 1sbn 0.4 162 133 0
Uracil-DNA Glycosylase/inhibitor 1udh 1udic 1udid 0.5 156 144 0
Uracil-DNA Glycosylase/inhibitor 1akz 1ughc 1ugh 0.28 185 159 0
Kallikrein A/pancreatic trypsin inhibitor 2pka 1bpi 2kaid 0.7 158 137 0
β-trypsinogen/bovine pancreatic trypsin inhibitor 2ptn 6pti 2ptcd 1.2 150 151 0
Subtilisin BPN/subtilisin inhibitor 1sup 3ssi 3sicd 0.61 147 169 0
B. Antibody-antigen complexes
IgG1 HyHel-5 Fab fragment/lysozyme 1bqlc 1dkj 1bql 0.84 155 136 0
IgG1 Fv fragment/lysozyme 1jhlc 1ghl 1jhld 0.49 149 116 0
Jel42 Fab fragment/histidine phosphocarrier protein 2jelc 1poh 2jeld 0.73 158 128 0
Fab HyHel-5/lysozyme 3hflc llza 3hfl 0.6 160 141 0
1gG1 HyHel-10 Fab fragment/lysozyme 3hfmc 1lza 3hfm 0.56 164 149 0
C. Other complexes
Actin/deoxyribonuclease I 1atnc 3dni 1atnd 0.5 155 136 0
Glycerol kinase/GSF III 1glac 1f3g 1glad 0.5 168 138 0
HIV-2 protease with peptide inhibitor (dimer) 2mipc 2mipc 2mip 0.6 190 168 0
Human growth hormone/receptor 3hhrc 1hgu 3hhrd 1.2 185 178 0 a 4-letter PDB code for the crystal structures used in this study.
b The RMSD (Å) of the Cα atoms of unbound the receptor and ligand after superposition onto the co-crystallized complex structure. c Crystal structure is taken from bound complex.
d Crystal structure is selected from 641 protein-protein interfaces.
e Hits are defined as candidate structures with all main chain atoms RMSD ≦ 2.0 Å from the crystal complex.
2.6 Results on bounded and unbound complexes
The ATRIPPI model was evaluated on 17 bound and unbound complexes with different atom and
residue types (Table 1) to discriminate the native
state from 2,500 near-native structures by using the
scoring matrices (Figures 2 and 3). The set consists
of 10 complexes selected from 641 dimer complexes and 7 complexes selected form other related works for comparing with other methods. We followed the method [25] to generate 2500 decoys, which are near the native structures, for each test complex in the data set.
Table 3. Average numbers of hits in top rank 200 using
ATRIPPI on 17 bound and 17 unbound complexes with different number of distance bins, atom types, and residue types
167 atom types 18 atom types 20 residue types
1 bin a 11 bins b 1 bin a 11 bins b 1 bin a 11 bins b Bound structure 117.82 150 0 143.41 103.58 7.76 Unbound structure 88.47 98.17 4.88 86.23 89.64 0
a Using only one distance bin and the cutoff is set to 6.0
b The distances observed into 0.5 Å bins ranging from 3.0
to 8.0 Å.
Table 4. Average numbers of hits in top 200 of using
ATRIPPI on 17 bound and unbound complexes with different distance-bin sizes
Bound complex Unbound complex
Complex name Interval size (0.5 Å) Interval size (1.0 Å) Interval size (0.5 Å) Interval size (1.0 Å) enzyme-inhibitor 132.2 144.1 90 95.5 antibody-antigen 110.4 157.2 97.6 133.6 other complexes 172.2 174.5 115.2 114.2 total 138.3 155.1 98.2 101.5
Hits are defined as candidate structures with RMSD ≦ 2.0 Å from the native crystal complexes.
Table 2 shows the performance of ATRIPPI
results on 17 bound and unbound complexes with different atom/residue types. The 167-atom-type model (ATRIPPI) is the best and the residue-based approach is the worst based on the number of top rank 200 structures whose root-mean-square
derivation (RMSD) < 2.0 Å on Cα coordinates
between selected structures and the native structure. The 167-atom-type model is better than 18-atom-type model and is much better than 20-residue-18-atom-type model because the ATRIPPI model can consider atom-atom interactions and incorporates specific interactions (e.g. electrostatic interactions, van der Waals, and hydrogen bonds). Conversely, the 18-atom-type model can not reflect residue-residue interactions (such as the aromatic-aromatic interactions). The residue-based model is often
unable to reflect the specific interactions. The ATRIPPI model considers not only residue-residue interactions but also atom-atom interactions.
Figure 5 shows the correlations between the
ATRIPPI potentials and the root mean square deviation (RMSD) between the native structure and decoy structures for five antibody-antigen complexes in bound systems. The RMSD values of the native structures are zero. Most of near-native structures are ranked within top rank 10. These results show that our energy function is able to identify native and near-native structures from lots of decoy structures.
Figure 5. Binding energy vs. RMSD (abscissa) for the
top 2,500 structures of each antibody-antigen complexes in bound systems: (a) 1bql, (b) 1jhl, (c) 2jel, (d) 3hfl and (e) 3hfm.
2.7 Distance bins and Interval sizes
The number of distance bins and the interval sizes are important factors for improving the
discrimination of the ATRIPPI model (Tables 3 and
4) for protein-protein interaction predictions. The
ATRIPPI model using 11 bins performed well when the atom-atom interaction models (i.e. 167 and 18
atom types) were applied (Table 3). The results
shows that the atomic pair scores using a cutoff distance with a suitable interval is able to reflect electrostatic effects, hydrogen bonding, and van der Waals contacts. Interestingly, the residue-based model obtained good results when only one bin was
applied. Table 4 shows that the ATRIPPI model with
large interval size (1.0 Å) outperformed the ATRIPPI model with small interval size (0.5 Å). For each antigen-antibody complex, the ATRIPPI model
yielded that the average number of near native structure in top 200 structures is greater than 150 in 17 bound complexes. Bound and unbound protein structures frequently differ in the conformations of some side chains in the binding site. Generally, all models yielded better performance on bound complexes than on unbound complexes.
3. Materials and Methods
Figure 1 shows the framework of our ATRIPPI
model for atom-atom and residue-residue preferences derived from protein-protein interfaces of 641 dimer complexes. The protein-protein binding site and contact residues were first identified for each complex in the data set. Based on the defined distance bins and atom-type representations, we calculated the atom-atom and residue-residue interactive frequencies. Finally, the frequencies of the atom-atom and residue-residues interacting preferences were studied and transformed into interactive scores based on Boltzmann distribution. The ATRIPPI model was then evaluated on 34 test complexes to distinguish between the native and near native structures from incorrect structures in the decoy set.
3.1 Interacting interfaces and distance bins
For each complex in the data set, we identified interacting interfaces and contact residues (and atoms) of two interacting chains. Contact residues, whose any heavy atoms should be within a threshold
(Rc) to any heavy atoms of another chain, were
considered as in the interacting sites of the protein-protein interface in a complex. Each chain must have more than 5 contact residues and the number of interacting contact-residue pairs more than 25 to make sure that the contact between the proteins was
reasonably extensive [29]. Based on different Rc, we
obtained various the sizes of interacting sites forming different distributions of the resulting
potentials. When the Rc is less than 4 Å, the
atom-atom interactions is able to describe the specific interactions (e.g. hydrogen bonds and disulfide
bonds). Conversely, an extension of Rc to larger
distances is abele to incorporate the influences of van der Waals interactions and residue-residue interactions. We divided distances observed into 0.5 Å bins ranging from 3.0 to 8.0 Å. The total number of distance bins is 11 by considering the contacts between atom types in the 0.0–3.0 Å are placed in a separate bin. The interval size and number of distance bins were decided based on various parameter tests.
3.2 Interaction preferences
The atom-atom contact frequencies observed in the protein-protein complex structures are assumed to obey a Boltzmann distribution. We followed previous work [15] to define the interaction preferences and scores between atom types i and j as
)
)
(
)
(
ln(
)
(
d
f
d
f
kT
d
S
ref ij ij=
−
where i and j denote atom types, respectively; k is the Boltzmann’s constant; T is the absolute
temperature; fij(d) and fref (d) are the observed and
reference probability, respectively, of the occurrences of atom types i and j contacting at the
distance bin d. The score is smaller than zero (Sij < 0)
if the observed probability is greater than the reference probability. The preference of an
interacting atom i, j pair is high when Sij < 0. By
contrast, Sij > 0 if fij < fref. Using a set of known 3D protein complex structures from data set C (i.e. the 641 protein-protein interfaces), we can make observations of atom-atom contacts in a particular distance bin. We compute the frequencies of observing atom type i and atom type j in a particular
distance bin from 641 dimer complexes. The fij (d) is
defined as
,
)
(
)
(
)
(
1∑
==
DB k ij ij ijk
N
d
N
d
f
N ij(d) is the number of atom type i and j in a
particular distance bin d; DB is the number of the distance bins. In this work, DB is 11. The denominator is the total number of atom types i, j contacts for all distance bins. The reference state is often built on the basis of the quasichemical
approximation.Here, the reference state is defines as
,
2
/
)
1
(
)
(
)
(
1 1+
×
=
∑∑
= = ≤ =n
n
d
f
d
f
n i i i j j ij refwhere n is the number of atom types (n is 167 in this paper).
4. Conclusions
This study demonstrates the robustness and feasibility of the ATRIPPI model with 167-atom types for protein-protein interactions. This model is able to yield the advantages of atom-based and residue-based interactions. The atom-based interaction is an effective means of assessing specific interactions, including hydrogen bonds, electrostatic interactions, and disulfide bonds. The residue-based interaction is able to reflect the aromatic-aromatic interactions. The ATRIPPI model with different distance bins is sensitive to binding affinity and is able to effectively identify the native and near-native structures from thousands of decoy structures for 34
test complexes. These results suggest that the ATRIPPI model is robust and provides biological meanings to support protein-protein interactions.
5. Acknowledgments
J.-M. Yang was supported by National Science Council and partial support of the ATU plan by MOE. Authors are grateful to both the hardware and software supports of the Structural Bioinformatics Core Facility at National Chiao Tung University.
6. References
[1] M. P. Cary, G. D. Bader, and C. Sander, "Pathway information for systems biology," FEBS Letters, vol. 579, pp. 1815-1820, 2005.
[2] T. Ito, T. Chiba, R. Ozawa, M. Yoshida, M. Hattori, and Y. Sakaki, "A comprehensive two-hybrid analysis to explore the yeast protein interactome,"
Proceedings of the National Academy of Science of the USA, vol. 98, pp. 4569-4574, 2001.
[3] A. Pandey and M. Mann, "Proteomics to study genes and genomes," Nature, vol. 405, pp. 837-846, 2000. [4] C. von Mering, R. Krause, B. Snel, M. Cornell, S. G.
Oliver, S. Fields, and P. Bork, "Comparative assessment of large-scale data sets of protein-protein interactions," Nature, vol. 417, pp. 399-403, 2002. [5] R. Jansen, H. Yu, D. Greenbaum, Y. Kluger, N. J.
Krogan, S. Chung, A. Emili, M. Snyder, J. F. Greenblatt, and M. Gerstein, "A Bayesian networks approach for predicting protein-protein interactions from genomic data," Science, vol. 302, pp. 449-453, 2003.
[6] P. Aloy and R. B. Russell, "Interrogating protein interaction networks through structural biology,"
Proceedings of the National Academy of Science of the USA, vol. 99, pp. 5896-5901, 2002.
[7] L. Lu, A. K. Arakaki, H. Lu, and J. Skolnick, "Multimeric threading-based prediction of protein-protein interactions on a genomic scale: application to the Saccharomyces cerevisiae proteome," Genome
Research, vol. 13, pp. 1146-1154, 2003.
[8] Y.-C. Chen, H.-C. Chen, and J.-M. Yang, "DAPID: A 3D-domain annotated protein-protein interaction database," Genome Informatics, vol. 17, pp. 206-215, 2006.
[9] Y.-C. Chen, Y.-S. Lo, W.-C. Hsu, and J.-M. Yang, "3D-partner: a web server to infer interacting partners and binding models," Nucleic Acids
Research, pp. W561-W567, 2007.
[10] L. R. Matthews, P. Vaglio, J. Reboul, H. Ge, B. P. Davis, J. Garrels, S. Vincent, and M. Vidal, "Identification of potential interaction networks using sequence-based searches for conserved protein-protein interactions or "interologs"," Genome
Research, vol. 11, pp. 2120-2126, 2001.
[11] P. Aloy, M. Pichaud, and R. B. Russell, "Protein complexes: structure prediction challenges for the 21st century," Current Opinion in Structural Biology, vol. 15, pp. 15-22, 2005.
[12] R. Chen and Z. Weng, "Docking unbound proteins using shape complementarity, desolvation, and electrostatics," Proteins, vol. 47, pp. 281-94, 2002. [13] F. Glaser, D. M. Steinberg, I. A. Vakser, and N.
Ben-Tal, "Residue frequencies and pairing preferences at protein-protein interfaces," Proteins, vol. 43, pp. 89-102, 2001.
[14] C. Zhang, G. Vasmatzis, J. L. Cornette, and C. DeLisi, "Determination of atomic desolvation energies from the structures of crystallized proteins,"
J Mol Biol, vol. 267, pp. 707-26, 1997.
[15] M. J. Sippl, "Knowledge-based potentials for proteins," Current Opinion in Structural Biology, vol. 5, pp. 229-235, 1995.
[16] C. J. Camacho, D. W. Gatchell, S. R. Kimura, and S. Vajda, "Scoring docked conformations generated by rigid-body protein-protein docking," Proteins, 2000. [17] J. Fernandez-Recio, M. Totrov, and R. Abagyan,
"Soft protein-protein docking in internal coordinates," Protein Sci, vol. 11, pp. 280-91, 2002. [18] J. G. Mandell, V. A. Roberts, M. E. Pique, V.
Kotlovyi, J. C. Mitchell, E. Nelson, I. Tsigelny, and L. F. Ten Eyck, "Protein docking using continuum electrostatics and geometric fit," Protein Eng, vol. 14, pp. 105-13, 2001.
[19] R. Norel, F. Sheinerman, D. Petrey, and B. Honig, "Electrostatic contributions to protein-protein interactions: fast energetic filters for docking and their physical basis," Protein Sci, vol. 10, pp. 2147-61, 2001.
[20] P. N. Palma, L. Krippahl, J. E. Wampler, and J. J. Moura, "BiGGER: a new (soft) docking algorithm for predicting protein interactions," Proteins, vol. 39, pp. 372-84, 2000.
[21] C. Zhang, G. Vasmatzis, J. L. Cornette, and C. DeLisi, "Determination of atomic desolvation energies from the structures of crystallized proteins,"
Journal of Molecular Biology, vol. 267, pp. 707-726,
1997.
[22] G. Moont, H. A. Gabb, and M. J. E. Sternberg, "Use of pair potentials across protein interfaces in screening predicted docked complexes," Proteins:
Structure, Function, and Bioinformatics, vol. 35, pp.
364-373, 1999.
[23] D. F. Hsu, Advanced Data Mining Technologies in
Bioinformatics, 2006.
[24] H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov, and P. E. Bourne, "The Protein Data Bank," Nucleic Acids Res, vol. 28, pp. 235-42, 2000.
[25] J. J. Gray, S. Moughon, C. Wang, O. Schueler-Furman, B. Kuhlman, C. A. Rohl, and D. Baker, "Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations," Journal of Molecular Biology, vol. 331, pp. 281-299, 2003.
[26] D. J. Barlow and J. M. Thornton, "Ion-pairs in proteins," J Mol Biol, vol. 168, pp. 867-85, 1983. [27] S. Kumar and R. Nussinov, "Salt bridge stability in
monomeric proteins," J Mol Biol, vol. 293, pp. 1241-55, 1999.
[28] S. K. Burley and G. A. Petsko, "Aromatic-aromatic interaction: a mechanism of protein structure stabilization," Science, vol. 229, pp. 23-8, 1985. [29] J. Park, M. Lappe, and S. A. Teichmann, "Mapping
protein family interactions: intramolecular and intermolecular protein family interaction repertoires in the PDB and yeast," Journal of Molecular Biology, vol. 307, pp. 929-938, 2001.