國立交通大學
電子物理系
碩 士 論 文
單一原子石墨層電子結構的計算模擬及大型矩陣對
角化技術研究
Simulation of the electronic structure of graphene nanostructures and
studies of the techniques of large scale matrix diagonalization
研 究 生:陳勇達
指導教授:鄭舜仁 教授
Simulation of the electronic structure of graphene nanostructures and
studies of the techniques of large scale matrix diagonalization
研 究 生:陳勇達 Student:Yung-Da Chen 指導教授:鄭舜仁 教授 Advisor:Shun-Jen Cheng 國 立 交 通 大 學 電子物理研究所 碩士論文 A Thesis
Submit to Department of Electrophysics College of Science
National Chiao Tung University in Partial Fulfillment of the Requirements
for the Degree of Master
in
Electrophysics
April 2009
HsinChu, Taiwan, Republic of China
感謝鄭舜仁老師兩年來不厭其煩的細心教導,讓我在研究上能
夠更上層樓,也讓我了解固態領域的淵博,並且從而學習到嚴謹
的態度,了解每個式子更深層的意涵,也感謝老師在各個方面對
我的教導,受益匪淺。感謝關肇正老師、邱博文老師、簡紋濱老
師於口試時提出的寶貴意見。
在學兩年期間,非常感謝書楷學長在各個方面給我的幫忙與意
見,也感謝彥廷學長、志彬學長帶領我進研究室,還有下來拿便
當的上瑜學長;感謝同窗虔震、文廷、瑞甫你們不時的幫忙與熱
烈討論,解開我許多疑惑;感謝學弟們克銘、禹淮、徐燁、浤鈞
你們在實驗室的幫忙,讓我能好好完成論文。感謝我的父母支持
我,讓我一路完成學業;感謝采玲,豐富了我的感情生活,一路
在我艱苦的時候支持我。最後,感謝在我碩士生活中曾經幫過我
的每一位,讓我有個美好的碩士生涯。
術研究
學生:陳勇達 指導教授:鄭舜仁 博士
國立交通大學電子物理研究所碩士班
摘 要
在這份論文中,我們將計算奈米尺度的單一原子石墨層(graphene)的電子結
構並研究大規模矩陣(Large scale matrix)對角化的技術,首先我們介紹
Tight-Binding 的基本理論,建立 K 空間與實空間矩陣之後,我們建構出單
一原子石墨層的奈米結構(graphene & graphene nanoribbon)的 Tight-Binding
矩陣,並只計算出單一原子石墨層與 nanoribbon 的能帶結構。接著再介紹
大規模稀疏矩陣(Large scale sparse matrix)對角化技術中的 LANCZOS 以及
使用最多時間研究的 ARPACK 演算法,應用在簡單的一維簡諧振子問題並
利用有限差分法(finite difference)計算其能階,檢驗其準確度,會隨著矩陣
增大而增加其準確度,使用一般配置 8G 記憶體的電腦,目前 ARPACK 可
studies of the techniques of large scale matrix diagonalization
Student : Yung-Da Chen advisor:Shun-Jen Cheng
Department of Electrophysics National Chiao Tung University
ABSTRACT
In this thesis, we theoretically study the electronic structures of graphene nanostructures and the techniques of numerical diagonalization of large scale matrix, i.e., Lanczos and Arpack eigensolvers. In the first part of this thesis, the electronic structures of infinite two-dimensional(2D) graphene systems and one-dimensional(1D) graphene nano-ribbons are calculated by using one-orbital tight binding theory. The general Hamiltonian matrices for the 2D and 1D
graphene systems are explicitly derived. The electronic structures of the graphene nano-systems are calculated by carrying out direct diagonalization. According to the study, we identify the localization of electron density at the zigzag edge in the zigzag ribbon. In the second part, we review the basic theory and algorithm of Lanczos and ARPACK eigensolvers for the
diagonalization of large scale sparse matrix. The ARPACK package is applied to solve the problem of 1D simple harmonic oscillation. The maximum size of matrix diagonalized by the package is 8 million by 8 million on PC with 8G memory. Typically, the high accuracy 0.00002 % for the numerically calculated ground states can be achieved.
致謝 ……… I 中文摘要 ……… II 英文摘要 ……… III 目錄 ……… IV 第一章、導論 ……… 1 1.1 graphene 簡介 ……… 1 1.2 研究動機 ……… 1 1.3 章節概要 ……… 2 第二章、Tight-binding Method ……… 3 2.1 Tight-binding 理論 ……… 3 2.2 k-space Hamiltonian ……… 5
2.3 Real space Hamiltonian ……… 11
第三章、數值結果 ……… 13 3.1 graphene ……… 13 3.2 graphene nanoribbon ……… 17 第四章、數值方法 ……… 24 4.1 Lanczos Method ……… 25 4.2 Arpack Method ……… 30 第五章、結論與未來工作 ……… 44 參考文獻 ……… 46 附錄 A ……… 47 附錄 B ……… 50
第一章、導論
1.1 graphene 簡介 單層石墨(graphene)為一個單一原子厚度、外觀為蜂巢狀結構的石墨薄 膜,這樣的材料從 2004 年英國的曼徹斯特(Manchester)大學的 Novoselov K.S 發表了可以利用力學的方法製作出單層石墨(graphene)[1,2]之後,開始引起 大家廣泛的注意,其中,在單層石墨中的電子必須用滿足相對論的 Diracfermion 去描述,在導電帶與價電帶交會的 Dirac point 上,能量與動量有線
性的關係,且電子在單一石墨層上群速度移動接近光速的三百分之一,相
當高的電子傳輸速度有可能成為新一代的電晶體材料。量子霍爾效應
(quantum hall effect)[3,4],也是單層石墨發現有趣的特性之一,包含了室溫
下便能觀測到的整數量子霍爾效應,以及 2007 年有文章宣稱觀測到分數量 子霍爾效應[5,6,7],也是最近新穎材料的研究當中最令人訝異的,有如此特 性的材料,開啟了各種研究的大門。 1.2 研究動機 主要的研究動機,是發展大規模矩陣的計算工具,以利後人能夠以現有 電腦速度與規模,用於計算由原子數決定矩陣大小的電子結構,對於奈米 材料的計算中,Tight-Binding 方法去計算能帶理論時,在以往大約只能算 到幾奈米大小,若現今引入此技術,大約能夠計算數十奈米,甚至到一百 奈米的大小,大幅度增加計算尺度,所以此技術對研究是非常重要的。在
開始發展之後,便能使用此計算方法及此數值計算軟體去模擬新穎材料單 層石墨的計算,以及單層石墨奈米緞帶(graphene nanoribbon),了解此兩種 材料的電子結構。 1.3 章節概要 第一章簡單介紹完單層石墨之後,第二章便會開始介紹本篇論文的主要 理論的計算方法 Tight-Binding 法,針對於物理上的解釋,以及滿足 Bloch 理論的波函數,進而推導出 K 空間的 Hamiltonian,這部份是計算塊材主要 的理論。但是針對實際上材料大小並非無窮大,所以在實際材料上必須考 慮到材料邊界的效應,便產生在實空間中所需的理論,進而產生實空間的 Hamiltonian。也由於在實空間中,矩陣的大小會隨著材料增大而增加,因 此在數值方法中,如何縮減矩陣元素,甚至是縮減矩陣大小,以增加數值 運算的大小,提升目前電腦規格能夠計算的範圍,便是一個很重要的課題。 所以第三章會介紹數值方法常用的 Lanczos[8,9]與 ARPACK[9,10],其中
Lanczos 是將矩陣縮減成三對角線矩陣(tridiagonal matrix),而 ARPACK 不
僅僅縮減矩陣,還利用到稀疏矩陣的記錄方式,以及切割矩陣的大小,用
以分段計算。而第四章開始使用 Tight-Binding 方法計算單層石墨(graphene)
與單層石墨奈米緞帶(graphene nanoribbon) [11,12]的電子結構,一窺究竟。
第二章、Tight-Binding Method
這一章我們主要介紹的是我們基本的理論架構, 如何將單電子在晶格 中的漢米爾量(Hamiltonian)化簡,其滿足 Bloch 函數的波函數形式,使用 Tight-Binding 方法需要有哪些近似?皆會在這一章探討。 2.1 Tight-Binding Method 從量子力學中,可以了解在單電子單原子的電子能量與波函數的關係, 為一特徵值方程,如(2.1.1)( )
2 2( )
( )
( )
0 2 H r V r r r m α α α α ϕ = −⎡⎢ ∇ + ⎤⎥ϕ =ε ϕ ⎣ ⎦ h r r r r (2.1.1) : atomic orbital α 圖 2.1.1、單原子單電子位能 這是一個在量子力學中已知答案的問題,我們了解氫原子系統(單原子單電 子)的能量與波函數,但在固態物理中,並沒有非常了解單電子在多原子中 的行為,如果,我們能用單原子系統描述多原子系統,便可以簡化在固態 物理中複雜的情形。 在固態的系統中,我們知道它是由原子以週期性來排列的。換句話說,電子或其他帶電荷的粒子在固態的系統中所受到的位能也是成週期性的排 列如公式(2.1.2)式
( )
(
)
U rr =U rr+Rr (2.1.2) R =n a1 1ˆ +n a2ˆ2 +n a3 3ˆ r( )
U rr 是來自晶格的位能,而不是外加的位能。Rr是一個滿足晶格週期的向 量,其中n n n1, 2, 3是整數,而a a aˆ ˆ1, 2,ˆ3是晶格向量。根據 Bloch’s theorem,當電 子在週期性的位能中,它的波函數可以表示成 Bloch’s function,如(2.1.3)式( )
ik r( )
kr
e
u
kr
ψ
=
r r⋅ rr
rr
(2.1.3)(
)
( )
k ku
rr
r
+
R
r
=
u
rr
r
在方程式中 ik r er r⋅ 所表示的是電子在晶體中具有平面波的特性,而ukr( )
rr 是一個 週期為Rr的函數。所以,系統 Hamiltonian 可以分成單原子系統與微擾項, 其中微擾來自於位能影響。如(2.1.4)式( )
2 2 2 H U r m = − h ∇ + r( )
(
( )
( )
)
2 2 2m V r U r V r ⎛ ⎞ = −⎜ ∇ + ⎟+ − ⎝ ⎠ h r r r( )
0 H U r = + Δ r (2.1.4)圖 2.1.2、晶格位能(不連續),Rj 代表 unit cell 所在的位置 將(2.1.4)代入薛丁格方程,可得:
( )
0( )
( )
( )
k k k Hψr rr =⎡⎣H + ΔU rr ⎤⎦ψr rr =Eψr rr (2.1.5) 2.2 k-space Hamiltonian 在此這樣的位能下,滿足(2.1.3)的波函數如下:[13,14](
)
1 1 ( ) j N ik R j k j r e r R N ψ ⋅ φ = =∑
r r − r r r r (2.2.1) 滿足晶格週期性的波函數,可看成 Wannier 函數φ(
r−Rj)
r r 乘上相位 ik Rj e ⋅ r r 的線 性組合,其中 N 是總原子數, 1 N 是歸一化常數,其意義為我們利用很多 個相同的單位晶胞去表達整個空間,兩兩不同晶胞之間距離為Rj r ,分布在 第 j 個單位晶胞內 Wannier 函數φ(
r −Rj)
r r (用來表達電子在單一晶胞中的行 為),再做線性疊加,即總波函數ψkr( )rr 。 要表達電子在單位晶胞內只有單原子中的行為,Wannier 函數可以利用原子軌域的線性疊加 LCAO(Linear Combination of Atomic Orbital),如(2.2.2)
( )
r Cα α( )r α φ r =∑
ϕ v (2.2.2) 代入(2.1.4),重新整理可得: , ( ) ( ) k r Cα αk r α ψr r =∑
Φ r v (2.2.3) , 1 1 ( ) j ( ) N ik R j k j r e r R N α α ϕ ⋅ = Φ r v ≡∑
r r v− r (2.2.4)將(2.2.3)、(2.2.4)代入(2.1.4),並乘上 * ,k( )r α′ Φ r v 取積分,可得: ( ) * , , 1 ( ) ( ) ( ) ( ) j j N N ik R R j j k k j j C e r R H r R dr C E r r dr N α α α α α α α α ϕ ϕ ′ ⋅ − ∗ ′ ′ ′ ′ − − = Φ Φ
∑
∑∑
r r r∫
v r v r r∑
∫
r v r v r (2.2.5) 處理右手邊的積分時,假設不同原子之間波函數不重疊,並利用不同軌域 之間互相正交(2.2.6)。( ) ( )
* jα r jα r dr j j α α ϕ′ ′ ϕ =δ δ′ ′∫
r r r (2.2.6) 處理左手邊積分時,考慮任意一顆原子週遭環境所產生的影響皆相同,則 可以消去一個總和(summation),便可以得到下式(2.2.7): ( ) * ( ) ( ) j j ik R R j j j Cα e α r R H α r R dr C Eα αα α α ϕ ϕ δ ′ ⋅ − ′ ′ ′ ′ − − =∑ ∑
r r r∫
v r v r r∑
(2.2.7) 經過一些簡單的計算與代換,可得( )
C Hα αα k ECα α ′ ′ =∑
r (2.2.8)( )
* ( ) ( ) i i ik d i d Hαα′ k ≡∑
e ⋅∫
ϕα′ r H′ ϕα r′−d dr′ r r r r v v r v (2.2.9) 其中,di r 為其他原子與參考原子的距離。當我們將一個一個原子慢慢組合成 晶體,並考慮原子間的交互作用時,可以將參考原子週遭的原子依距離分 成第一近鄰(Nearest Neighbor)、第二近鄰(2NN)、第三近鄰(3NN)…等,考 慮愈多,精確度愈高,計算便會更複雜。 將(2.2.8)用矩陣表示,如(2.2.11) x x s s x p p x x x x x C C s H s s H p s H C C p H s p H p p H E H s H p H Cα Cα α α α α α α ⎡ ⎤ ⎡ ⎤ ⎡ ⎤ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ = ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎣ ⎦ ⎣ ⎦ ⎣ ⎦ L L M M O M M M L (2.2.11) 這是單位晶胞中具有一個原子的矩陣。讓我們討論更一般的狀況,如果一個單位晶胞中有 n 個原子,則該單 位晶胞的電子波函數應與每個原子有關,其 Wannier 函數應修正為[13,14]:
( )
ik n ( ) n n n n r C eα τ α r α φ r =∑
r r⋅ ϕ v−τr (2.2.12) 圖 2.2.1、單位晶胞有 n 個原子示意圖 (2.2.12)中ϕnα為第 n 顆原子的α 軌域,τn r 為相對於晶格向量 j Rr 的位置向量, 並且乘上相對於Rj r 的相位。再將 Wannier 函數代入滿足 Bloch 函數的波函 數,可得: , ( ) n ( ) k n k n r Cα α r α ψr r =∑
Φ r v (2.2.13) , 1 1 ( ) jn ( ) N ik R n jn jn j n n k j r e r R R R N α α ϕ τ ⋅ = Φ r v ≡∑
r r v− r r = r +r (2.2.14) 將(2.2.13)、(2.2.14)代入 (2.1.4),並乘上 * ,k( )r α′ Φ r v 取積分,可得: , ( ) n , ( ) , ( ) n , ( ) n k n k n k n k n n r H Cα r dr r E Cα r dr α α α α α α ∗ ∗ ′ ′ ′ ′ Φ∑
Φ = Φ∑
Φ∫
r v r v r∫
r v r v r (2.2.14) 等式右邊,假設兩兩不同原子之間的波函數重疊的範圍很小,趨近於零, 且不同軌域之間互相正交(2.2.15):( ) ( )
* jα r jα r dr j j α α ϕ ′ ′ ϕ =δ δ′ ′∫
r r r (2.2.15) 所以可得( ) * 1 ( ) ( ) j n j n N N ik R R n n j n n j n n nn n j j n C e r R H r R dr C E N τ τ α α α α αα α α ϕ τ ϕ τ δ δ ′ ′ ⋅ − − + ′ ′ ′ ′ ′ ′ ′ − − − − =
∑
∑∑
r r r r r∫
v r r v r r∑
(2.2.16) 利用r′ ≡ −r Rj′−τn′ r r v v 與 ; ( ) ( ) i jn j n j n j n dr ≡ ΔRr ′ ′ = Rr −τr − Rr ′−τr′ ,此時所有相對位置關係已 轉成原子之間的近鄰關係,則可得下式 ; n n n C Hα α α EC α α ′ ′ ′ ′ =∑
(2.2.17)( )
* ; ( ) ( ) i i ik d n n n n i d H α α′ ′ k ≡∑
er r⋅∫
ϕ′ ′α r H′ ϕα r′−d dr′ r r v v r v (2.2.18) (2.2.16)經過整理,可得到 Hamiltonian 的通式:( )
(
*)
; * 0 ( ) ( ) ( ) ( ) i i n n n n n nn ik d n n i d H k r U r dr e r U r d dr α α α α α αα α α ε ϕ ϕ δ δ ϕ ϕ ′ ′ ′ ′ ′ ′ ⋅ ′ ′ ≠ ′ ′ ′ ≡ + Δ ′ ′ ′ + Δ −∫
∑
r r∫
r r v v v r v v v (2.2.19) 若表達成矩陣形式,如下: ( ) ( ) ( ) ( ) ( )( ) ( )( ) ( ) ( ) ( ) ( ) ( )( ) ( )( ) 1, 1, 1, 1, 1, 2, 1, 2, 1, 1, 1, 1, 1, 2, 1, 2, 2, 1, 2, 1, 2, 2, 2, 2, 2, 1, 2, 1, 2, 2, 2, 2, x x x x x x x x x x x x x x x x s H k s s H k p s H k s s H k p C p H k s p H k p p H k s p H k p s H k s s H k p s H k s s H k p p H k s p H k p p H k s p H k p ⎡ ⎤ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎣ ⎦ L L L L L M M O M M O L L L L L M M O M M O M M O ( ) ( ) ( ) ( ) ( ) ( ) ( ) ( ) x x x x s s p p s s p p k C k C k C k C k E C k C k C k ⎡ ⎤ ⎡ ⎤ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ = ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎣ ⎦ ⎣ ⎦ M M M M M M (2.2.20) 此矩陣大小為Nα×Nα ,由原子個數與軌域個數決定。若依照不同原子將此 矩陣元素分類,則此複雜的矩陣可看成下式: 11 12 1 1 21 22 2 21
2
1
2
atom
H
H
C
C
H
H
C
E C
⎡
⎤ ⎡ ⎤
⎡ ⎤
⎢
⎥ ⎢ ⎥
⎢ ⎥
=
⎢
⎥ ⎢ ⎥
⎢ ⎥
⎢
⎥ ⎢ ⎥
⎢ ⎥
⎣
⎦ ⎣ ⎦
⎣ ⎦
L
L
L
M
M
M
O
M
M
(2.2.21) 其中H11與H22代表 on-site 能量,非對角線H12與H21為不同原子之間的hopping 項,這矩陣表示了每個原子之間的關係與其交互作用,但在複雜的 Hamiltonian 中並不了解如何將式子填入矩陣中。若今有兩種一維的原子結 構如圖(2.2.2) 圖 2.2.2、上序列為一維相同原子的排列,下序列為一維相異原子的排列, NN(Nearest Neighbor)為近鄰之意,Vm
( )
k r 為第 m 近鄰的交互作用項。 若考慮相同原子(單位晶胞中有一顆原子),將 Hamiltonian 依照原子間距 區分為單原子能量、第一近鄰(Nearest Neighbor)d1 v 、第二近鄰(2NN) 2 dv 、第 三近鄰(3NN)d3 v 等等,所以(2.2.19)可以寫成(2.2.22)式( )
1( ) ( )
2H k
r
=
E
α⋅
δ
αα′+
V k
r
+
V k
r
+
L
(2.2.22)( )
m V kr 如(2.2.23)列舉兩項如下( )
( )
( )
( )
( )
( )
1 1 2 2 * 3 1 , 1 , 1 1 * 3 2 , 2 , 2 2,
( )
(
)
,
( )
(
)
ik d d ik d dV k
t
d
e
t
d
r
U
r
d
d r
V k
t
d
e
t
d
r
U
r
d
d r
α α α α α α α α α α α αϕ
ϕ
ϕ
ϕ
⋅ ′ ′ ′ ⋅ ′ ′ ′≡
≡
Δ
−
⋅
≡
≡
Δ
−
⋅
∑
∫
∑
∫
r v v r v vr
r
r
v
v
v
r
r
r
v
v
v
(2.2.23) 其中tα α, ′( )
dm r 為實驗或使用第一原理所決定的參數若單位晶胞中有兩顆原子,2x2 的 Hamiltonian 則如下圖(2.2.3)所示 圖 2.2.3、增加近鄰數對矩陣的影響 屆此已可以將任意結構的半導體利用週期性條件將 Hamiltonian 矩陣化,只 要知道原子間交互作用的參數、晶體結構,便能決定每一個矩陣元素的數 值,在進行對角化,便可以得到相對應的特徵值與特徵向量,稍後在第三 章,我們將會針對單位晶胞有兩顆原子的單層石墨(graphene)當為示範。
( )
( ) ( )
11 2( )
1( ) ( )
3( )
1 3 22 2 E V k V k V k H k V k V k E V k ⎡ + + ⎤ ⎢ ⎥ = ⎢ + + ⎥ ⎣ ⎦ r r r r r r r( )
11( )
2( )
1( )
( )
1 22 2 E V k V k H k V k E V k ⎡ + ⎤ ⎢ ⎥ = ⎢ + ⎥ ⎣ ⎦ r r r r r( )
( )
11 1( )
1 22 E V k H k V k E ⎡ ⎤ ⎢ ⎥ = ⎢ ⎥ ⎣ ⎦ r r r 只考慮最近鄰 考慮更多近鄰 2N 3N 單位晶胞有兩顆原子2.3 Real space Hamiltonian 如果只計算一部份材料,在任意方向上都不具有週期性位能時,波函數 便與 K 無關,Bloch 理論便不適用,所以此時薛丁格方程為:
( )
( )
Hψ rr =Eψ rr (2.3.1) 然而總波函數可以由每一個單電子單原子波函數組成,每一個單電子單原子可以由原子軌域的線性疊加 LCAO(Linear Combination of Atomic Orbital)
所組成,因此其展開形式便假設為:
( )
( )
1 N j j j r Cα α r α ψ ϕ = =∑∑
r r (2.3.2) 將(2.3.2)代入(2.3.1)並乘上 *( )
jα r ϕ ′ ′ r 取積分,可得( )
( )
( )
( )
* * 1 1 N N j j j j j j j j Cα α r H α r dr Cα α r E α r dr α α ϕ′ ′ ϕ ϕ ′ ′ ϕ = = =∑∑
∫
r r r∑∑
∫
r r r (2.3.3) 假設兩兩不同原子之間的波函數重疊的範圍很小,趨近於零,且不同軌域 之間互相正交:( ) ( )
* jα r jα r dr j j α α ϕ′ ′ ϕ =δ δ′ ′∫
r r r (2.3.4) (2.3.3)考慮(2.3.4)後,再將 H 拆開,可得:( )
( )
* 1 ; 1 N j j j j j j j N j j j j j C r U r dr EC C H EC α α α α α α α α α α α α α ε δ δ′ ′ ϕ′ ′ ϕ ′ ′ = ′ ′ ′ ′ = ⎡ + Δ ⎤= ⎣ ⎦ ⇒ =∑∑
∫
∑∑
r r r (2.3.5)( )
( )
* ; j j j j j j H α α′ ′ ε δ δα ′ α α′ ϕ ′ ′α r Uϕα r dr ⇒ = +∫
r Δ r r (2.3.6) 將(2.3.6)用矩陣表達1, 1, 1, 1, 1, 2, 1, 2, 1, 1, 1, 1, 1, 2, 1, 2, 2, 1, 2, 1, 2, 2, 2, 2, 2, 1, 2, 1, 2, 2, 2, 2, x x x x s x x x x x x p x x s x x x x x x p s H s s H p s H s s H p C p H s p H p p H s p H p C s H s s H p s H s s H p C p H s p H p p H s p H p C ⎡ ⎤ ⎢ ⎥ ⎡ ⎢ ⎥ ⎢ ⎢ ⎥ ⎢ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎣ ⎢ ⎥ ⎢ ⎥ ⎣ ⎦ L L L L L M M O M M O M L L L L L M M O M M O M M O M M x x s p s p C C C E C ⎤ ⎡ ⎤ ⎥ ⎢ ⎥ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ = ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥⎦ ⎢⎣ ⎥⎦ M M M (2.3.8) 至此已經介紹了在 Tight-Binding 法中,如何將複雜的 Hamiltonian 轉為單原 子 Hamiltonian 與晶格微擾項,並將其展開為矩陣進行運算,接下來利用 Graphene 來計算。
第三章、單層石墨、單層石墨奈米緞帶
本章節將會展示對基本的單層石墨以及單層石墨奈米緞帶 (graphene nanoribbon) 的計算結果。接下來就介紹一下單層石墨的基本特性。 3.1 Graphene 單一石墨層是由碳以六角形晶格(Hexagonal lattice)所組成[11],如圖 3.1.1 所示,其晶格向量為:( )
(
)
1 3, 3 , 2 3, 3 2 2 a a ar = ar = − (3.1.1) a為兩碳原子間距 1.42Α o ,三個同心圓由內到外為第一近鄰、第二近鄰、第 三近鄰,所以單一石墨層的單位晶胞中有兩顆原子。且經由計算,倒晶格空間(Reciprocal Space)的倒晶格向量(Reciprocal lattice vector)為
( )
(
)
1 2 2 2 1, 3 , 1, 3 3 3 b b a a π π = = − r r (3.1.2) 圖 3.1.1、graphene 結構 圖 3.1.2、graphene k 空間結構 在 k 空間中, 有重要物理意義的點在 K 點上,也就是布里淵區的六角形頂點上,這被稱為 Dirac Point,其 k 空間座標為: 2 2 , 3 3 3 K a a π π ⎛ ⎞ = ⎜ ⎟ ⎝ ⎠ (3.1.3) 第一近鄰的三個向量分別是(依據不同材料有不同向量)
( )
(
)
(
)
1 1, 3 , 2 1, 3 , 3 1, 0 2 2 a a dr = dr = − dr =a − (3.1.4) 所以便能開始使用第二章 Tight-Binding 法所推導出的 Hamiltonian 公式 (2.2.20)如下( )
(
*)
* ; 0 ( ) ( ) i ( ) ( ) i ik d n n n n n nn n n i d H α α′ ′ k ε α ϕ ′ ′α r Uϕα r dr δ δ′ αα′ e ⋅ ϕ ′ ′α r Uϕα r d dr ≠ ′ ′ ′ ′ ′ ′ ≡ +∫
Δ +∑
r r∫
Δ − r r v v v v v r v 相對原子關係di r 為(3.1.4)式的三個向量。 其中所使用到的條件有 1. 每個單位晶胞有兩顆原子2. 單軌域近似(One orbital approximation (Pz))
3. 只考慮第一近鄰(Nearest Neighbor) 產生矩陣大小為 2x2 的矩陣如下 1 1 2 2 1, 1, 1, 2, 2, 1, 2, 2, z z z z p p z z z z z z z z p p C C p H p p H p E p H p p H p C C ⎡ ⎤ ⎡ ⎤ ⎡ ⎤ = ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎣ ⎦ ⎣ ⎦ ⎣ ⎦ (3.1.5) 由於 graphene 本身是Pz主導,可以只取Pz軌域,矩陣元素的參數由[17]可得 * 1,p Hz 1,pz = 2,p Hz 2,pz =εα +
∫
ϕnα( )rv′ ΔUϕnα( )r drv v′ ′≡Ep =0 (3.1.6) 並因有效鍵結為Pz所產生的π鍵,因此針對非對角線上的元素,定義 * 2pz( )r U 1pz(r d dri) t 3.033eV ϕ ′ Δ ϕ ′− ′≡ = −∫
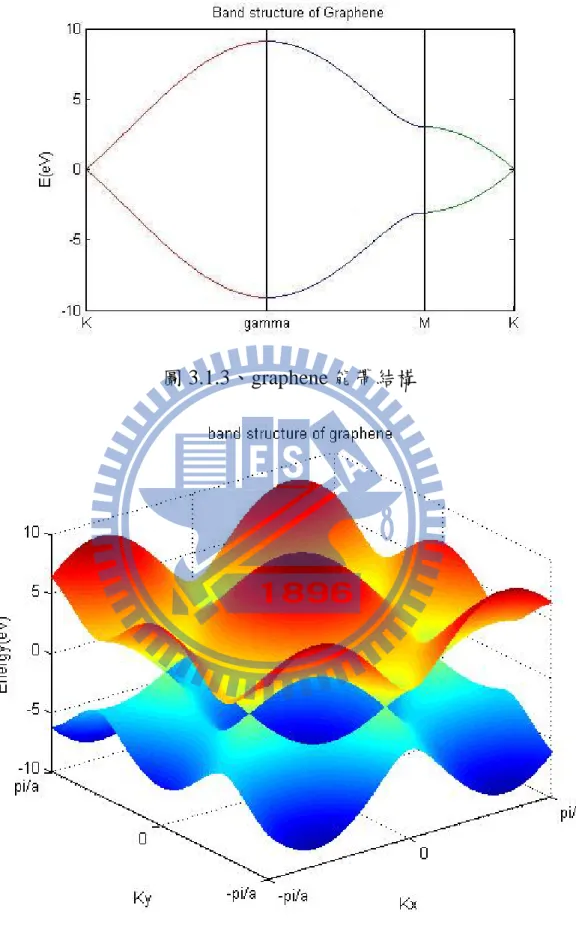
v v r v (3.1.7) 並考慮以二號原子為中心,與周圍有三顆一號原子有鍵結,所以3 1 2 2 3 1, 2, 2 cos 2 x x a ik ik d ik a ik d ik d z z y p H p =t e⎣⎡ ⋅ +e ⋅ +e ⋅ ⎤⎦=t e⎢⎡ − + e ⎜⎜⎛k a⎟⎟⎞⎥⎤ ⎢ ⎝ ⎠⎥ ⎣ ⎦ r r r r r r (3.1.8) Hamiltonian 便為 2 2 3 2 cos 2 3 2 cos 2 x x x x a ik ik a p y a ik ik a y p E t e e k a H t e e k a E − − ⎡ ⎡ ⎛ ⎞⎤⎤ + ⎢ ⎢ ⎜⎜ ⎟⎟⎥⎥ ⎢ ⎢⎣ ⎝ ⎠⎥⎦⎥ = ⎢ ⎥ ⎡ ⎛ ⎞⎤ ⎢ + ⎥ ⎢ ⎜ ⎟⎥ ⎢ ⎜ ⎟ ⎥ ⎢ ⎝ ⎠⎥ ⎣ ⎦ ⎣ ⎦ (3.1.9) 此結果可從[16]中驗證,在此使用到的 t=-3.033eV、Ep=0 參數參考[17]。 所計算出來的能量為 2 3 3 3
1 4 cos cos 4 cos
2 2 2
p y x x
E± =E ±t + ⎛⎜k a⎟⎞ ⎛⎜⎜k a⎟⎟⎞+ ⎛⎜⎜k a⎞⎟⎟
圖 3.1.3、graphene 能帶結構
3.2 graphene nanoribbon
計算完 graphene 之後,若是帶狀結構的 graphene nanoribbon,也就是一
個方向無窮延伸,另一個方向有限,所以在 x 方向上具有週期性,y 方向上
不具有週期性,所以 Tight-Binding 需要修正 k 只存在 kx 方向。即
(
x, 0)
kr= k (3.2.1)
因此若考慮傳輸方向為 zigzag,如下圖之結構
圖 3.2.1、graphene nanoribbon 結構,傳輸方向為 zigzag
在單位晶胞中原子數有八個原子,個別定為一號到八號,所以為 8*8 大小 的矩陣,並且每個原子只考慮 Pz 軌域,而每個原子最近鄰的向量為
( )
(
)
(
)
1 0,1 , 2 3, 1 , 3 3, 1 2 2 a a dr =a dr = − − dr = − (3.2.2) 考慮各個原子之間的鍵結關係之後,並依據(2.2.20),矩陣元素如下 * , z , z n ( ) n ( ) p 0 1, 2,...,8 i p H i p =εα +∫
ϕα rv′ ΔUϕα r drv v′ ′≡E = i= (3.2.3)3 2 ; 1 † * ; 0 1, 3, 5, 7 , 1, 1, , ( ) ( ) 2, 4, 6 jn j n t ik d ik d ik R z z z z n n jn j n ik d j te te i i p H i p i p H i p e r U r R dr te i α α ϕ ϕ ′ ′ ⋅ ⋅ ⋅Δ ′ ′ ′ ′ − ⋅ ′≠ ⎧ + = ⎪ ′ ′ ′ + = + = Δ − Δ = ⎨ = ⎪⎩
∑
∫
r r r r r r r r 64444447444444r 8 v v v (3.2.4) 3 2 3 2 1 3 1 2 3 2 1 3 1 2 3 2 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 ik d ik d p ik d ik d ik d p ik d ik d ik d p ik d ik d ik d p ik d ik d ik d p ik d ik d p E te te te te E te te E te te te te E te te E te te te te E ⋅ ⋅ − ⋅ − ⋅ − ⋅ ⋅ ⋅ ⋅ − ⋅ − ⋅ − ⋅ ⋅ ⋅ ⋅ − ⋅ − ⋅ + + + + + + r r r r r r r r r r r r r r r r r r r r r r r r r r r r r r r r 1 3 1 2 3 2 0 0 0 0 0 0 0 0 0 0 0 0 ik d ik d ik d ik d p ik d ik d p te te E te te te te E − ⋅ ⋅ ⋅ ⋅ − ⋅ − ⋅ ⎡ ⎤ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ + ⎥ ⎢ ⎥ + ⎢ ⎥ ⎣ ⎦ r r r r r r r r r r r r (3.2.5) 因為 ribbon 本身只有 x 方向上具有週期性,依據(3.2.1)(
)
(
)
(
)
(
)
(
)
(
)
(
)
(
)
2 cos / 2 0 0 0 0 0 0 2 cos / 2 0 0 0 0 0 0 2 cos / 2 0 0 0 0 0 0 2 cos / 2 0 0 0 0 0 0 2 cos / 2 0 0 0 0 0 0 2 cos / 2 0 0 0 0 0 0 2 cos / 2 p x x p p x x p p x x p p x E t k a t k a E t t E t k a t k a E t t E t k a t k a E t t E t k a ⎡ ⎤ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ (3.2.6)其中參數部分也是使用[17],經由計算,可以得到下圖結果,
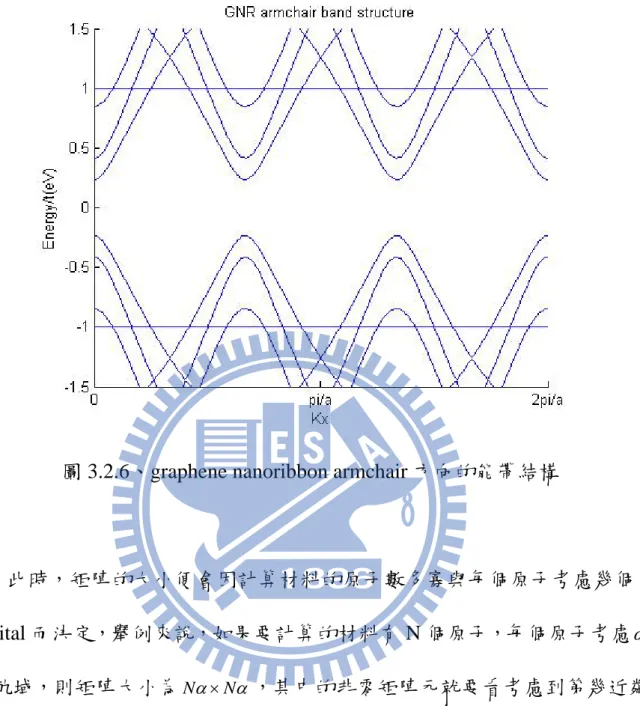
圖 3.2.2、graphene nanoribbon zigzag 方向的能帶結構,為靠近零的八個能量。
圖 3.2.3、graphene nanoribbon zigzag 方向的能帶結構,各個原子在 Kx=pi/a
圖 3.2.4、graphene nanoribbon zigzag 方向的能帶結構,各個原子在 Kx=0 的
若考慮傳輸方向為 armchair,如下圖之結構
圖 3.2.5、graphene nanoribbon 結構,傳輸方向為 armchair
在單位晶胞中原子數有十四個原子,個別定為一號到十四號,一樣只考慮 Pz 軌域,鄰近向量考慮如下
( )
(
)
(
)
1 1, 0 , 2 1, 3 , 3 1, 3 2 2 a a dr =a dr = − − dr = − (3.2.7) 所以為一 14*14 大小的矩陣,考慮各個原子之間的鍵結關係之後,並依據 (2.2.19)與(2.2.20),矩陣元素如下 * , z , z n ( ) n ( ) p 0 1, 2,...,14 i p H i p =εα+∫
ϕα rv′ ΔUϕα r drv v′ ′≡E = i= (3.2.3) armchair 2 1 a 3 4 6 5 7 8 10 9 11 12 14 13 3 dr 1 dr 2 dr(3.2.8) 將矩陣進行對角化之後,可得到能帶圖。 1 2 3 1 3 2 1 3 1 2 3 1 2 2 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 ik d ik d p ik d ik d p ik d ik d ik d p ik d ik d ik d p ik d ik d ik d p ik d E te te te E te te E te te te te E te te E te te te te ⋅ ⋅ − ⋅ − ⋅ − ⋅ − ⋅ − ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ − ⋅ − r r r r r r r r r r r r r r r r r r r r r r r r r r r r 3 1 3 2 1 3 1 2 3 1 2 3 2 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 ik d ik d p ik d ik d ik d p ik d ik d ik d p ik d ik d ik d p ik d ik d ik d p i E te te E te te te te E te te E te te te te E te te − ⋅ ⋅ − ⋅ − ⋅ − ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ − ⋅ − ⋅ − ⋅ − r r r r r r r r r r r r r r r r r r r r r r r r r r r r 3 2 1 3 1 2 3 1 2 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 ik d k d ik d p ik d ik d ik d p ik d ik d p ik d ik d p E te te te te E te te E te te te E − ⋅ ⋅ − ⋅ ⋅ ⋅ ⋅ ⋅ ⋅ − ⋅ − ⋅ ⎡ ⎤ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎣ ⎦ r r r r r r r r r r r r r r r r r r r r
圖 3.2.6、graphene nanoribbon armchair 方向的能帶結構 此時,矩陣的大小便會因計算材料的原子數多寡與每個原子考慮幾個 orbital 而決定,舉例來說,如果要計算的材料有 N 個原子,每個原子考慮α 個軌域,則矩陣大小為Nα×Nα ,其中的非零矩陣元就要看考慮到第幾近鄰 而決定,然而,目前電腦記憶體大小仍舊無法計算過大材料,因此,一個 好的計算方法便很重要,所以接下來要介紹一個處理大型矩陣對角化的演 算法。
第四章、數值方法
現今電腦科技蓬勃發展,記憶體容量急速上升,用於一般電腦綽綽有 餘,但適用於科學計算方面仍然稍嫌貧瘠,如何以有限的電腦資源解決無 窮的基底需求的問題,是數值計算中的一大課題。在這章節,我們將討論 如何化簡一個大型矩陣的方法。 大型矩陣的解決方法 一般而言,處理物理系統時常將算子(operator)使用基底轉為矩陣,利用 矩陣對角化求得能量 E,進而進行物理討論,但如果今天數值離散化的方法 如果不夠精確,往往勢必將增加基底,以增加其精確度。目前(2009)的電腦 記憶體雖然已經便宜又好用,對於物理系統而言,卻還是只能處理有限大 小的問題,因此,一個好的處理大型矩陣的方法便很重要。以下,將介紹 如何解決這樣的問題。 相似變換(similar transformation) 若矩陣 A 能夠找到一個么正(unitary)矩陣 U,使得 −1 A = UBU ,則可稱 A 相似於 B (A is similar to B , A ~ B),滿足相似變換的矩陣 A 與 B 有下列性 質。 1. A 與 B 具有相同特徵值 2. tr(A)=tr(B) 3. det(A)=det(B) 4. rank(A)=rank(B)因此,如果 A 的特徵值很不好解,若能使用相似變換將 A 轉換成 B, 而 B 是比較好求特徵值的,則為相同解 A 的特徵值。 其中解決大型矩陣知名的方法有二: 1. 若 A 是對稱(赫密特)矩陣 Lanczos 法: AU = UT T 為 tridiagonal 矩陣(對角線三條矩陣) 2. 若 A 是非對稱(非赫密特)矩陣 Arnoldi 法: T AU = UH + fe
H 為上海森堡矩陣(upper Heisenberg matrix),f 為殘餘向量
若 A 為對稱,則 Arnoldi 法會變成 Lanczos 法。 透過這兩個方法,可以將 A 矩陣轉換為 H 與 T,並且使用 QR 分解法 與高斯消去法各別去求 H 與 T 特徵值相較於 A 簡單的多。但為何能這樣子 做呢?以下介紹這兩個方法。 4.1 Lanczos Method 首先,考慮標準特徵値問題 μ = Ax x (4.1.1) A 為一個 N x N 的矩陣,μ為特徵值,X 為特徵向量,如果我們從任意向
量v1開始,在這 Lanczos 演算法中,我們會利用連續的向量,當做新的基底
{
2 1}
{
}
1, 1, 1,..., 1 1, 2, 3,..., p p − = v Av A v A v u u u u (4.1.2) 1 v 是 N 維的任意向量,並且vi的個數為 p 個。這一連串獨立的向量ui所定 義的子空間稱為 Krylov 子空間,特徵空間可以被這些向量當做基底所展 開,事實上,在 Lanczos 方法中,我們使用vi的線性組合,形成互相正交的 集合ui,並重新展開(或重新表示)解在該子空間,這個方法的目的就是去減 少 A 的矩陣元素,成為三條對角線的矩陣 J,而 J 對角化是非常簡單的。矩 陣 J 是被定義為 1, 2,..., p 1, 2,..., p ⎡ ⎤ ⎡ ⎤ ⋅⎣ ⎦ ⎣= ⎦⋅ A u u u u u u T (4.1.3) 其中向量ui是以行向量排成N×p的矩陣,稱之為 U,且 1 1 1 2 2 2 3 3 3 4 1 1 p p p a b b a b b a b b a b b a − − ⎡ ⎤ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ = ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎣ ⎦ T O O O (4.1.4) 矩陣其他部分為零,那麼ui要如何得到才可以使 H 轉成 T? 首先,由任意向量v1歸一化後當作第一個起始向量u1 我們將 A 乘上u1為第一新向量,由於向量正交性,所以新向量必須與u1正 交,即 1 = 1−a1 1=( −a1) 1 w Hu u H u (4.1.5) 其中w2為暫時向量,而1 1 1 a = u Hu+ (4.1.6) 1 a 也就是Hu1與u1的內積。由於所得新向量尚未歸一,所以歸一化常數為 2 1 1 1 b = w w+ (4.1.7) 所以第一個正交向量為 1 2 1 b =w u (4.1.8) 現在我們開始建構u3,讓我們定義一個暫時向量w2 2 = 2−a2 2−b2 1 =( −a2) 2−b2 1 w Hu u u H u u (4.1.9) 其中 2 2 2 a = u Hu+ (4.1.10) 2 b b1由前面計算可知為正交歸一係數,所以 2 2 2 2 b = w w+ (4.1.11) 所以第二個正交新向量為 2 3 2 b = w u (4.1.12) 相同方法可求得 3 = 3−a3 3−b3 2−c3 1 w Hu u u u (4.1.13) 注意,我們定義c 1 3 0 + =u Hu ≡ ,因此,此迭代程序便導致為一個三項關係式。 在迭代 N 次後,正交的新向量Hup只與兩個向量有關係,即up與up−1,得 1 1 p+ = p−ap p−bp p− w Hu u u (4.1.14) 且
2 1 1 1 , 1 1 1 p p p ap p p + + + = + + + = + + b w w u Hu (4.1.15) 我們使用此方法決定其他三條對角線(tridiagonal)矩陣 T 的其他元素。 所以 1, 2,..., p 1, 2,..., p ⎡ ⎤ ⎡ ⎤ ⋅⎣ ⎦ ⎣= ⎦⋅ ⇒ = ⇒ + = A u u u u u u T AU UT U AU T (4.1.16) 此為一個相似變換(similar transformation),其中一種特性為 A 與 T 有相同 特徵値,便可以利用 Lanczos 方法去找出ui使 A 能夠轉成 T,讓對角化簡單 化,這即為 Lanczos 方法的精神。 接下來,我們現在就可以將此演算法公式化: 1. 選擇開始向量u1,選擇是任意的,唯一條件是必須歸一化。 2. 計算a1:a1 1 1 + = u Au 3. 計算b1:
[
] [
]
2 1 ( 1 ) 1 ( 1 ) 1 b = A−aI u + A−aI u 4. 導出向量u2: 1 1 2 1 ( a ) b − = A I u u 5. 計算a2:a2 2 2 + = u Au 6. 計算b2:[
] [
]
2 2 ( 2 ) 2 1 1 ( 2 ) 2 1 1 b = A−a I u −bu + A−a I u −bu 7. 導出向量u3: 2 2 2 1 3 2 ( a ) b b − − = A I u u u 重新運算第 5~7 步驟,當 i 從 1 到 n。1 1 1 2 2 2 3 3 3 4 1 1 p p p a b b a b b a b b a b b a − − ⎡ ⎤ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ = ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎣ ⎦ T O O O 因此,得到(3.1.4)的 T 之後,很明顯的解 T 會比解 A 還要簡單,依照 Gauss 消去法,可以求解出特徵值為a a a a1, 2′ ′ ′, 3, 4,L,a′p,其中 2 1 1 , 2, , m m m m b a a m p a − − ′ = − = ′ L (4.1.17) 在經過 Lanczos 法化簡矩陣之後,矩陣元素個數從 2 n 成為3n,大大縮小了 記憶體使用量,化簡之後也比較好求近似的特徵值。 數值穩定性 針對 Lanczos 演算法,若經過精確計算,可以證明使用這一連串的向量式可 以形成正交基底,並且特徵值與特徵向量可以有相當好的近似原矩陣 A, 但事實上,正因為三對角線(Tridiagonal)矩陣 T 的關係,使正交性快速的減 低,T 的特徵值是相近於 A 的特徵值,並且在某些情況之下,有可能產生 的新基底是與舊基底線性相關(linear dependent)的,所以 T 的特徵值也有可 能並不接近 A 的特徵值,因此,Lanczos 演算法仍然不是很穩定。 Lanczos 所需改善的地方: 1.避免正交性的損失。 2.使現有基底的正交。 3.移除假的特徵值。
因此,基本的 Lanczos 的演算法中,做為大型矩陣計算方面,需要再做許多 修改,接下來介紹的 ARPACK,克服了某些 Lanczos 的問題。 4.2 ARPACK 簡介 ARPACK 是一個用來求解大規模特徵值問題的軟體。ARPACK 是由 Fortran77 所寫的副程式的集合,主要可以解決來自各應用領域的大規模對 稱、非對稱與廣義特徵值問題。可以求特定數量的特徵值,其中包含最大 或最小實部特徵值、最大或最小虛部特徵值、以及最大或最小振幅的特徵
值。ARPACK 軟體是基於隱式重開始 Arnoldi/Lanczos 方法(Implicitly
Restarted Arnoldi/Lanczos Method)。Arnoldi/Lanczos 是用來求解大規模 n*n
矩陣的幾個特徵值的重要方法,特別適合用於大規模稀疏矩陣的特徵值問 題。隱式重開始 Arnoldi 方法採用了隱式位移 QR 法與 k 階 Arnoldi 分解結 合的策略,再配合隱式重開始的方法,節省標準 Arnoldi 和 Lanczos 方法中 存在的數值困難和儲存困難(需大量儲存基底),紀錄矩陣的方式是採用稀疏 矩陣,也就是只紀錄矩陣不為零的位置與大小,進而達到處理大規模特徵 值問題的需求。所以 ARPACK 本身是適合使用在矩陣元素中,為零元素很 多的矩陣,例如解決固態物理中所使用到的 Tight-Binding Method、解決微
分方程的有限差分法(Finite Difference Method),都是矩陣中具有相當多為零
主要功能包括:
1. 求解單雙精度實對稱矩陣特徵值問題
2. 求解雙精度複數矩陣特徵值問題(Hermitian & non-Hermitian)
3. 廣義特徵值問題
4. 奇異值分解(SVD)問題
最新軟體版本可上 http://www.caam.rice.edu/software/ARPACK/ 自行下載使
Implicit Restart Arnoldi Method (IRAM)
圖 4.2.1、IRAM 流程圖
Arnoldi Factorization
在介紹 IRAM 之前,首先要瞭解 k 階 Arnoldi Factorization。先考慮一個
一般特徵值問題,定義:如果待求矩陣 ∈ n n× C A ,且滿足 T k = k k+ k k AU U H f e (4.2.1) 其中 n k k C × ∈ U 為正交歸一的行向量,稱為 Arnoldi 向量,且U fHk k =0, × ∈ k k k C
H 是上海森堡矩陣(upper Heisenberg matrix),為下對角線以上皆有值
的矩陣,eTk =
(
L 0 1 0 L)
在第 k 個位置才有 1 其它為零,此關係為 A 的k 階 Arnoldi Factorization,為 Krylov 子空間的一種應用。若 A 是 Hermitian,
為 A 的 k 階 Lanczos 分解,所對應的行向量Uk稱為 Lanczos 向量。理論上 來說,解決上海森堡矩陣會比直接解原始矩陣來的有效率。 使用 Arnoldi 分解時,需要儲存大量的 Krylov 子空間的基底,且理論上 需要與 A 行向量個數一樣多的基底才會收斂,但電腦模擬計算是使用有限 記憶體,如果計算矩陣很大,往往在收斂以前就已達記憶體上限,所以必 須利用重開始 Arnoldi 分解,減少記憶基底的數目,經由每次疊代改變起始 向量,改變相對產生的 Krylov 子空間,直到 Krylov 子空間將所有有興趣的 特徵值與特徵空間包起來,進而達到收斂的結果。至於如何改變起始向量 呢? Filter polynomials 假設在n n× 矩陣 A 中有特徵值μi與特徵向量xi,而有興趣的是前 k 個特 徵值與特徵向量,並且基本假設為使用特徵向量展開起始向量v1如下列形 式: 1 1 1 k m i i i i i i k C C = = + =
∑
+∑
v x x (4.2.2) 若 f 是任意的多項式,乘上起始向量,則可有: 1 1 1 ( ) ( ) ( ) k m i i i i i i i i k f C f μ C f μ = = + =∑
+∑
A v x x (4.2.3) 選擇 f 使 f(μi) (i= +k 1,..., )n 都比 f(μi) (i=1,..., )k 還要小,則乘上v1後會使想要 的特徵向量的係數變大,不想要的特徵向量係數變小,則稱 f 為 Filter polynomials,如果在 A 的特徵值μ μ1... m中,μk+1...μm是 p 個不感興趣的,若 f與 A 有關,則 f 為 A 的特徵多項式
(
1) (
)
( )= −μk+ L −μm f A A I A I (4.2.4) 利用此 Filter 多項式,碰到後 p 個不感興趣的向量時,此多項式會變成 零,即可將起始向量中,不感興趣的特徵向量成分刪掉,使起始向量展延 的 Krylov 子空間為我們想要的特徵空間,但由於此多項式為一矩陣向量乘 積,直接處理將此 Filter 多項式乘上向量,稱為顯式重開始(Explicit Restarting)。若矩陣很大時,顯式重開始勢必會花費許多的時間,因此,隱 式重開始便因應而生。Implicit Restart Arnoldi Method
針對顯式重開始的不方便,因此發展了隱式重開始,先將 Arnoldi 分解 從 k 階變為 m=k+p 階(前 k 個有興趣,後 p 個沒興趣),再進行隱式位移 QR 法,達到分類的效果,使矩陣能夠只解出想要的特徵值。將(3.2.1)右邊乘上 Q 後可以修改如下: T m m m m m + = + + + AU U H f e Q (4.2.5) 其中 m+ = m U U Q,H = Q H Qm+ H m ,Q = Q Q1, 2,L,Qp,Qj為矩陣Hm−μjI的 QR 分解,此時利用 p 次隱式位移 QR 分解法取所求得 m + U 的第一行向量如下
(
)
(
)(
)
(
)(
)(
)
(
)
(
)
(1) (1) 1 1 1 1 (2) (1) (2) 2 1 2 1 1 (3) (2) (3) 3 1 3 2 1 1 ( ) ( 1) ( ) 1 1 1 m m m m m m p p p m m p p μ μ μ μ μ μ μ μ + + + + + + + − = → = − = → = − − = → = − − − = → = − − U U Q u A I v U U Q u A I A I v U U Q u A I A I A I v U U Q u A I A I v M L (4.2.6)由此可以看出,藉由隱式位移 QR 分解,可以得到與顯式重開始相同效 果的基底,詳細推導內容請參見附錄。由於 T m e Q的前 k-1 項為零,所以此 m 階的前 k 項可以等價改寫成 k 階 Arnoldi 分解,或等同只取前 k 項,本質上 是一樣的: T k k k k k + = + ++ + AU U H f e (4.2.7) 其中,殘餘向量 k k k 1βˆk mσ + + + = + f U e f ,此時的 k 階 Arnoldi 分解,可以包含 此 m 階 Arnoldi 分解中,有興趣的特徵值所展延的特徵空間,並且起始向量 也將不想要的特徵值過濾掉,進而再增加 p 階使 Arnoldi process 產生下一個 新的 m 階 Arnoldi 分解,再進行隱式位移 QR 法,直到收斂為止。利用所得 到的該 Krylov 子空間的基底向量與顯式重開始v1所產生的空間相同的特 性,便可以大大減少產生 Krylov 子空間所需要的計算量。此為 ARPACK 重 要的精神。 根據上述方法,可編成下列演算法:
(
)
( )
( )
[
]
1 2 : , , , , - ; 1, 2, 3,... 1. , ,... ; 2. 1, 2,..., , T m m m m m m p m Input withan m step Arnoldi Factorization For l until convergence
Computer and select set of p shifts based upon or perhaps other information
For j p Factor σ μ μ μ σ = + = = = A U H f AU U H f e H H Q R
(
)
(
)
(
)
1 ; ; ; ; _ ˆ 3. ; 4. 1: ,1: ; 1: ,1: ; 5. -m j H H m m m m m k k k m k k m k m T k k k k k qr End For n k k kBeginning with the k step Arnoldi factorization
apply p additional step of the Arnoldi process to obtain a new m step
μ β σ + − ← ← ← ← + ← ← = + H I H Q H Q U U Q q q Q f v f U U H H AU U H f e _ T k k k k k Arnoldi factorization End For = + AU U H f e 圖 4.1.2、演算法 與 Lanczos 法的差別: 1. 起始向量經由疊代修正到最適合起始向量 2. 重開始以增加其基底正交性 3. 修改起始向量,求出想要的特徵值,避免假的特徵值出現
特徵空間、Krylov 子空間與稀疏矩陣紀錄方法
眾所皆知的,一個向量可以展延(span)成一維空間,二個獨立向量可以
展延(span)成二維空間,以此類推,當有 N 個獨立向量時,便可以展延成無
限維空間,在物理上稱為 Hilbert 空間(Hilbert space)。
特徵空間是用來描述一個由 A 的特徵值相對應特徵向量所形成的空
間,稱為特徵空間。若要用另一個方法去解決此特徵問題,則必須 Krylov
子空間維度能等於 A 的特徵空間,才可在 Krylov 子空間中找到 A 的所有特
徵值。
在先前介紹的 Arnoldi 法與 Lanczos 法所使用的空間稱為 Krylov 子空
間,其定義為
{
2}
1, 1, 1,... v K = v Av A v 其中v1為自行決定的起始向量,也就是當起始向量決定時,Krylov 子空 間便決定了。使用 Krylov 子空間最大的特性是,可以只紀錄矩陣中不為零 的部份,因為要產生此空間只需要矩陣乘上向量而產生的基底向量,而基 底向量中的元素皆是由 A 不為零的元素所產生,所以,只要使用 Krylov 子 空間,便可以使用稀疏矩陣(sparse matrix)的記錄方式,進而節省記憶體的 儲存空間,便能在有限的電腦資源做到更大的運算。4.2.2 Test Result 我們現在就拿簡單例子二維諧振子來測試使用結果,這是電子處在拋物 線位能下的薛丁格方程式 圖 3.2.1、位能與參數
( )
( )
2 2 2 2 2 1 2 2 d m x x E x m dx ω ψ ψ ⎡ ⎤ − + = ⎢ ⎥ ⎣ ⎦ h (4.2.1) 該例子的能量大家熟知為 1 2 E n ω = + h (4.2.2)我們是使用有限差分法(finite difference method)[15]將微分方程轉成矩陣,
再利用 ARPACK 去執行對角化所產生的結果,其正解應為 0.5、1.5、2.5...
圖 4.2.1、矩陣 10 萬*10 萬的特徵值
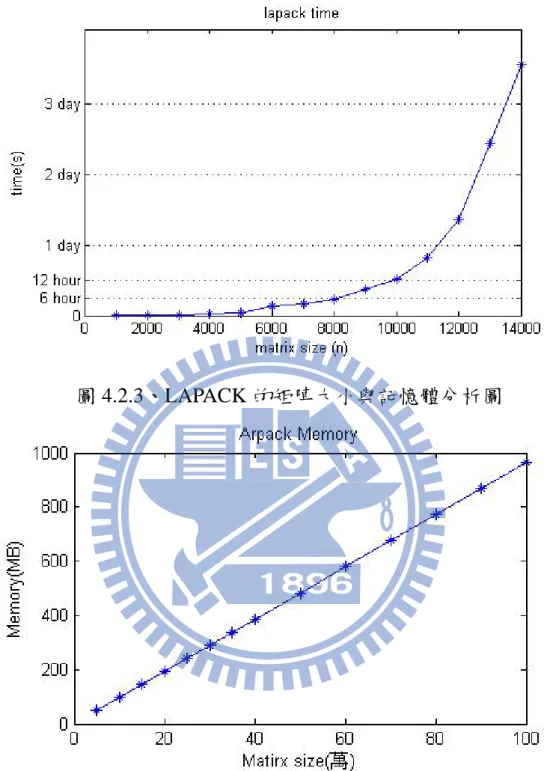
圖 4.2.3、LAPACK 的矩陣大小與記憶體分析圖
圖 4.2.5、LAPACK 的矩陣大小與記憶體分析圖 可以看出以 ARPACK 與 LAPACK 相比之下,記憶體大小大幅度的降低,計 算大小大幅度的增加。 ARPACK 的所有參數皆列在附錄 A 中,其中最主要需要更改的參數如 下所示: 參數 功能描述 n 矩陣大小 nev 求解特徵值的個數 ncv Arnoldi 分解長度,即基底數量;預設值=4*nev which 指定求解特徵值的特性(參考附錄 A) iparam(3) 允許疊代的最大次數;預設值=3*n 其中,主要控制運算時間長短的兩個參數為 ncv 與 iparam(3),增加基底
數 ncv 能降低運算時間,其主要是將待解矩陣分割成小矩陣的大小,ncv 越 大,所產生小矩陣的大小越大,疊代次數會較少,但是每次疊代後解小矩 陣的時間越多;反之,ncv 越小,所產生的小矩陣的大小越小,解小矩陣的 時間越少,但疊代次數會較多。舉例來說:若待解矩陣為 1000*1000 的大 小,ncv 假設是 200,則每次的小矩陣為 200*200,可能的疊代次數假設是 5 次,即解 5 次 200*200 大小的矩陣;若 ncv 假設是 10,則每次的小矩陣 為 10*10,可能的疊代次數假設是 100 次,即解 100 次 10*10 的矩陣。 疊代次數(iparam(3))只需要修改到滿足程式未判定疊代次數不夠即可。 圖 4.2.6、ARPACK 參數修改對時間的影響,前項參數為 ncv(basis)=4*nev, 後項參數為 iparam(3) =3*N
在經過一連串的測試之後,針對求一維簡諧振子最低的 15 個能量,測
試出基底數量取 12*nev (nev=15)是可以節省最多時間的,在此提供給大家
參考。但是一般來說,不同的矩陣對於參數的優化並沒有一個固定的公式,
端看不同矩陣的稀疏程度、特徵值的結構與個數的而決定,因此,針對任
第五章、總結與未來工作
總結: 由 ARPACK 的計算結果,可以了解到我們現在已經能計算超過十萬大 小的矩陣,遠遠超過 LAPACK 再目前能計算的一萬,並且節省大量時間, 然而其精確度也是非常高的,可以看到矩陣大小十萬的精確度已達 0.0002%,在計算更大的矩陣時,應會更準確。在投資非常多時間之後,我 們已有一個可信賴的大型對角化軟體,在未來,便能計算更多的材料,這 是一個非常有用的軟體。 在單層石墨方面,我們也已經能計算出一些基本的能帶結構與其物理的 結果,雖然目前物理方面的討論仍不足,但是在未來,相信是可以給更多 的物理分析結果,以了解單層石墨更多的物理特性。 未來工作: 在單層石墨方面,目前大小還沒有算的很大,所以接下來的工作是計算 非常龐大的單層石墨,以及計算 zigzag 與 armchair 方向的奈米帶狀單層石 墨不同邊界的結果,或是算很大的島狀結構的單層石墨,都是屬於我們未 來的課題,而且目前都是只有計算單電子模型,故未來還可以引入多電子 模型,並加入電子間交互作用項,甚至是加入鎳基板[18]等等。至於 ARPACK 方面,還可以擴充不同的計算結構,目前是能計算實數矩陣最大最小值,之後,可以慢慢增加計算複數矩陣,或是加入能平行處理矩陣的
參考文獻
[1]Novoselov, K.S. et al. "Electric Field Effect in Atomically Thin Carbon Films", Science, Vol 306 (5696), p. 666-669, 2004
[2] Novoselov, K.S. et al. "Two-dimensional atomic crystals", PNAS, Vol 102 (30), p. 10451-10453, January 26, 2005
[3] Mark Wilson, Phys. Today 59 January 21 (2006)
[4] A.K. Geim & K.S. Novoselov, Nature Materials 6, 183-191 (2007)
[5] J. R. Williams, L. C. DiCarlo, and C. M. Marcus, Science 317, 638 (2007) [6] D. A. Abanin and L. S. Levitov, Science 317, 641 (2007)。
[7]Barbaros Özyilmaz et al.,Phys. Rev. Lett. 99, 166804 (2007)
[8]Marek Korkusinski,”Application of the effective band-orbital model to semiconductor nanostructure”
[9]Gene Howard Golub, Charles F. Van Loan,”Matrix computations”(1996)
[10]R. B. Lehoucq, D. C. Sorensen, C. Yang,” ARPACK Users' Guide:Solution of Large Scale Eigenvalue Problems with Implicitly Restarted Arnoldi Methods”(1997)
[11]A. H. Castro Neto, F. Guinea, N. M. R. Peres, K. S. Novoselov and A. K. Geim, Rev. Mod. Phys. 81, 109 (2009)
[12] K. Nakada, M. Fujita, G. Dresselhaus, M. S. Dresselhaus, Phys. Rev. B 54, 17954 - 17961 (1996)
[13] Peter Y. Yu,Manuel Cardona,”Fundamentals of Semiconductors:Physics and Materials Properties”(springer-Verlag 1996)
[14] D. J. Chadi and M. L. Cohen Calculations of the Valence Bands phys. stat. sol. (b) 68, 405 (1975)
[15] L. V. Fausett, “Applied Numerical Analysis Using Matlab”, Prentice-Hall(1999) [16] R. Saito, M. Fujita, G. Dresselhaus, M.S. Dresselhaus , Phys. Rev. B 46, 1804 - 1811
(1992)
[17] R. Saito, G. Dresselhaus, M.S. Dresselhaus ,”Physical Properties of Carbon Nanotubes”,(Imperial College Press, Convent Garden, London 1998)
[18]A. Varykhalov, J. Sa´nchez-Barriga, A. M. Shikin, C. Biswas, E. Vescovo, A. Rybkin, D. Marchenko, and O. Rader, Phys. Rev. Lett 101 157601(2008)
附錄 A:隱式位移 QR 法
(
)
(
)
(
)
(
)
(
)
1 1 1 1 1 1 1 1 1 1 1 1 1 1 (1) (1) (1) (1) 1 (1) (1) (1) 1 1 1 1 1 1 ( ) Arnoldi , , = 0 0 * T m m m m m T m m m m m m m T m m m m T m m m m H m m m m m H T m m m m m μ μ μ μ μ μ + + = + → − = − + − = → − = + → − = + → = + ≡ + ≡ ≡ AU U H f e A I U U H I f e H I Q R H QR A I U U Q R f e A I U Q U Q R Q f e Q AU U H f b H R Q I U U Q b e Q Q L 對 進行位移 分解 回到 分解的形式 其中 (1) (1) (1) 1 1 (1) 1 1 1 (1) (1) 1 1 1 1 : * * 0 : ( det( ) 0) * * * * 0 * * * -0 0 * * m m m m m mone step of single shifted QR algorithm
proof easily
singular
non singular singular
μ μ μ − ⎡ ⎤ = ⎢ ⎥ ⎣ ⎦ − = = = H H H H I R Q H H Q R R Q 此時 表示不重要的數字 為 的特徵值,故左邊為 而 必為 ,則 必為 (1) 1 1 1 (1) (1) 1 1 1 1 1 1 0 0 0 0 * * * * * * * * 0 * * * 0 0 0 0 * * * * * * * * * 0 * * * 0 0 0 0 m m m μ μ μ μ − ⎡ ⎤ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎣ ⎦ ⎡ ⎤ ⎢ ⎥ ⎢ ⎥ ∴ − = = ⎢ ⎥ ⎢ ⎥ ⎣ ⎦ ⎡ ⎤ ⎢ ⎥ ⎡ ⎤ ⎢ ⎥ → = + = = ⎢ ⎥ ⎢ ⎥ ⎣ ⎦ ⎢ ⎥ ⎣ ⎦ H I R Q H H R Q I
![圖 2.1.2、晶格位能(不連續),Rj 代表 unit cell 所在的位置 將(2.1.4)代入薛丁格方程,可得: ( ) 0 ( ) ( ) ( ) k k kHψrrr=⎡ ⎣ H + Δ U r r ⎤⎦ ψ r r r = E ψ r r r (2.1.5) 2.2 k-space Hamiltonian 在此這樣的位能下,滿足(2.1.3)的波函數如下:[13,14] ( ) 1( )1 jNik Rk jjrerRψN⋅φ == ∑ r r −rrr r (2.2.1) 滿足晶格週](https://thumb-ap.123doks.com/thumbv2/9libinfo/8367030.177275/11.892.122.809.409.769/方程=⎡⎣H+ΔU⎤⎦ψr=ψ下滿⋅φ=.webp)