Short Communication
Chromosome 17p13.3 deletion syndrome: aCGH characterization, prenatal
findings and diagnosis, and literature review
Chih-Ping Chen a,b,c,d,e,f *, Tung-Yao Chang g, Wan-Yuo Guo h, Pei-Chen Wu g, Liang-Kai Wang a,
Schu-Rern Chern b, Peih-Shan Wu i, Jun-Wei Su a,j, Yu-Ting Chen b, Li-Feng Chen a and Wayseen
Wang b,k
a Department of Obstetrics and Gynecology, Mackay Memorial Hospital, Taipei, Taiwan b Department of Medical Research, Mackay Memorial Hospital, Taipei, Taiwan
c Department of Biotechnology, Asia University, Taichung, Taiwan
d School of Chinese Medicine, College of Chinese Medicine, China Medical University, Taichung, Taiwan e Institute of Clinical and Community Health Nursing, National Yang-Ming University, Taipei, Taiwan f Department of Obstetrics and Gynecology, School of Medicine, National Yang-Ming University, Taipei, Taiwan
g Taiji Fetal Medicine Center, Taipei, Taiwan
h Department of Radiology, Taipei Veterans General Hospital, Taipei, Taiwan i Gene Biodesign Co. Ltd, Taipei, Taiwan
j Department of Obstetrics and Gynecology, School of Medicine, College of Medicine, Taipei Medical University, Taipei, Taiwan
k Department of Obstetrics and Gynecology, China Medical University Hospital, Taichung, Taiwan l Department of Bioengineering, Tatung University, Taipei, Taiwan
* Correspondence to: Chih-Ping Chen, MD
Department of Obstetrics and Gynecology, Mackay Memorial Hospital 92, Section 2, Chung-Shan North Road, Taipei, Taiwan
Tel: +886-2-25433535; Fax: +886-2-25433642, +886-2-25232448 E-mail: [email protected]
Highlights
We present chromosome 17p13.3 deletion syndrome in a fetus. The fetus has lissencephaly and multiple ultrasound anomalies. We discuss the genotype-phenotype correlation.
We review the associated prenatal findings of 17p13.3 deletion.
Abstract
We report molecular cytogenetic characterization of 17p13.3 deletion syndrome by array comparative genomic hybridization (aCGH), fluorescence in situ hybridization (FISH) and quantitative polymerase chain reaction (qPCR) in a fetus with lissencephaly, corpus callosum dysgenesis, ventriculomegaly, microcephaly, intrauterine growth restriction (IUGR), polyhydramnios and single umbilical artery. aCGH analysis revealed a 3.17-Mb deletion at 17p13.3, or arr [hg19] 17p13.3 (0-3,165,530)1. The qPCR assays revealed a maternal origin of the deletion. Metaphase FISH analysis detected absence of the LIS1 probe signal on the aberrant chromosome 17. The karyotype was 46,XX,del(17)(p13.3). We review the literature of chromosome 17p13.3 deletion syndrome with prenatal findings and diagnosis, and suggest that prenatal ultrasound detection of central nervous system anomalies such as lissencephaly, corpus callosum dysgenesis/agenesis, ventriculomegaly and microcephaly associated with IUGR, polyhydramnios, congenital heart defects, abdominal wall defects and renal abnormalities should include a differential diagnosis of chromosome 17p13.3 deletion syndrome.
Keywords: chromosome 17p13.3 deletion; Miller-Dieker lissencephaly syndrome; MRI; prenatal
diagnosis; ultrasound
Abbreviations
MDLS: Miller-Dieker lissencephaly syndrome; OMIM: Online Mendelian Inheritance in Man; CNS: central nervous system; IUGR: intrauterine growth restriction; del: deletion;
MRI: magnetic resonance imaging; aCGH: array comparative genomic hybridization; FISH: fluorescence in situ hybridization; BAC: bacterial artificial chromosome; qPCR: quantitative polymerase chain reaction; STRs: short tandem repeats;
1. Introduction
Chromosome 17p13.3 deletion syndrome or Miller-Dieker lissencephaly syndrome (MDLS; OMIM 247200) is a contiguous gene deletion syndrome that is characterized by CNS anomalies of cerebral agyria/pachygyria or type I lissencephaly, ventriculomegaly, corpus callosum dysgenesis/agenesis and microcephaly, seizures, facial dysmorphism of prominent forehead and occiput, bitemporal narrowing, furrowed brow, small nose, anteverted nostrils, low-set ears, prominent lip and micrognathia, hypoplastic male external genitalia, IUGR, mental retardation and extracranial anomalies of cardiac defects, omphalocele and genitourinary abnormalities (Chen et al., 2010, 2011; Chitayat et al., 1997; Dieker et al., 1969; Dobyns et al., 1984, 1991; Dobyns and Das, 2009; Lin et al., 2009; Miller, 1963). Isolated lissencephaly is characterized by a smooth brain with agyria or pachygyria (Dobyns and Das, 2009). Subcortical band heterotopia is characterized by a subcortical band of heterotopic gray matter presenting beneath the cortex and being separated from the cortex by a thin white matter (Dobyns and Das, 2009).
MDLS is caused by cytogenetic visible deletion or microdeletion of 17p13.3 with haploinsufficiency of PAFAH1B1 or LIS1, whereas isolated lissencephaly and subcortical band heterotopia can be caused by heterozygous mutation in PAFAH1B1 (Dobyns et al., 1993; Neer et al., 1993; Reiner et al., 1993). With the advent of fetal ultrasound and MRI, it is possible to prenatally identify cranial and extracranial abnormalities associated with MDLS such as widespread agyria, abnormal sylvian fissure and insula, ventriculomegaly, corpus callosum dysgenesis/agenesis, microcephaly, IUGR, polyhydramnios, congenital heart defects, genitourinary anomalies, micrognathia and omphalocele (Fong et al., 2004; Ghai et al., 2006). Here, we present our experience of aCGH characterization of chromosome 17p13.3 deletion in a fetus with lissencephaly and multiple abnormalities detected by prenatal ultrasound.
2. Clinical description
A 40-year-old, gravida 2, para 1, woman presented with multiple sonographic abnormalities in the third trimester. Her husband was 43 years old. She and her husband were healthy, and there was no family history of congenital malformations. At 19 weeks of gestation, the woman underwent amniocentesis because of advanced maternal age, and the result revealed a karyotype of 46,XX. The pregnancy was uneventful until 31 weeks of gestation when polyhydramnios occurred. Level II ultrasound examination revealed a singleton fetus with IUGR, single umbilical artery,
microcephaly, polyhydramnios, ventriculomegaly, and abnormal sulcal development with absence of gyri and sulci and a shallow sylvian fissure (Fig. S1). Fetal MRI confirmed corpus callosum dysgenesis and lissencephaly (Fig. S2). At 32 weeks of gestation, the fetus was delivered and died at birth.
3. Methods for detection 3.1. Array-CGH
Whole-genome aCGH analysis on the DNA extracted from cord blood and parental bloods was performed using NimbleGen ISCA Plus Cytogenetic Array (Roche NimbleGen, Madison, WI, USA). The NimbleGen ISCA Plus Cytogenetic Array has 630,000 probes and a median resolution of 15-20 kb across the entire genome according to the manufacturer's instruction. Parental bloods were also collected, and the samples were subjected to aCGH analysis using the same Array kit. The DNA from the cells was extracted first. It was done by following the manufacturer's protocol of QIAamp DNA Mini kit (Qiagen, Inc., Valencia, CA, USA). Then, the 0.5μg of the extracted DNA was labeled in Cy5 dye compared with equivalent amount of normal female gDNA (G1521, Promega) labeled in Cy3 dye to perform the aCGH experiment. The experiment was performed according to the procedures recommended from Roche NimbleGen ISCA plus Cytogenetic Array's user guide. The data was finally represented by using Nexus 6.1 (BioDiscovery, Hawthorne, CA,
USA).
3.2. Conventional cytogenetic analysis
Routine cytogenetic analysis by G-banding techniques at the 550 bands of resolution was performed. Placental tissue was collected, and the sample was subjected to chorionic villi cell culture. Twenty cells of chorionic villi cells were investigated. Parental bloods were collected, and the samples were subjected to lymphocyte culture. Twenty cells of lymphocytes were investigated in each parent.
3.3. qPCR
qPCR analysis was performed on the DNA extracted from cord blood and parental bloods as described elsewhere (Chen et al., 2000). Briefly, primers specifically flanking STR markers on chromosome 17p region such as D17S695 (17p13.3), D17S2181 (17p13.3) and D17S969 (17p12) were applied to undertake polymorphic marker analysis and parental origin determination of the deletion.
3.4. FISH
Metaphase FISH analysis was performed on cultured chorionic villi cells using Vysis Miller-Dieker region/isolated lissencephaly LSI LIS1/RASA probes (Abbott Laboratories, IL, USA), according to the standard FISH protocol described elsewhere (Chen et al., 2013).
4. Results
A 1,560-g female fetus was delivered with microcephaly, high forehead, hypertelorism, depressed nasal bridge, small nose, anteverted nostrils, up-slanting palpebral fissures, micrognathia and fifth finger clinodactyly. Whole-genome aCGH analysis on the DNA extracted from cord blood revealed a 3.17-Mb deletion at 17p13.3, or arr [hg19] 17p13.3 (0-3,165,530)1 (Fig. 1). The deleted region encompasses 71 genes including 36 OMIM genes of DOC2B, RPH3AL, FAM57A,
GEMIN4, RNMTL1, NXN, TIMM22, ABR, TUSC5, YWHAE, CRK, MYO1C, INPP5K, PITPNA, SLC43A2, SCARF1, RILP, RPRF8, MIR22, WDR81, SERPINF2, SERPINF1, RPA1, RTN4RL1, DPH1, OVCA2, MIR132, MIR212, HIC1, SMG6, SRR, TSR1, SGSM2, MNT, PAFAH1B1 and OR1D2. Whole-genome aCGH analysis on the DNA extracted from parental bloods revealed no
genomic imbalance. Cytogenetic analysis of cultured chorionic villi cells revealed a terminal deletion of 17p13.3. The karyotype was 46,XX,del(17)(p13.3) (Fig. 2). The parental karyotypes were normal. The qPCR assays showed only the paternal allele in the fetus on the informative markers of D17S695 (17p13.3) and D17S2181 (17p13.3), indicating a distal 17p deletion from maternal origin (Fig. 3) (Table 1). Metaphase FISH analysis on cultured chorionic villi cells showed absence of LIS1 probe signal on the aberrant chromosome 17, indicating haploinsufficiency of the LIS1 gene (Fig. 4).
5. Discussion
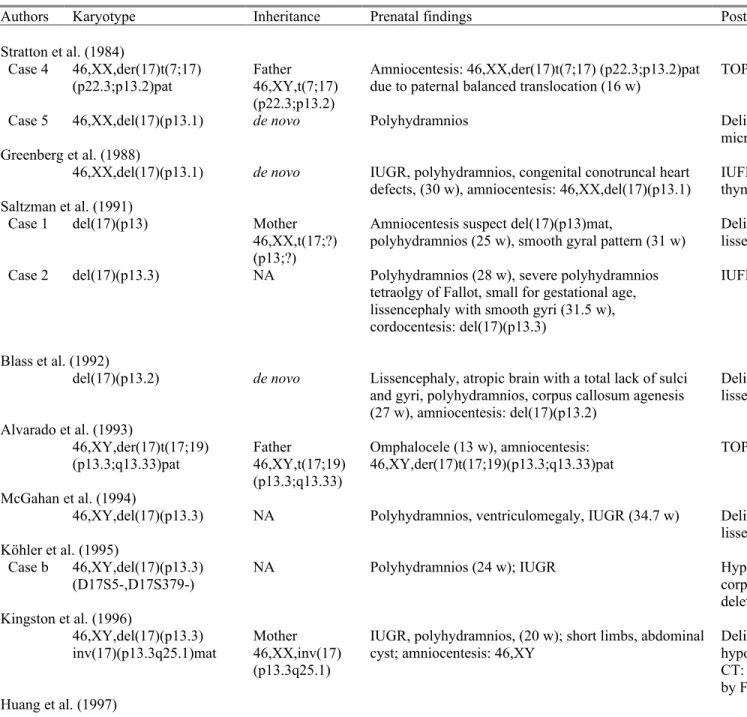
The present case prenatally manifested lissencephaly, corpus callosum dysgenesis, ventriculomegaly, microcephaly, IUGR, polyhydramnios and single umbilical artery. To date, at least 28 cases of chromosome 17p13.3 deletion syndrome with prenatal findings and diagnosis have been reported (Table 2) (Alvarado et al., 1993; Blaas et al., 1992; Chen et al, 2010; Fong et al., 2004; Greenberg et al. 1988; Grosso et al., 2008; Herman and Siegel, 2008; Huang et al., 1997; Joyce et al., 2002; Kim et al., 2011; Kingston et al., 1996; Köhler et al., 1995; Lenzini et al., 2007; Lin et al., 2009; McGahan et al., 1994; Saltzman et al., 1991; Sermer et al, 1987; Stratton et al, 1984; Thomas et al., 2004; van Zelderen-Bhola et al., 1997). Table 1 shows that the sonographic
abnormalities associated with chromosome 17p13.3 deletion syndrome include CNS anomalies such as lissencephaly, corpus callosum dysgenesis/agenesis, ventriculomegaly and microcephaly, IUGR, polyhydramnios, congenital heart defects, omphalocele and renal abnormalities.
Among the 29 cases including our case with prenatal findings and diangosis on Table 1, polyhydramnios, IUGR and ventriculomegaly are the most common associated sonographic abnormalities and account for 66% (19/29), 62% (18/29) and 59% (17/29), respectively. Lissencephaly and corpus callosum dysgenesis/agenesis were detected prenatally in 41% (12/29) and 17% (5/29), respectively, and all were found in the third trimester. Conotruncal heart detects were prenatally detected in 14% (4/29), and the associated conotruncal heart detects may lead to a diagnosis suspicious of DiGeorge syndrome (Chen et al, 2010; Greenberg et al. 1988; Saltzman et al., 1991; Thomas et al., 2004). Omphalocele and renal anomalies were prenatally detected in only 7% (2/29) (Alvarado et al., 1993; Chitayat et al., 1997; Fong et al., 2004; Lenzini et al., 2007; van Zelderen-Bhola et al., 1997). Among the 29 cases of chromosome 17p13.3 deletion syndrome with associated prenatal findings on Table 1, about 16 cases (55%) had prenatal genetic analysis of which nine cases (31%; 9/29) had prenatally detected chromosome 17 aberration because of familial translocation and/or ultrasound abnormalities (Alvarado et al., 1993; Blaas et al., 1992; Greenberg et al. 1988; Lenzini et al., 2007; Lin et al., 2009; Saltzman et al., 1991; Stratton et al, 1984; Thomas et al., 2004), and seven cases (including the present case) (24%; 7/29) had prenatally diagnosed normal karyotype at amniocentesis because of various indications such as advanced maternal age, habitual abortions and ultrasound abnormalities (Chitayat et al., 1997; Fong et al., 2004; Huang et al., 1997; Joyce et al., 2002; Kingston et al., 1996; van Zelderen-Bhola et al., 1997). Table 1 shows that at least 45% (13/29) of the cases with chromosome 17p13.3 deletion syndrome and associated prenatal findings had parental cryptic translocations (Alvarado et al., 1993; Fong et al., 2004; Grosso et al., 2008; Joyce et al., 2002; Thomas et al., 2004; van Zelderen-Bhola et al., 1997) or parental chromosome 17 inversions (Fong et al., 2004; Kingston et al., 1996). It has been estimated that about 80% of the patients with MDLS have a de novo deletion involving 17p13.3, and approximately 20% have a 17p13.3 deletion inherited from a parental cryptic rearrangement (Dobyns et al., 2009).
Ghai et al. (2006) suggested that in pregnancies with fetal MDLS, the common ultrasound findings include widespread agyria, abnormal sylvian fissure and insula, ventriculomegaly(usually
mild), corpus callosum dysgenesis, microcephaly, IUGR and polyhydramnios, and less common findings include micrognathia, congenital heart disease, genitourinary anomalies and omphalocele. Fong et al. (2004) suggested that mild ventriculomegaly may be the first sign of abnormal or delayed brain maturation, and abnormal sulcal development should be suspected and investigated by ultrasound at 24-26 weeks of gestation, and fetal MRI and FISH analysis of 17p13.3 deletion are helpful for identification of MDLS in the fetus. Fetal cerebral sulci can be demonstrated by ultrasound as early as 18 weeks of gestation, and most main sulci can be demonstrated at 30-32 weeks of gestation (Cohen-Sacher et al., 2006; Monteagudo and Timor-Tritsch, 1997; Toi et al. 2004). The present case demonstrates that ventriculomegaly, abnormal sulcal development, corpus callosum dysgenesis, microcephaly, IUGR, polyhydramnios and single umbilical artery can be associated with fetuses carrying chromosome 17p13.3 deletion syndrome. Prenatal identification of a combination of those sonographic abnormalities should include a detailed investigation of lissencephaly by ultrasound and fetal MRI, and aCGH is helpful for molecular characterization of 17p13.3 deletion.
The present case had a 3.17-Mb deletion encompassing the genes of YWHAE, CRK and
PAFAH1B1. PAFAH1B1 (OMIM 601545) encodes platelet-activating factor acetylhydrolase
isoform 1B subunit. Reiner et al. (1993) suggested that PAFAH1B1 is involved in a signal transduction pathway that is important for cerebral development, and haploinsufficiency of
PAFAH1B1 causes MDLS. Point mutations or deletion mutations of the PAFAH1B1 have been
associated with lissencephaly 1 and subcortical laminar heterotopia (OMIM 607432) (Chong et al., 1996; Leventer et al., 2001; Lo Nigro et al., 1997; Pilz et al., 1999; Sicca et al., 2003; Torres et al., 2004). YWHAE (OMIM 605066) encodes 14-3-3 protein which plays important roles in neuronal migration (Toyo-oka et al., 2003), in periventricular heterotopias and corpus callosum hypoplasia (Mignon-Ravix et al., 2010), and in cardiac channel activity and cardiomyopathy (Allouis et al., 2006; Chang et al., 2013; Choe et al., 2006). CRK (OMIM 164762) is an oncogene that encodes v-crk avian sarcoma virus CT10 oncogene homologue which is an adaptor protein that has a role in cell proliferation and migration (Feller et al., 1998; Tzuda et al., 2002).
Cardoso et al. (2003) suggested that deletion of CRK and YWHAE in combination with deletion of PAFAH1B1 contributes to the more severe form of lissencephaly seen only in patients with MDLS. Nagamani et al. (2009) investigated five patients with 17p13.3 microdeletion involving
YWHAE but not PAFAH1B1 and concluded that microdeletion of 17p13.3 involving YWHAE is
associated with facial dysmorphisms, growth restriction and cognitive impairment, and CRK plays a role in growth restriction. Bruno et al. (2010) investigated eight patients with 17p13.3 microdeletion and five patients with 17p13.3 microduplication and concluded that YWHAE and
CRK are candidate genes for autism (duplications), facial dysmorphisms (deletions), growth
restriction (deletions) and limb malformations (deletions and duplications). Schiff et al. (2010) studied four patients with 17p13.3 microdeletion involving YWHAE but distal to PAFAH1B1 and found a distinct phenotype of mild mental retardation, moderate to severe growth restriction, white matter abnormalities and developmental defects in brain and eye. Tenney et al. (2011) reported two patients with 17p13.3 deletion involving YWHAE but not PAFAH1B1 and identified a clinical syndrome with macrocephaly, small stature, dysmorphic features, generalized epilepsy, developmental delay and non-specific white matter changes. Østergaard et al. (2012) reported a boy with a 284-kb microdeletion of 17p13.3 involving CRK but not YWHAE and PAFAH1B1, and identified a distinct phenotype of mental retardation, postnatal growth restriction, facial dysmorphisms, clinodactyly and syndactyly.
In summary, we present molecular cytogenetic characterization and associated prenatal abnormalities in a fetus with chromosome 17p13.3 deletion syndrome. We review previously reported cases of chromosome 17p13.3 deletion syndrome with prenatal findings and diagnosis, and we discuss the genotype-phenotype correlation of the genes of YWHAE, CRK and PAFAH1B1 that are deleted within 17p13.3 in this case.
Acknowledgements
This work was supported by research grants NSC-99-2628-B-195-001-MY3 and NSC-101-2314-B-195-011-MY3 from the National Science Council and MMH-E-102-04 from Mackay Memorial Hospital, Taipei, Taiwan.
Appendix A. Supplementary data
References
Allouis, M., et al. 2006. 14-3-3 is a regulator of the cardiac voltage-gated sodium channel Nav1.5. Circ. Res. 98, 1538-1546.
Alvarado, M., Bass, H.N., Caldwell, S., Jamehdor, M., Miller, A.A., Jacob, P. 1993. Miller-Dieker syndrome. Detection of a cryptic chromosome translocation using in situ hybridization in a family with multiple affected offspring. Am. J. Dis. Child. 147, 1291-1294.
Blaas, H.-G., Eik-Nes, S.H., Kiserud, T., van der Hagen, C.B., Smedvig, E. 1992. Lissencephaly, type I. TheFetus.net 1992-03-06-01. Available at http://sonoworld.com/TheFetus/page.aspx?id=125. Access: May 10, 2013.
Bruno, D.L., et al. 2010. Further molecular and clinical delineation of co-locating 17p13.3 microdeletions and microduplications that show distinctive phenotypes. J. Med. Genet. 47, 299-311.
Cardoso, C., et al. 2003. Refinement of a 400-kb critical region allows genotypic differentiation between isolated lissencephaly, Miller-Dieker syndrome, and other phenotypes secondary to deletions of 17p13.3. Am. J. Hum. Genet. 72, 918-930.
Chang, B., et al. 2013. 14-3-3ε gene variants in a Japanese patient with left ventricular noncompaction and hypoplasia of the corpus callosum. Gene 515, 173-180.
Chen, C.-P., Chern, S.-R., Wang, W. 2000. Rapid determination of zygosity and common aneuploidies from amniotic fluid cells using quantitative fluorescent polymerase chain reaction following genetic amniocentesis in multiple pregnancies. Hum. Reprod. 15, 929-934.
Chen, C.-P., et al. 2010. Ventriculomegaly, intrauterine growth restriction, and congenital heart defects as salient prenatal sonographic findings of Miller-Dieker lissencephaly syndrome associated with monosomy 17p (17p13.2pter) in a fetus. Taiwan. J. Obstet. Gynecol. 49, 81-86.
Chen, C.-P., et al. 2011. Prenatal diagnosis of a de novo 17p13.1 microduplication in a fetus with ventriculomegaly and lissencephaly. Taiwan. J. Obstet. Gynecol. 50, 554-557.
Chen, C.-P., et al. 2013. Chromosome 18p deletion syndrome presenting holoprosencephaly and premaxillary agenesis: prenatal diagnosis and aCGH characterization using uncultured amniocytes. Gene, in press.
Chitayat, D., et al. 1997. Omphalocele in Miller-Dieker syndrome: expanding the phenotype. Am. J. Med. Genet. 69, 293-298.
Choe, C.-U., et al. 2006. C-terminal HERG (LQT2) mutations disrupt IKr channel regulation through 14-3-3ε. Hum. Mol. Genet. 15, 2888-2902.
Chong, S.S., et al. 1996. Point mutations and an intragenic deletion in three ILS patients confirm LIS1 as the lissencephaly causative gene in isolated lissencephaly sequence and Miller-Dieker syndrome. Am. J. Hum. Genet. 59(Suppl.), A23.
Cohen-Sacher, B., Lerman-Sagie, T., Lev, D., Malinger, G. 2006. Sonographic developmental milestones of the fetal cerebral cortex: a longitudinal study. Ultrasound Obstet. Gynecol. 27, 494-502.
Dieker, H., Edwards, R.H., ZuRhein, G., Chou, S.M., Hartman, H.A., Opitz, J.M. 1969. The lissencephaly syndrome. In: Bergsma, D., ed. The Clinical Delineation of Birth Defects: Malformation Syndromes. National Foundation-March of Dimes, New York, p.53-64.
Dobyns, W.B., Stratton, R.F., Greenberg, F. 1984. Syndromes with lissencephaly. I: Miller-Dieker and Norman-Roberts syndromes and isolated lissencephaly. Am. J. Med. Genet. 18, 509-506.
Dobyns, W.B., Curry, C.J.R., Hoyme, H.E., Turlington, L., Ledbetter, D.H. 1991. Clinical and molecular diagnosis of Miller-Dieker syndrome. Am. J. Hum. Genet. 48, 584-594.
Dobyns, W.B., Reiner, O., Carrozzo, R., Ledbetter, D.H. 1993. Lissencephaly: a human brain malformation associated with deletion of the LIS1 gene located at chromosome 17p13. JAMA 270, 2838-2842.
Dobyns, W.B., Das, S. 2009. LIS1-associated lissencephaly/subcortical band heterotopia. In GeneReviewsTM [Internet]. Pagon, R.A., Bird, T.D., Dolan, C.R., Stephens, K., Adam, M.P., eds. Seattle (WA): University of Washington, Seattle; 1993-. Available at http://www.ncbi.nlm.nih.gov/books/NBK5189/. Update: Mar 3, 2009. Access: May 7, 2013.
Feller, S.M., et al. 1998. Physiological signals and oncogenesis mediated through Crk family adapter proteins. J. Cell Physiol. 177, 535-552.
Fong, K.W., Ghai, S., Toi, A., Blaser, S., Winsor, E.J., Chitayat, D. 2004. Prenatal ultrasound findings of lissencephaly associated with Miller-Dieker syndrome and comparison with pre- and postnatal magnetic resonance imaging. Ultrasound Obstet. Gynecol. 24, 716-723.
Ghai, S., Fong, K.W., Toi, A., Chitayat, D., Pantazi, S., Blaser, S. 2006. Prenatal US and MR imaging findings of lissencephaly: review of fetal cerebral sulcal development. Radiographics 26, 389-405. Greenberg, F., et al. 1988. Prenatal diagnosis of deletion 17p13 associated with DiGeorge anomaly. Am. J.
Med. Genet. 31, 1-4.
Grosso, S., et al. 2008. Bilateral periventricular nodular heterotopia and lissencephaly in an infant with unbalanced t(12;17)(q24.31; p13.3) translocation. Dev. Med. Child Neurol. 50, 473-476.
Herman, T.E., Siegel, M.J. 2008. Miller–Dieker syndrome, type 1 lissencephaly. J. Perinatol. 28, 313-315. Huang, H.-C., Bautista, S.L., Chen, B.-S., Chang, K.-P., Chen, Y.-J., Wuu, S.W. 1997. Miller-Dieker
syndrome with microdeletion of chromosome 17p13.3: report of one case. Zhonghua Min Guo Xiao Er Ke Yi Xue Hui Za Zhi 38, 472-476.
Joyce, C.A., Dennis, N.R., Howard, F., Davis, L.M., Thomas, N.S. 2002. An 11p;17p telomeric translocation in two families associated with recurrent miscarriages and Miller-Dieker syndrome. Eur. J. Hum. Genet. 10, 707-714.
Kim, Y.J., et al. 2011. Miller-Dieker syndrome with der(17)t(12;17) (q24.33;p13.3)pat presenting with a potential risk of mis-identification as a de novo submicroscopic deletion of 17p13.3. Korean J. Lab. Med. 31, 49-53.
Kingston, H.M., Ledbetter, D.H., Tomlin, P.I., Gaunt, K.L. 1996. Miller-Dieker syndrome resulting from rearrangement of a familial chromosome 17 inversion detected by fluorescence in situ hybridisation. J. Med. Genet. 33, 69-72.
Köhler, A., Hain, J., Müller, U. 1995. Clinical and molecular genetic findings in five patients with Miller-Dieker syndrome. Clin. Genet. 47, 161-164.
Lenzini, E., D'Ottavio, G., Città, A., Benussi, D.G., Petix, V., Pecile, V. 2007. Prenatal diagnosis of Miller-Dieker syndrome by ultrasound and molecular cytogenetic analysis. Clin. Genet. 72, 487-489.
Leventer, R.J., Cardoso, C., Ledbetter, D.H., Dobyns, W.B. 2001. LIS1 missense mutations cause milder lissencephaly phenotypes including a child with normal IQ. Neurology 57, 416-422.
Lin, C.-Y., et al. 2009. Prenatal diagnosis of monosomy 17p (17p13.3pter) associated with polyhydramnios, intrauterine growth restriction, ventriculomegaly and Miller-Dieker lissencephaly syndrome in a fetus. Taiwan. J. Obstet. Gynecol. 48, 408-411.
Lo Nigro, C., Chong, S.S., Smith, A.C.M., Dobyns, W.B., Carrozzo, R., Ledbetter, D.H. 1997. Point mutations and an intragenic deletion in LIS1, the lissencephaly causative gene in isolated lissencephaly sequence and Miller-Dieker syndrome. Hum. Mol. Genet. 6, 157-164.
McGahan, J.P., Grix, A., Gerscovich, E.O. 1994. Prenatal diagnosis of lissencephaly: Miller-Dieker syndrome. J. Clin. Ultrasound 22, 560-563.
Mignon-Ravix, C., et al. 2010. Deletion of YWHAE in a patient with periventricular heterotopias and pronounced corpus callosum hypoplasia. J. Med. Genet. 47, 132-136.
Miller, J.Q. 1963. Lissencephaly in 2 siblings. Neurology 13, 841-850.
Monteagudo, A., Timor-Tritsch, I.E. 1997. Development of fetal gyri, sulci and fissures: a transvaginal sonographic study. Ultrasound Obstet. Gynecol. 9, 222-228.
Nagamani, S.C.S., et al. 2009. Microdeletions including YWHAE in the Miller-Dieker syndrome region on chromosome 17p13.3 result in facial dysmorphisms, growth restriction, and cognitive impairment. J. Med. Genet. 46, 825-833.
Neer, E.J., Schmidt, C.J., Smith, T. 1993. LIS is more. Nat. Genet. 5, 3-4.
Østergaard, J.R., Graakjær, J., Brandt, C., Birkebæk, N.H. 2012. Further delineation of 17p13.3 microdeletion involving CRK. The effect of growth hormone treatment. Eur. J. Med. Genet. 55, 22-26. Pilz, D.T., et al. 1999. Subcortical band heterotopia in rare affected males can be caused by missense
mutations in DCX (XLIS) or LIS1. Hum. Mol. Genet. 8, 1757-1760.
Reiner, O., et al. 1993. Isolation of a Miller-Dieker lissencephaly gene containing G protein beta-subunit-like repeats. Nature 364, 717-721.
Saltzman, D.H., Krauss, C.M., Goldman, J.M., Benacerraf, B.R. 1991. Prenatal diagnosis of lissencephaly. Prenat. Diagn. 11, 139-143.
Schiff, M., et al. 2010. Further delineation of the 17p13.3 microdeletion involving YWHAE but distal to PAFAH1B1: four additional patients. Eur. J. Med. Genet. 53, 303-308.
Sermer, M., Benzie, R.J., Pitson, L., Carr, M., Skidmore, M. 1987. Prenatal diagnosis and management of congenital defects of the anterior abdominal wall. Am. J. Obstet. Gynecol. 156, 308-312.
Sicca, F., et al. 2003. Mosaic mutations of the LIS1 gene cause subcortical band heterotopia. Neurology 61, 1042-1046.
Stratton, R.F., Dobyns, W.B., Airhart, S.D., Ledbetter, D.H. 1984. New chromosomal syndrome: Miller-Dieker syndrome and monosomy 17p13. Hum. Genet. 67, 193-200.
Tenney, J.R., Hopkin, R.J., Schapiro, M.B. 2011. Deletion of 14-3-3ε and CRK: a clinical syndrome with macrocephaly, developmental delay, and generalized epilepsy. J. Child Neurol. 26, 223-227.
Thomas, M.A., Duncan, A.M.V., Bardin, C., Kaloustian, V.M.D. 2004. Lissencephaly with der(17)t(17;20) (p13.3;p12.2)mat. Am. J. Med. Genet. 124, 292-295.
Toi, A., Lister, W.S., Fong, K.W. 2004. How early are fetal cerebral sulci visible at prenatal ultrasound and what is the normal pattern of early fetal sulcal development? Ultrasound Obstet. Gynecol. 24, 706-715. Torres, F.R., Montenegro, M.A., Marques-de-Faria, A.P., Guerreiro, M.M., Cendes, F., Lopes-Cendes. I.
2004. Mutation screening in a cohort of patients with lissencephaly and subcortical band heterotopia. Neurology 62, 799-802.
Toyo-oka, K., et al. 2003. 14-3-3ε is important for neuronal migration by binding to NUDEL: a molecular explanation for Miller-Dieker syndrome. Nat. Genet. 34, 274-285.
Tsuda, M., Tanaka, S., Sawa, H., Hanafusa, H., Nagashima, K. 2002. Signaling adaptor protein v-Crk activates Rho and regulates cell motility in 3Y1 rat fibroblast cell line. Cell Growth Differ. 13, 131-139. van Zelderen-Bhola, S.L., et al. 1997. Prenatal and postnatal investigation of a case with Miller-Dieker
syndrome due to a familial cryptic translocation t(17;20)(p13.3;q13.3) detected by fluorescence in situ hybridization. Prenat. Diagn. 17, 173-179.
Figure Captions
Fig. 1. Whole-genome aCGH analysis on cord blood shows a 3.17-Mb deletion at 17p13.3, or arr [hg19] 17p13.3 (0-3,165,530)1. (A) Chromosomal view and (B) zoom in view.
Fig. 2. A karyotype of 46,XX,del(17)(p13.3).
Fig. 3. Representative electrophoretograms of qPCR assays using informative polymorphic markers of D17S695 (17p13.3), D17S2181 (17p13.3) and D17S969 (17p12). The makers D17S969 is outside the deleted region and shows two peaks of equal fluorescent activity from two different parental alleles in the fetus. The makers D17S695 (17p13.3) and D17S2181 (17p13.3) are within the deleted region and show only one peak of fluorescent activity from the paternal allele in the fetus, indicating that the deletion is from maternal origin.
Fig. 4. Metaphase FISH analysis on chorionic villi cells using Vysis Miller-Dieker region/isolated lissencephaly LSI LIS1 (spectrum orange)/RASA (spectrum green) probes shows a LIS1 signal and a RASA signal on a chromosome 17, and absence of the LIS1 signal on the aberrant chromosome del(17)(p13.3), indicating haploinsufficiency of the LIS1 gene.
Appendix A. Supplementary data
Fig. S1. Prenatal ultrasound at 31 weeks of gestation shows abnormal sulcal development with absence of gyri and sulci and a shallow sylvian fissure.
Fig. S2. (A) Trans-axial view, (B) sagittal view and (C) coronal view of fetal brain MRI at 31 weeks of gestation show a smooth brain with absence of age-matched cortical sulci/gyri, thin gray matter and white matter, small brain volume, and mild ventriculomegaly.
Table 1. Molecular results using polymorphic DNA markers specific for 17p*
Markers Location Father Mother Fetus
D17S695 17p13.3 746,152-746,356 198,198 194,208 198
D17S2181 17p13.3 1,540,534-1,540,748 220,220 212,212 220 D17S969 17p12 11,844,966-11,845,097 136,140 124,140 124,136 * Alleles (basepair sizes) are listed below each individual.
Table 2. Reported cases of chromosome 17p13.3 deletion syndrome with prenatal findings and diagnosis
Authors Karyotype Inheritance Prenatal findings Postnatal findings
Stratton et al. (1984)
Case 4 46,XX,der(17)t(7;17)
(p22.3;p13.2)pat Father46,XY,t(7;17)
(p22.3;p13.2)
Amniocentesis: 46,XX,der(17)t(7;17) (p22.3;p13.2)pat
due to paternal balanced translocation (16 w) TOP at 20 w, micrognathia, left renal cystic dysplasia
Case 5 46,XX,del(17)(p13.1) de novo Polyhydramnios Delivery at term, 2,950 g, MDLS facial trait, seizures,
microcephaly, CT: lissencephaly Greenberg et al. (1988)
46,XX,del(17)(p13.1) de novo IUGR, polyhydramnios, congenital conotruncal heart
defects, (30 w), amniocentesis: 46,XX,del(17)(p13.1) IUFD at 34 w, 905 g, conotruncal cardiac defect, DORV, thymic hypoplasia mimicking DiGeorge syndrome Saltzman et al. (1991)
Case 1 del(17)(p13) Mother
46,XX,t(17;?) (p13;?)
Amniocentesis suspect del(17)(p13)mat,
polyhydramnios (25 w), smooth gyral pattern (31 w) Delivery at 36 w, 51b 7oz, MDLS facial trait, CT: lissencephaly, DNA study: monosomy 17p13
Case 2 del(17)(p13.3) NA Polyhydramnios (28 w), severe polyhydramnios
tetraolgy of Fallot, small for gestational age, lissencephaly with smooth gyri (31.5 w), cordocentesis: del(17)(p13.3)
IUFD at 36 w
Blass et al. (1992)
del(17)(p13.2) de novo Lissencephaly, atropic brain with a total lack of sulci
and gyri, polyhydramnios, corpus callosum agenesis (27 w), amniocentesis: del(17)(p13.2)
Delivery at 36 w, 2,550 g, MDLS facial trait, MRI: lissencephaly, corpus callosum agenesis, feeding problems Alvarado et al. (1993)
46,XY,der(17)t(17;19)
(p13.3;q13.33)pat Father46,XY,t(17;19)
(p13.3;q13.33)
Omphalocele (13 w), amniocentesis:
46,XY,der(17)t(17;19)(p13.3;q13.33)pat TOP
McGahan et al. (1994)
46,XY,del(17)(p13.3) NA Polyhydramnios, ventriculomegaly, IUGR (34.7 w) Delivery at 37.4 w, 2,060 g, MDLS facial trait, CT and MRI:
lissencephaly, corpus callosum dysgenesis, colpocephaly Köhler et al. (1995)
Case b 46,XY,del(17)(p13.3)
(D17S5-,D17S379-) NA Polyhydramnios (24 w); IUGR Hypotonia at birth, seizures at 7 m, MRI: lissencephaly, corpus callosum dysgenesis; microsatellite analysis: 17p13.3 deletion, paternal origin
Kingston et al. (1996)
46,XY,del(17)(p13.3)
inv(17)(p13.3q25.1)mat Mother 46,XX,inv(17) (p13.3q25.1)
IUGR, polyhydramnios, (20 w); short limbs, abdominal
cyst; amniocentesis: 46,XY Delivery 2,300 g, short limbs, syndactyly, cleft palate, hypospadias, undescended testes, MDLS facial trait, seizures, CT: lissencephaly, death at 2 y 7 m, del(17)(p13.3) confirmed by FISH
46,XX.ish del(17)
(p13.3p13.3)(D17S379-) de novo Amniocentesis: 46,XX (16 w) due to AMA; IUGR, polyhydramnios (32 w, 36 w) Delivery at term, 2,020 g, microcephaly, MDLS facial trait, PDA, CT and MRI: lissencephaly, corpus callosum hypoplasia, del(17)(p13.3) confirmed by FISH, seizures, hypotonia, psychomotor retardation at 18 m
van Zelderen-Bhola et al. (1997) 46,XY.ish der(17)t(17;20) (p13.3;q13.3)pat [(c197-2, c197-4, c197-9)-, wcp20+] Father 46,XY,t(17;20) (p13.3;q13.3)
Polyhydramnios, enlarged left kidney, right renal agenesis, ventriculomegaly (29 w); amniocentesis: 46,XY
Delivery at 31 w, 1,345 g, MDLS facial trait, camptodactyly, small nails, MRI: lissencephaly, right renal agenesis, left renal dysplasia, VSD, cryptic translocation t(17;20) confirmed by FISH Joyce et al. (2002) Case 1 46,XX,der(17)t(11;17) (p15.5;p13.3)mat (D17S379-) Mother 46,XX,t(11;17) (p15.5;p13.3) Polyhydramnios, amniocentesis: 46,XX (30 w); ventriculomegaly, corpus callosum agenesis (32 w); IUGR
Delivery at 34 w, 1,520 g, MDLS facial trait, ultrasound: corpus callosum agenesis, lissencephaly, del(17)(p13.3) in the proband and cryptic t(11;17) translocation in the mother confirmed by FISH, death at 1 d
Case 2 46,XX,der(17)t(11;17) (p15.5;p13.3)mat (D17S379-) Mother 46,XX,t(11;17) (p15.5;p13.3)
Amniocentesis: 46,XX due to previous spontaneous
miscarriages; ventriculomegaly (23 w); IUGR Delivery at 36 w, microcepahly, MDLS facial trait, del(17)(p13.3) in the proband and cryptic t(11;17) translocation in the mother confirmed by FISH, death at 14 m
Fong et al. (2004)
Case 1* 46,XY.ish del(17)
(p13.3p13.3)(D17S379-) de novo Omphalocele, borderline ventriculomegaly (20 w, 24 w, 36 w); amniocentesis: 46,XY (20 w) Delivery at 37 w, 2,750 g, MDLS facial trait, omphalocele, MRI: lissencephaly, corpus callosum hypogenesis, colpocephaly, 17p13.3 microdeletion by FISH, seizures, developmental delay at 4 m, death at 23 m
Case 2 46,XX,del(17)(p13) .ish del(17)(p13.3p13.3) (D17S379-) Father 46,XY,inv(17) (p13.1q25.3)
Ventriculomegaly, abnormal cortical sulcal ultrasound findings (23.4 w; 35 w); fetal MRI: lissencephaly (32 w)
Delivery at term with neonatal death at 1 d
Case 3 46,XY.ish der(17)t(9;17)
(q34.3;p13.3)mat (LIS1-) Mother46,XX,t(9;17) (q34.3;p13.3)
Polyhydramnios, ventriculomegaly, abnormal brain cortical sulcal ultrasound findings (26.3 w); fetal MRI: ventriculomegaly, lissencephaly (28 w)
IUFD at 34 w
Case 4 46,XY.ish del(17)
(p13.3p13.3)(LIS1-) de novo Ventriculomegaly, abnormal cortical sulcal ultrasound findings (30 w); IUGR, corpus callosum dysgenesis; fetal MRI: lissencephaly, ventriculomegaly, corpus callosum dysgenesis (31 w)
TOP at 33 w, stillbirth, craniofacial dysmorphism
Case 5 46,XY.ish der(17)t(3;17)
(q29;p13)pat (LIS1-) Father46,XY,t(3;17)
(q29;p13)
Ventriculomegaly, abnormal cortical sulcal ultrasound
findings (30.6 w); IUGR Delivery at 35 w, seizures, developmental delay at age 1 y, MRI: lissencephaly, ventriculomegaly
Case 6 46,XY,del(17)(p13)
.ish del(17)(p13.3p13.3) (LIS1-)
de novo Abnormal cortical sulcal ultrasound findings (31.6 w);
polyhydramnios, micrognathia
Delivery at 32 w, CHD, seizures, developmental delay, death at age 11 m, MRI: lissencephaly
Case 7 46,XX,r(17)(p13q25)
.ish del(17)(p13.3p13.3) (LIS1-)
de novo Ventriculomegaly, abnormal cortical sulcal ultrasound
findings (33 w); IUGR, polyhydramnios Delivery at 35 w, death at age 3 m, MRI: lissencephaly, ventriculomegaly
46,XY,der(17)t(17;20)
(p13.3;p12.2)mat Mother46,XX,t(17;20)
(p13.3;p12.2)
DORV, D-transposition of the great vessels, small left and right pulmonary arteries, PDA, small left ventricle, IUGR, ventriculomegaly (30 w); amniocentesis: an abnormal chromosome 17
Delivery at 39 w, 2,335 g, MDLS facial trait, umbilical hernia, small and undescended testes, MRI: lissencephaly, colpocephaly, corpus callosum agenesis, death at 10 d Lenzini et al. (2007)
46,XX.ish der(17)t(17;18)
(p13;p11.2)(LIS1-) de novo Polyhydramnios, IUGR, ventriculomegaly, corpus callosum dysgenesis, lissencephaly, pachygyria, hypoechogenic cerebral parenchyma, equinovarus foot, hyperechoic renal parenchyma (29 w); amniocentesis and cordocentesis: 46,XX.ish der(17)t(17;18) (p13;p11.2)(LIS1-); FISH and microsatellite analysis: 4-Mb 17p13.3 deletion, maternal origin of der(17)
IUFD at 38 w, agyria, ventricular dilation, left talipes equinovarus Grosso et al. (2008) 46,XY,der(17)t(12;17) (q24.31;p13.3)mat Mother 46,XX,t(12;17) (q24.31;p13.3)
Polyhydramnios, IUGR (33 w) Delivery at 33 w, 1,560 g, MDLS facial trait, hypotonia,
seizures, MRI: lissencephaly, corpus callosum hypoplasia Herman and Siegel (2008)
del(17)(p13) NA Polyhydramnios, IUGR, poor fetal movement (41 w) Delivery at 41 w, 1,910 g, MDLS facial trait, MRI:
lissencephaly, corpus callosum dysgenesis,colpocephaly Lin et al. (2009)
46,XY,del(17)(p13.3) de novo IUGR, polyhydramnios, ventriculomegaly, abnormal
sulcal development with absence of gyri and sulci and shallow sylvian fissure (31 w); amniocentesis: 46,XY,del(17)(p13.3); fetal MRI: lissencephaly (34 w)
Delivery at 35 w, 1,346 g, MDLS facial trait, MRI: lissencephaly, growth retardation, developmental delay, seizures at 10 m
Chen et al. (2010)
46,XX,del(17)(p13.2) de novo Ventriculomegaly, IUGR, ASD, tetralogy of Fallot (36
w) Delivery at 38 w, 2,308 g, facial dysmorphsim mimicking DiGeorge syndrome, tetralogy of Fallot, MRI: lissencephaly, colpocephaly, corpus callosum agenesis, developmental delay, growth retardation, seizures
Kim et al. (2011) 46,XX,der(17)t(12;17) (q24.33;p13.3)pat Father 46,XY,t(12;17) (q24.33;p13.3)
IUGR, ventriculomegaly Delivery at 35 w, 1,680 g, MDLS facial trait, PDA,
hypotonia, MRI: lissencephaly, corpus callosum agenesis, seizures
Present case
46,XX,del(17)(p13.3) de novo Amniocentesis: 46,XX (19 w) due to AMA;
polyhydramnios, IUGR, ventriculomegaly,
microcephaly, corpus callosum agenesis, lissencephaly, single umbilical artery (31 w)
Delivery at 32 w, 1,560 g, death at birth, MDLS facial trait, clinodactyly, aCGH on cord blood: 3.17-Mb deletion at 17p13.3, placenta: 46,XX,del(17)(p13.3)
*: as Chitayat et al., (1997), NA: not available, d: days, w: weeks, m: months, y: years, MDLS: Miller-Dieker lissencephaly syndrome, IUFD: intrauterine fetal demise, IUGR: intrauterine growth restriction, TOP: termination of pregnancy, CHD: congenital heart defects, CT: computed tomography, AMA: advanced maternal age, ASD: atrial septal defect, VSD: ventricular septal defect, DORV: double-outlet right ventricle, PDA: patent ductus arteriosus