and sharing with colleagues.
Other uses, including reproduction and distribution, or selling or
licensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of the
article (e.g. in Word or Tex form) to their personal website or
institutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies are
encouraged to visit:
Drug design for hemagglutinin: Screening and molecular dynamics from
traditional Chinese medicine database
Tung-Ti Chang
a,b, Mao-Feng Sun
a,c, Hsin-Yi Chen
d, Fuu-Jen Tsai
d,e, Calvin Yu-Chian Chen
a,d,f,*
aLaboratory of Computational and Systems Biology, School of Chinese Medicine, China Medical University, Taichung 40402, Taiwan b
Department of Chinese Pediatrics, China Medical University Hospital, Taiwan
c
Department of Acupuncture, China Medical University Hospital, Taiwan
d
Department of Bioinformatics, Asia University, Taichung 41354, Taiwan
e
Department of Medical Genetics, Pediatrics and Medical Research, China Medical University Hospital and College of Chinese Medicine, China Medical University, Taichung 40402, Taiwan
fComputational and Systems Biology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
1. Introduction
Influenza viruses can be categorized into three subtypes, namely A, B and C, and among subtypes, influenza A virus is the most virulent and can infect humans, horses, pigs, mammals and birds (Webster et al., 1992). In the past, several major pandemics had been caused by influenza A virus, including the 1918 Spanish flu (H1N1), the 1957 Asian flu (H2N2) and the 1968 Hong Kong flu (H3N2) (Nicholls, 2006). Most recently, the Swine flu (2009/H1N1 virus) had caused a global pandemic and resulted in more than 18,000 death over the world as dated on August 2010 (WHO, 2010). The surface of influenza A virus contains two glycoproteins— hemagglutinin and neuraminidase. Hemagglutinin can be further divided into 15 different serotypes (H1–H15) (Suzuki and Nei, 2002). Of all serotypes, H1, H2 and H3 are the most commonly
circulated. During the viral replication cycle, hemagglutinin (HA) has a critical role in the binding of virus to host surface sialic acid residues and fusion of the viral membrane to the host plasma membrane (Salomon and Webster, 2009). In terms of structure, HA is a trimer with each mature subunit composed of a HA1 chain responsible for sialic acid binding and a HA2 chain responsible for membrane fusion (Wharton et al., 1995).
Currently, neuraminidase inhibitors, oseltamivir and zanamivir are still the front line anti-viral for treating influenza infection, but cases of oseltamivir resistant pandemic 2009 H1N1 viruses have been reported. M2 channel inhibitors, amantadine and rimantadine, that were effective in the past, are not recommended by WHO in 2009 H1N1 influenza pandemic due to drug resistance issues (Chen
et al., 2009b; Cheng et al., 2009). To develop new anti-viral for
treating influenza infection, we focused our attention on HA, specifically on subtype H1. Based on the before mentioned influenza viral replication strategy, a HA inhibitor could block viral entry into cells, thereby preventing generation of new viral progeny. In recent years, drug design using molecular simulations has gained impor-tance and popularity. Molecular simulations have been conducted to investigate protein dynamics and protein–ligand interactions on
A R T I C L E I N F O Article history:
Received 5 September 2010
Received in revised form 28 October 2010 Accepted 11 November 2010
Available online 1 February 2011 Keywords:
H1N1 Influenza virus Docking
Molecular dynamics
Traditional Chinese medicine (TCM) database
A B S T R A C T
Since the outbreak of Swine flu, H1N1 virus had caused a global pandemic and resulted in more than 18,000 deaths over the world as dated on August 2010. Influenza virus surface glycoprotein, hemagglutinin, holds a critical role in mediating viral entry into host cell. Currently, there is no available hemagglutinin binding inhibitor, and already, influenza virus strains resistant to front-line antivirals have been discovered. Here, we report two rosmaricine derivatives might be potent leads which screening from the world largest traditional Chinese medicine database. The docking results shown the rosmaricine derivatives bind influenza hemagglutinin with high affinity based on binding energy. Both rosmaricine_5 and rosmaricine_16 have high binding energy and have hydrogen bonding with hemagglutinin sialic acid binding site residues, Glu83, Asp103 and Asn104. These interactions persist throughout the molecular dynamics simulation and keep the rosmaricine derivatives in the hemagglutinin sialic acid binding site. From the molecular dynamics results show that a potential hemagglutinin inhibitor should have a protonated amine for interacting with Asp103 and hydroxyl groups for interacting with Glu83 and Asn104. Overall, we suggest our two de novo compounds can become novel antivirals for treating influenza infection.
ß2011 Published by Elsevier B.V. on behalf of Taiwan Institute of Chemical Engineers.
* Corresponding author. Current address: Computational and Systems Biology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA.
Tel.: +1 617 353 7123.
E-mail addresses:[email protected],[email protected](C.-C. Chen).
Contents lists available atScienceDirect
Journal of the Taiwan Institute of Chemical Engineers
j o u r n a l h o m e p a g e : w w w . e l s e v i e r . c o m / l o c a t e / j t i c e
1876-1070/$ – see front matter ß 2011 Published by Elsevier B.V. on behalf of Taiwan Institute of Chemical Engineers.
different topics (Bhargavi et al., 2010; Cambria et al., 2010; Cao and Wang, 2010; Chang et al., 2010; Chen, 2010; da Cunha et al., 2010;
Huang et al., 2010a,b; Kahlon et al., 2010; Koshy et al., 2010).
Previously, we had conduced in silico studies on influenza hemagglutinin (Chen et al., 2009a, 2010). As a continuation, we performed molecular dynamics simulation to further investigate ligand–H1 interaction. In the past, we have successfully conducted drug design by combining traditional Chinese medicine (TCM) and both ligand-based and structural-based drug design (Chen et al.,
2009a, 2010; Huang et al., 2010a,b,c). We hope new success can be
discovered by employing TCM on treating H1N1 infection. 2. Methods
2.1. Molecular modeling of H1N1 hemagglutinin
The hemagglutinin amino acid sequence of H1N1 was obtained from GenBank, source sequence accession CY061805. The protein
template used for homology modeling was taken from Protein Data Bank, entry 1RVX (Gamblin et al., 2004). A sequence alignment of the target and template, performed using the Clustal W program
(Larkin et al., 2007), showed high percent of sequence identity
(80.4%) and similarity (89.5%) (Fig. 1) which supported using the template for homology modeling. The MODELLER program (Eswar
et al., 2006) implanted in Build Homology Models module of
Discovery Studio 2.5 was used to build molecular model of hemagglutinin. The final model was evaluated by using Rama-chandran graph and Verify-3D to ensure model quality.
2.2. Docking
A total of 20,000 natural products downloaded from the TCM Database@Taiwan (http://tcm.cmu.edu.tw/) were docked into the sialic acid binding site of hemagglutinin using LigandFit module of Discovery Studio 2.5 (Venkatachalam et al., 2003). Prior to docking, CHARMm (Chemistry at Harvard Macromolecular Mechanics)
[()TD$FIG]
Fig. 1. Sequence alignment between the H1 crystal structure template 1RVX and the H1 target sequence. Sequence identity and similarity are 80.4% and 89.5%, respectively (Genbank accession ID: CY061805; protein ID: ADG08430).
T.-T. Chang et al. / Journal of the Taiwan Institute of Chemical Engineers 42 (2011) 563–571 564
force field was applied to the structure. The docking site was defined by the binding location of N-acetyl-D-glucosamine in the
hemagglutinin template structure.
Ligand conformations generated in Monte Carlo simulation were placed into the binding site using a shape-based matching method. The protein was held rigid throughout docking while the ligands were allowed to orientate inside the binding pocket for sampling different binding poses. DREIDING force field was applied for calculating ligand–receptor interaction energies. Candidate ligand poses were energetically minimized using Smart Minimizer, which incorporates both Steepest Descent and Conjugate Gradient algorithm. Ligand poses were scored and ranked by binding energy. DockScore, potential mean force (PMF) and potential mean force 04 (PMF04) were also calculated for readers’ interests. Compound N-acetyl-D-glucosamine (NAG) was
used as control and was re-docked into the binding site for assessing binding affinity. The binding energy of NAG was used for filtering TCM compounds.
2.3. De novo design
After completing the docking screens, TCM compounds with binding energy higher than NAG were chosen for de novo design. The De Novo Evolution module of receptor–ligand interactions in Discovery Studio 2.5 was used for generating derivatives. In De Novo Evolution, the Ludi algorithm was employed to generate interaction sites within the ligand docking location and to identify fragments that form favorable interactions with the interaction sites. Fragments that best complement the interaction sites were fused or linked to the existing TCM scaffolds.
To screen out compounds that may not be orally active drug, the Lipinski’s Rule of Five was applied to the TCM de novo products. Derivatives that have molecular weight greater than 500 Da or contain excess number of hydrogen bond donors or acceptors or exceed the octanol–water partition coefficient were excluded for further analysis. Derivatives that passed the screen were docked back to the binding site to evaluate binding affinity and receptor– ligand interactions.
2.4. Molecular dynamics simulation
Selected TCM candidates, in complex with hemagglutinin, were taken for molecular dynamics simulation. CHARMm force field was applied to the protein complexes prior to simulation. Each protein– ligand system was first energetically minimized with 500 steps of Steepest Descent and 500 steps of Conjugate Gradient method. After minimization, the system was heated from 50 K to 310 K in a time interval of 50 ps. Following, the equilibration step was conducted for 200 ps in 310 K. The final production run was conducted in NVT ensemble at 310 K for 20 ns. Each MD simulation was conducted with SHAKE algorithm that constraints bonding to hydrogen atoms. The time step was set for 0.001 ps. A snapshot was taken at every 2.5 ps. A total of 4000 snapshots were taken for a ligand–hemagglutinin complex.
The root mean square deviation (RMSD) of the protein and the ligand were computed for accessing protein stability. Hydrogen bond analysis was performed for analyzing key receptor–ligand interactions. The distance for hydrogen bond was set as 2.5 A˚. Energy trajectory was also computed for investigating thermody-namic stability of the protein–ligand complex.
[()TD$FIG]
3. Results and discussions
The sequence identity and similarity between the target and the template sequence are 80.4% and 89.5%, respectively (Fig. 1). To validate the modeled H1 structure, Ramachandran plot
(Ramachandran et al., 1963) was used to examine the
compati-bility of dihedral angles of protein backbone atoms. The results show that 91.53% of residues are found in the favorable regions while only 8.47% of residues are in the disallowed or semi-allowed regions (Fig. 2). In addition to Ramanchandran plot, we also performed Verify-3D to evaluate protein fitness. Verify-3D checks the influenza hemagglutinin structure by evaluating the compat-ibility of the HA sequences with the protein 3D structure (Luthy
et al., 1992). For our result, all the key residues in the HA binding
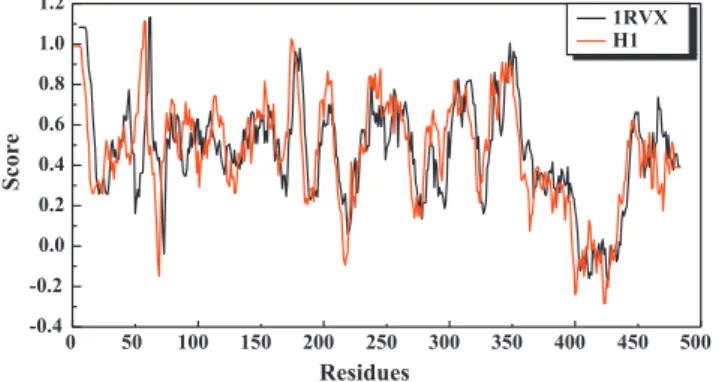
site (Arg238, Asn104 and Asn81) have Verify-3D scores greater than 1 (Fig. 3). These results further support the usage of the HA model.
3.1. Docking results
Results from docking and scoring of the TCM compounds and TCM derivatives are shown inTables 1 and 2, respectively. The top de novo products, rosmaricine_5 and rosmaricine_16, are deri-vatives of rosmaricine, which could be isolated from leaves of rosemary (Rosmarinus officinales) (Calabrese et al., 2000). Rose-mary has hepatoprotective and antitumerogenic activity and is also an antioxidant (al-Sereiti et al., 1999); furthermore, other rosmaricine derivatives have also been studied as potential antitumor agents (Boido et al., 1994). Previously, natural compounds have been reported for antiviral anti-inflammatory and anti-oxidant effects (Chang et al., 2009; Lai et al., 2009); in addition, compounds isolated from Ferula assafoetida have been shown for antiviral activity (Lee et al., 2009). Our study adds to the pool of natural compounds that have been shown to possess therapeutic potential.
The structures of the top two derivatives and the control are shown inTable 3. The docking poses of NAG, rosmaricine_5 and rosmaricine_16 are shown inFig. 4(a)–and (c). Both rosmaricine_5 and rosmaricine_16 share addition of a hydroxyl substituted benzene ring and have very similar docking poses (Fig. 4(a) and (b)). The only difference is that rosmaricine_5 can establish two hydrogen bonds to Glu83 via the two added hydroxyl groups. 3.2. Molecular dynamics simulation
To examine the interactions between TCM compound and hemagglutinin, we selected the top two derivatives and the
control for molecular dynamics simulations. The difference between docking poses and the snapshots taken at the end of molecular simulations are shown in Fig. 4(b), (d) and (f). As shown inFig. 4(a) and (d), the control, NAG, has dramatic changes in binding conformation after the simulation run. The control
Table 3
The scaffold of control, original compounds, and derivatives. Table 1
The docking results of TCM compounds. The top 20 compounds of the database are shown here. Name BE DS PMF PMF04 Camptothecin 120.76 41.925 80.81 20.42 Catharanthine 119.98 60.341 31.84 16.72 Magnoflorine 117.94 46.071 57.63 30.84 Colchamine 101.23 47.745 66.07 18.37 Heliotrine 100.46 78.17 50.39 23.26 Rosmaricine 100.44 79.093 71.28 24.89 Yohimbine 98.33 74.02 48.18 29.04 Uncarine_A 98.09 66.364 25.43 14.99 Peimisine 97.98 79.552 69.12 15.76 Piperine 92.84 44.495 46.23 7.04 Reserpine 90.85 69.175 94.81 42.55 Mesembrine 88.81 67.956 21.52 14.63 Physostigmine 87.48 70.325 47.06 17.66 Evocarpine 84.17 37.048 41.51 0.04 Stephanine 83.54 73.257 56.86 25.32 pingbeimine_C 83.09 77.365 73.74 23.6 Cyclostachine_A 81.18 46.609 72.71 16.68 Corydine 79.43 83.07 58.75 28.79 Cinchonine 76.92 76.956 34.42 19.04 Papaverine 65.89 50.186 51.75 18.44 N-acetyl-D-glucosamine* 52.81 49.69 59.89 29.43
BE: binding energy. DS: dock score.
PMF: potential of mean force. N-acetyl-D-glucosamine*: control.
Table 2
The docking results of top 5 derivatives.
Name BE DS PMF PMF04 Rosmaricine_5 131.739 93.56 31.55 97.921 Rosmaricine_16 120.762 88.38 27.92 96.528 Songbeisine_6 108.648 75.46 16.9 72.679 Rhynchophylline_8 98.931 61.73 30.08 85.757 Scopolamine_32 98.397 61.05 24.68 90.164
BE: binding energy. DS: dock score.
PMF: potential of mean force. N-acetyl-D-glucosamine*: control.
[()TD$FIG]
0 50 100 150 200 250 300 350 400 450 500 -0.4 -0.2 0.0 0.2 0.4 0.6 0.8 1.0 1.2 Score Residues 1RVX H1Fig. 3. Verify-3D result. Majority of protein residues have verify-3D scores greater than 1. Regions where scores have negative values have been investigated and these regions are not close to the binding site residues.
T.-T. Chang et al. / Journal of the Taiwan Institute of Chemical Engineers 42 (2011) 563–571 566
initially interacts with Arg238 via the amine functional group, but this hydrogen bonding is later changed to between the carbonyl group and Arg238. An additional hydrogen bonding is also formed between the hydroxyl group of the glucosamine and Arg238. At end of the simulation run, NAG interaction to Asn104 is also found.
For rosmaricine_5, the binding conformation is largely con-served, as observed from docking diagram and the snapshot from 20 ns. However, significant changes in binding site residues are found (Fig. 4(b) and (e)). More hydrogen bond interactions are observed between rosmaricine_5 and Glu83, with all interactions concentrate between ligand hydroxyl groups and Glu83 carboxyl-ate side chain. Ligand hydrogen bond interactions to Asn104 and Asp103 are also observed; however the interacting locations are
different for the docking pose and for the result obtained from molecular dynamics simulation (Fig. 4(b) and (e)). As for rosmaricine_16, the binding conformation after the simulation run is similar to that from rosmaricine_5 (Fig. 4(c) and (f)). Similar to rosmaricine_5, more hydrogen bonds are formed to Glu83. However, ligand interaction to Asp103 is less predominant due to changes in Asp103 substructure conformation.
To analyze the trajectories, we calculated whole molecule RMSD and ligand RMSD for the hemagglutinin–ligand complexes and the ligands. All ligand–protein complexes reach equilibrium during the simulation run as evidenced in whole molecule RMSD and ligand RMSD (Fig. 5). The energy trajectories show that both derivative-hemagglutinin complexes are much more stable than the control after the simulation run (Fig. 6).
[()TD$FIG]
Fig. 4. The docking poses of the control and the top 3 derivatives in H1: (a) N-acetyl-D-glucosamine, (b) rosmaricine _5 and (c) rosmaricine_16. The snapshots taken at the end of molecular dynamics simulations are (d) N-acetyl-D-glucosamine, (e) rosmaricine _5 and (f) rosmaricine_16.
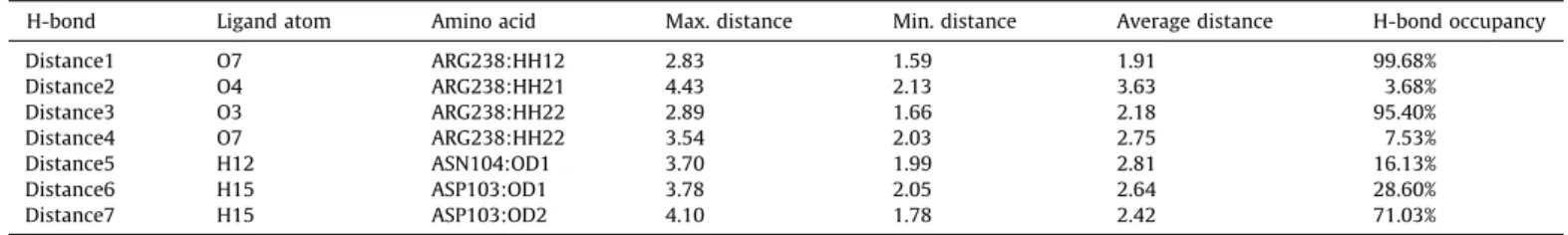
For all the hydrogen bonds observed from docking and molecular dynamics simulations, we summarized the hydrogen bond frequency (Tables 4–6) and computed the hydrogen bond distance trajectories (Figs. 7–9). For control, of all the interactions to Arg238, only two hydrogen bonds are continuously present and are under an average distance of 2.5 A˚ (Table 4). As for interaction to Asp103 and Asn104, all hydrogen bond distances fluctuate. Specifically, the interaction to Asp103 diminishes after 16 ns
Table 5
Hydrogen bond network formed between hemagglutinin and rosmaricine_5.
H-bond Ligand atom Amino acid Max. distance Min. distance Average distance H-bond occupancy
H-bond_1 O1 ARG238:HE 5.82 2.41 3.94 0.05%
H-bond_2 O1 ARG238:HH21 5.16 1.64 2.90 43.03%
H-bond_3 O1 ARG238:HH22 5.10 2.37 3.67 0.13%
H-bond_4 O1 ASN104:HD22 4.70 2.31 3.88 0.05%
H-bond_5 O28 ASN104:HD22 4.31 2.46 3.40 0.03%
H-bond_6 O28 ASN104:HD21 3.07 1.78 2.28 80.65%
H-bond_7 O31 ASN104:HD21 3.78 1.89 2.72 26.00%
H-bond_8 O56 ASN104:HD22 3.44 1.77 2.46 57.15%
H-bond_9 H29 GLU83:OE2 2.60 1.73 1.96 99.98% H-bond_10 H29 GLU83:OE1 4.17 2.12 3.45 1.75% H-bond_11 H32 GLU83:OE1 3.31 1.92 2.69 18.55% H-bond_12 H32 GLU83:OE2 3.85 1.76 2.45 60.55% H-bond_13 H50 ASP103:OD1 3.71 1.60 2.46 62.23% H-bond_14 H50 ASP103:OD2 4.39 1.79 3.11 23.40% H-bond_15 H51 ASP103:OD1 3.73 1.60 2.54 58.63% H-bond_16 H51 ASP103:OD2 4.35 1.80 3.01 33.50% H-bond_17 H52 ASP103:OD1 3.72 1.58 2.36 72.60% H-bond_18 H52 ASP103:OD2 4.24 1.70 2.91 37.68% H-bond_19 H55 GLU83:OE2 4.74 1.81 3.34 12.55% H-bond_20 H68 GLU83:OE2 2.46 1.66 1.93 100.00%
H-bond cutoff distance: 2.5 A˚.
[()TD$FIG]
0.5 1.0 1.5 2.0 2.5 3.0 3.5 0 2 4 6 8 10 12 14 16 18 20 0.0 0.5 1.0 1.5 2.0 2.5 3.0 3.5 4.0 RMSDNAG Xylopine_2 Rosmaricine_14
RMSD
Time (ns)
Fig. 5. The all atoms RMSD for N-acetyl-D-glucosamine (NAG) and top 2 candidates
in H1 complexes during 20 ns simulation (top). The ligand RMSD is shown at bottom.
[()TD$FIG]
0 2 4 6 8 10 12 14 16 18 20 -26000 -25750 -25500 -25250 -25000 -24750 -24500Total energy (kcal/mol)
Time (ns)
NAG Rosmaricine_5 Rosmaricine_16
Fig. 6. The total energy of hemagglutinin in complex with N-acetyl-D-glucosamine (NAG) and top 2 candidates during 20 ns of simulation.
Table 4
Hydrogen bond network formed between hemagglutinin and N-acetyl-D-glucosamine.
H-bond Ligand atom Amino acid Max. distance Min. distance Average distance H-bond occupancy
Distance1 O7 ARG238:HH12 2.83 1.59 1.91 99.68% Distance2 O4 ARG238:HH21 4.43 2.13 3.63 3.68% Distance3 O3 ARG238:HH22 2.89 1.66 2.18 95.40% Distance4 O7 ARG238:HH22 3.54 2.03 2.75 7.53% Distance5 H12 ASN104:OD1 3.70 1.99 2.81 16.13% Distance6 H15 ASP103:OD1 3.78 2.05 2.64 28.60% Distance7 H15 ASP103:OD2 4.10 1.78 2.42 71.03%
H-bond cutoff distance: 2.5 A˚.
T.-T. Chang et al. / Journal of the Taiwan Institute of Chemical Engineers 42 (2011) 563–571 568
(Fig. 7andsupplementary video). For rosmaricine_5, the initial interaction to Arg238 is unstable, and the separating distance could be as large as 5.82 A˚ (Table 5). The most stable interaction is the hydrogen bonding between rosmaricine_5 and Glu83 and Asn104 in which most hydrogen bonding persist throughout the simulation. The distance between Asn104 to rosmaricine_5 was relatively unchanged throughout the simulation (Fig. 8). This is in contrast to interaction to Asp103 in which the distance appears to be periodically changing over simulation time. For rosmari-cine_16, the ligand has very stable interaction to Glu83 and Asn104 (Table 6andFig. 9). However, the interaction to Asp103 is less stable and has large fluctuations in bonding distance
(Fig. 9).
After careful analysis of the docking and molecular dynamics simulation results, we observed that the ligands interact predomi-nantly to Glu83, Asp103 and Asn104. The main interacting functional groups found on the ligands are (1) the protonated primary amine, (2) the added hydroxyl groups and (3) the hydroxyl groups found on the main scaffold (Fig. 10). The protonated amine group interacts with Asp103 in the form of salt bridge interaction while the other hydroxyl groups act as hydrogen bond donors and bind to Glu83 and Asn104. We conclude that these functional domains found on rosmaricine derivatives are the key structures responsible for inhibitory activity. In addition, to inhibit influenza hemagglutinin, stable interactions to Glu83 and Asn104 appear to us as the basic requirements.
Table 6
Hydrogen bond network formed between hemagglutinin and rosmaricine_16.
H-bond Ligand atom Amino acid Max. distance Min. distance Average distance H-bond occupancy
H-bond_1 O31 ASN104:HD22 3.29 1.91 2.46 59.03%
H-bond_2 H29 GLU83:OE2 2.46 1.72 1.99 100.00% H-bond_3 H32 GLU83:OE2 2.41 1.68 1.96 100.00% H-bond_4 H50 ASP103:OD1 3.46 1.59 2.35 68.63% H-bond_5 H51 ASP103:OD1 3.55 1.56 2.44 59.78% H-bond_6 H52 ASP103:OD1 3.50 1.59 2.39 66.43% H-bond_7 H55 GLU83:OE2 2.56 1.71 1.97 99.90%
H-bond cutoff distance: 2.5 A˚.
[()TD$FIG]
Fig. 9. Time dependent hydrogen bond distance between rosmaricine_16 and hemagglutinin residues.
[()TD$FIG]
1.0 1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0 5.5 0 2 4 6 8 10 12 14 16 18 20 1.5 2.0 2.5 3.0 3.5 4.0 Distance O7 / R238:HH12 O4 / R238:HH21 O3 / R238:HH22 O7 / R238:HH22 Distance Time (ns)H12 / N104:OD1 H15 / D103:OD1 H15 / D103:OD2
Fig. 7. Time dependent hydrogen bond distance between N-acetyl-D-glucosamine and hemagglutinin residues: Arg 238, Asn 103 and Asp 104.
[()TD$FIG]
1 2 3 4 5 6 7 1 2 3 4 5 6 1 2 3 4 5 1 2 3 4 5 6 0 2 4 6 8 10 12 14 16 18 20 1 2 3 4 5 Distance O1 / R238:HE O1 / R238:HH21 O1 / R238:HH22 Distance O1 / N104:HD22 O28 / N104:HD22 O28 / N104:HD21 O31 / N104:HD21 O56 / N104:HD22 Distance H29 / E83:OE2 H29 / E83:OE1 H32 / E83:OE1 H32 / E83:OE2 DistanceH50 / D103:OD1 H50 / D103:OD2 H51 / D103:OD1 H51 / D103:OD2 H52 / D103:OD1 H52 / D103:OD2
Distance
Time (ns)
H55 / E83:OE2 H68 / E83:OE2
Fig. 8. Time dependent hydrogen bond distance between rosmaricine_5 and hemagglutinin residues.
4. Conclusion
The wide spread of influenza A (H1N1) virus had overwhelmed the health authorities worldwide with the number of infected cases. Influenza viral surface glycoprotein, hemagglutinin, has an important role in viral replication cycle and therefore, is a very potential therapeutic target. Here, we show two rosmaricine derivatives, rosmaricine_5 and rosmaricine_16, as potent inhibi-tors for blocking hemagglutinin binding to sialic acid residues. Both derivatives share similar binding poses and establish continuous hydrogen bonding to key binding site residues, Asn104 and Glu83. Structural analysis of rosmaricine derivatives suggests a hemagglutinin inhibitor should contain a protonated amine group for interacting with Asp103 and two hydroxyl groups substituted domains for interacting with Glu83 and Asn104. We anticipate our two derivatives can be further developed into next generation of antivirals for treating influenza infection.
Acknowledgements
The research was supported by grants from the National Science Council of Taiwan (NSC 99-2221-E-039-013), China Medical University (CMU98-TCM, CMU99-S-02) and Asia University (CMU98-ASIA-09). This study is also supported in part by Taiwan Department of Health Clinical Trial and Research Center of Excellence (DOH99-TD-B-111-004) and Taiwan Department of Health Cancer Research Center of Excellence (DOH99-TD-C-111-005). We are grateful to the National Center of High-performance Computing for computer time and facilities.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, atdoi:10.1016/j.jtice.2010.11.001.
References
al-Sereiti, M. R., K. M. Abu-Amer, and P. Sen, ‘‘Pharmacology of Rosemary (Rosmar-inus Officinalis Linn.) and Its Therapeutic Potentials,’’ Indian J. Exp. Biol., 37, 124 (1999).
Bhargavi, K., P. K. Chaitanya, D. Ramasree, M. Vasavi, D. K. Murthy, and V. Uma, ‘‘Homology Modeling and Docking Studies of Human Bcl-2l10 Protein,’’ J. Biomol. Struct. Dyn., 28, 379 (2010).
Boido, A., F. Savelli, and F. Sparatore, ‘‘Rosmaricine Derivatives as Potential Antitu-moral Agents,’’ Farmaco, 49, 111 (1994).
Calabrese, V., G. Scapagnini, C. Catalano, F. Dinotta, D. Geraci, and P. Morganti, ‘‘Biochemical Studies of a Natural Antioxidant Isolated from Rosemary and Its Application in Cosmetic Dermatology,’’ Int. J. Tissue React.: Exp. Clin. Aspects, 22, 5 (2000).
Cambria, M. T., D. Di Marino, M. Falconi, S. Garavaglia, and A. Cambria, ‘‘Docking Simulation and Competitive Experiments Validate the Interaction between the 2,5-Xylidine Inhibitor and Rigidoporus Lignosus Laccase,’’ J. Biomol. Struct. Dyn., 27, 501 (2010).
Cao, Z. X. and J. H. Wang, ‘‘A Comparative Study of Two Different Force Fields on Structural and Thermodynamics Character of H1 Peptide Via Molecular Dynamics Simulations,’’ J. Biomol. Struct. Dyn., 27, 651 (2010).
Chang, T. T., H. J. Huang, K. J. Lee, H. W. Yu, H. Y. Chen, F. J. Tsai, M. F. Sun, and C. Y. C. Chen, ‘‘Key Features for Designing Phosphodiesterase-5 Inhibitors,’’ J. Biomol. Struct. Dyn., 28, 309 (2010).
Chen, C. Y. C., ‘‘Virtual Screening and Drug Design for Pde-5 Receptor from Traditional Chinese Medicine Database,’’ J. Biomol. Struct. Dyn., 27, 627 (2010).
Chen, C. Y., Y. H. Chang, D. T. Bau, H. J. Huang, F. J. Tsai, C. H. Tsai, and C. Y. C. Chen, ‘‘Ligand-Based Dual Target Drug Design for H1N1: Swine Flu—a Preliminary First Study,’’ J. Biomol. Struct. Dyn., 27, 171 (2009a).
Chen, H., C. L. Cheung, H. Tai, P. Zhao, J. F. Chan, V. C. Cheng, K. H. Chan, and K. Y. Yuen, ‘‘Oseltamivir-Resistant Influenza A Pandemic (H1N1) 2009 Virus, Hong Kong, China,’’ Emerg. Infect. Dis., 15, 1970 (2009b).
Chen, C. Y., H. J. Huang, F. J. Tsai, and C. Y. C. Chen, ‘‘Drug Design for Influenza A Virus Subtype H1N1,’’ J. Taiwan Inst. Chem. Engrs., 41, 8 (2010).
Cheng, P. K., T. W. Leung, E. C. Ho, P. C. Leung, A. Y. Ng, M. Y. Lai, and W. W. Lim, ‘‘Oseltamivir- and Amantadine-Resistant Influenza Viruses a (H1N1),’’ Emerg. Infect. Dis., 15, 966 (2009).
da Cunha, E. F. F., E. F. Barbosa, A. A. Oliveira, and T. C. Ramalho, ‘‘Molecular Modeling of Mycobacterium Tuberculosis DNA Gyrase and Its Molecular Docking Study with Gatifloxacin Inhibitors,’’ J. Biomol. Struct. Dyn., 27, 619 (2010).
Eswar, N., B. Webb, M. A. Marti-Renom, M. S. Madhusudhan, D. Eramian, M. Y. Shen, U. Pieper, and A. Sali, ‘‘Comparative Protein Structure Modeling Using Modeller,’’ Curr. Protoc. Bioinform., Chapter 5, 6 Unit 5.
Gamblin, S. J., L. F. Haire, R. J. Russell, D. J. Stevens, B. Xiao, Y. Ha, N. Vasisht, D. A. Steinhauer, R. S. Daniels, A. Elliot, D. C. Wiley, and J. J. Skehel, ‘‘The Structure and Receptor Binding Properties of the 1918 Influenza Hemagglutinin,’’ Science, 303, 1838 (2004).
Huang, H. J., K. J. Lee, H. W. Yu, C. Y. Chen, C. H. Hsu, H. Y. Chen, F. J. Tsai, and C. Y. C. Chen, ‘‘Structure-Based and Ligand-Based Drug Design for Her 2 Receptor,’’ J. Biomol. Struct. Dyn., 28, 23 (2010a).
Huang, H. J., K. J. Lee, H. W. Yu, H. Y. Chen, F. J. Tsai, and C. Y. C. Chen, ‘‘A Novel Strategy for Designing the Selective Ppar Agonist by The ‘‘Sum of Activity’’ Model,’’ J. Biomol. Struct. Dyn., 28, 187 (2010b).
Kahlon, A. K., S. Roy, and A. Sharma, ‘‘Molecular Docking Studies to Map the Binding Site of Squalene Synthase Inhibitors on Dehydrosqualene Synthase of Staphylo-coccus Aureus,’’ J. Biomol. Struct. Dyn., 28, 201 (2010).
Koshy, C., M. Parthiban, and R. Sowdhamini, ‘‘100 Ns Molecular Dynamics Simulations to Study Intramolecular Conformational Changes in Bax,’’ J. Biomol. Struct. Dyn., 28, 71 (2010).
Larkin, M. A., G. Blackshields, N. P. Brown, R. Chenna, P. A. McGettigan, H. McWilliam, F. Valentin, I. M. Wallace, A. Wilm, R. Lopez, J. D. Thompson, T. J. Gibson, and D. G. Higgins, ‘‘Clustal W and Clustal X Version 2.0,’’ Bioinformatics, 23, 2947 (2007). Luthy, R., J. U. Bowie, and D. Eisenberg, ‘‘Assessment of Protein Models with
Three-Dimensional Profiles,’’ Nature, 356, 83 (1992).
Nicholls, H., ‘‘Pandemic Influenza: The Inside Story,’’ PLoS Biol., 4, 156 (2006). Ramachandran, G. N., C. Ramakrishnan, and V. Sasisekharan, ‘‘Stereochemistry of
Polypeptide Chain Configurations,’’ J. Mol. Biol., 7, 95 (1963).
[()TD$FIG]
Fig. 10. The three features for designing drugs targeting H1.
T.-T. Chang et al. / Journal of the Taiwan Institute of Chemical Engineers 42 (2011) 563–571 570
Salomon, R. and R. G. Webster, ‘‘The Influenza Virus Enigma,’’ Cell, 136, 402 (2009). Suzuki, Y. and M. Nei, ‘‘Origin and Evolution of Influenza Virus Hemagglutinin Genes,’’
Mol. Biol. Evol., 19, 501 (2002).
Venkatachalam, C. M., X. Jiang, T. Oldfield, and M. Waldman, ‘‘Ligandfit: A Novel Method for the Shape-Directed Rapid Docking of Ligands to Protein Active Sites,’’ J. Mol. Graph. Model., 21, 289 (2003).
Webster, R. G., W. J. Bean, O. T. Gorman, T. M. Chambers, and Y. Kawaoka, ‘‘Evolution and Ecology of Influenza A Viruses,’’ Microbiol. Rev., 56, 152 (1992).
Wharton, S. A., L. J. Calder, R. W. Ruigrok, J. J. Skehel, D. A. Steinhauer, and D. C. Wiley, ‘‘Electron Microscopy of Antibody Complexes of Influenza Virus Haemagglutinin in the Fusion Ph Conformation,’’ EMBO J., 14, 240 (1995).