中國醫藥大學機構典藏 China Medical University Repository, Taiwan:Item 310903500/32574
45
0
0
全文
(2) 中文摘要 先前的研究發現:E2 和 ERα會透過 PI3K-Akt signaling pathway 來抑制 LPS 所活化的 JNK1/2,進而抑制 IκB 降解與 NFκB 進核、阻 止 LPS 誘發 TNFα和 active caspase-3 的表現與心肌細胞凋亡。TLR4 mRNA 在其 3 端之非轉譯區(3’untranslated regions; 3’UTR)通常含 有 AU-rich element(ARE)。ARE 能與許多 RNA 結合蛋白(RNA-binding protein)作用調控 mRNA 穩定性。E2 和 ERα如何影響 TLR4 表現,並 且 LPS 刺激心肌細胞是否會造成 RNA 結合蛋白表現上升並影響 TLR4 表現還未知。因此本實驗利用 LPS 刺激心肌細胞觀察 RNA 結 合蛋白調控 TLR4 mRNA 表現和 E2/ERα減少 TLR4 的調控。 由實驗結果得知,LPS 透過磷酸化 JNK 顯著誘導 TLR4 表現, 增加細胞質中 HuR 的表現以加強 TLR4 mRNA 的穩定性。E2 和 ERα 則會減少細胞質中 HuR 表現量並降低 TLR4 mRNA 穩定性,並減少 TNF-α和 IL-6 在心肌細胞的表現。. I.
(3) Abstract Our previous results indicate that Akt mediates 17β-estradiol and/or estrogen receptor α to inhibit the LPS induced the JNK activity, TNFα protein expression, and exhibit the cardioprotective effects. TLR4 mRNAs often contain AU-rich elements (AREs) in their 3’untranslated regions (3’UTR) which have a high affinity for RNA-binding proteins. It is not known whether E2 and ERα affect the TLR4 mRNA stability and TLR4 protein expression through regulating the RNA-binding proteins, human antigen R (HuR), TTP and AUF-1 in myocardial cells. Therefore, we would like to investigate if the LPS induce these RNA-binding proteins to regulate TLR4 mRNAs of cardiomyocytes, and whether the E2/ERα reduces the TLR4 mRNA stability induced by LPS through the inhibition. of. RNA-binding. protein. expression.. Using. doxycycline(Dox)-induced Tet-On ERα H9c2 myocardic cell model, we want to identify whetherE2 and/or ERαmanipulate the LPS-induced TLR4 mRNA stability. The result of western blotting and RT-PCR assays demonstrated that LPS significantly increased the level of cytoplasmic HuR protein and the stability of TLR4 mRNA, and farther induced the TLR4 protein. II.
(4) expression in H9c2 cells. This effect was mediated through the phosphorylation of intracellular JNK. Interesting, E2 and ERα decreased the cytoplasmic HuR level and TLR4 mRNA stability, and farther decreased the level of HuR protein and IL-6 proteins induced by LPS in H9c2 cardiomyoblast cells.. III.
(5) 致謝 回想起碩士兩年的生活,我的心中充滿無限感謝!感謝 黃志揚 老師願意收我這個完全沒進過實驗室的學生,並且在兩年間給予我指 導與教誨。感謝國防大學 王錫崗教授、成功大學 李碧雪教授、 陳 恵珍老師、 郭薇雯老師在論文的審閱及校正,讓此論文得以順利完 成。 感謝忠榮學長、俊憲學長、宜君學姐、靜慧學姐、美伶學姐、悅 珊學姐、德勇學長、美學學姐、惠勵學姐、靜怡學姐、懿紅學姐、力 仁學長和雅雯指導我這甚麼都不懂的學妹,憶帆、凡妮、千育、志豪、 承訓、俊賢、昆霖、寬嘉,和你們一起當同學真的是最開心的事。實 驗中的不開心,因為你們的陪伴讓我可以繼續堅持。 最後,我要感謝我的家人。雖然我人在台中,是離彰化最近的孩 子。但兩年間我回家的次數可能還少於遠在台北的姐姐。因為你們的 支持,我才能完成我的學業。. IV.
(6) Index 中文摘要………………………………………………………………… 中文摘要…………………………………………………………………I ………………………………………………………………… Abstract ………………………………………………………………II ……………………………………………………………… 致謝 ……………………………………………………………………IV …………………………………………………………………… Index ……………………………………………………………………V …………………………………………………………………… Introduction …………………………………………………………… …………………………………………………………… 1 Aim………………………………………………… …………………………………………………… ……………………………………………………………………… …………………7 …… Materials and Methods ………………………………………………8 ……………………………………………… Materials …………………………………………………………… 8 Methods ……………………………………………………………13 …………………………………………………………… Result ………………………………………………………………… ………………………………………………………………19 ………………………………………………………… Discussion……………………………………………………………… ………………………………………………………………23 ……………………………………………………………… Reference…………………………………………… ……………………………………………………………… ………………………………………………………………27 ………………… Figures………………………………………………………………… …………………………………………………………………32 …………………………………………………………………. V.
(7) Introduction Sepsis is systemic physiological and pathological response to severe inflammation. The inflammatory response to infection or injury is a highly conserved and regulated reaction of the organism[1]. Previous research indicates that endotoxin, such as LPS, is an important pathogen responsible for. cardiovascular. disorders. [2].. Lipopolysaccharide. (LPS). is. high-molecular-weight complexes that are major components of the outer membranes of the cell walls of gram-negative bacteria. LPS is composed of oligosaccharide domain and lipid A domain. Oligosaccharide domains contain two subunits: O-specific side chain and core oligosaccharide. Lipid A, the anchor moiety of LPS, is the active component for its toxic activity to bind to TLR4 [3]. Toll-like receptors (TLRs) are type I transmembrane receptors that expressed on the cell membrane after stimulation. LPS induce TLR4 expression on the surface of cardiacmyocytes plays a role in cardiac dysfunction. Toll-like receptor 4 (TLR4) mediates myocardial dysfunction via NFκB-dependent mechanisms through an increase in TNFα and IL-6 production by themselves. However, TLR4 inhibition reduces the following signal pathway to improve cardiacmyocyte contractility after LPS application [4].. 1.
(8) Epidemiological research shows pre-menopausal women have lower rate of cardiovascular diseases than age-matched men in cardiovascular diseases. However, women after menopause have higher morbidity but estrogen-replacement therapy contributes to a low incidence of heart disease [5]. Steroid estrogen containing estrone (E1)、17β-estradiol (E2)、 estriol (E3). 17β-estradiol is the principle intracellular human estrogen and is more potent than estrone and estriol. It is the primary estrogen secreted prior to menopause. 17β-estradiol modulates cell proliferation and differentiation, development of reproductive system (such as uterus, vagina, breast development, male testis, epididymis, prostate development), maintain the bone density of skeletal system, and protect the cardiac system [6]. 17β-estradiol signaling is transduced though estrogen receptors which are members of the superfamily of steroid/thyroid hormone nuclear receptors [7]. Estrogen receptors (ERs) included six functional domains, termed A to F. The N-terminal, A/B domain is an agonist-independent transcriptional activation function domain-1 (AF-1) which can turn on transcription without estrogen binding. C domain is DNA binding domain (DBD). D domain is the hinge domain, which combine C and E domains. E domain is the ligand-binding domain (LBD) which can encompasses. 2.
(9) both an agonist-dependent transcriptional activation function domain-2 (AF-2) and a dimerization region. F domain is important for modulating transactivation and Protein-Protein interactions [8]. The molecular mechanisms of estrogen signalings can be separated into four pathways. (A) Classical ligand-dependent mechanism: The Class I members of the nuclear. steroid/thyroid. receptor. superfamily. is. ligand-dependent. mechanism of ER. The binding of ligand induces an activating conformational change within the ER and promotes homodimerization and high affinity binding to specific DNA response elements (EREs). Depending on the cell and promoter context, the DNA-bound receptor exerts either a positive or negative effect on expression of the downstream target genes. (B) Ligand-independent mechanism:The effects of elevated intracellular cAMP or polypeptide growth factors such as epidermal growth factor (EGF). and. insulin-like. growth. factor-1. (IGF-1). modify. the. phosphorylation state of the ER by cellular kinases and phosphorylated ER promotes homodimerization and high affinity binding to ERE to enhance their transcriptional activities (C) DNA binding-independent mechanism : E2-ER complexes alter. 3.
(10) transcription of genes containing alternative response elements such as AP-1 through association with other DNA-bound transcription factors. (D) Cell-surface (nongenomic) signaling:E2 activates a putative membrane associated binding protein, possibly a form of ER linked to intracellular signal transduction pathways that generate rapid tissue responses. The roles of coactivators and corepressors in ER signaling are discussed in the first minireview of this series [9]. There are two different forms of the estrogen receptor, ERα and ERβ. Each estrogen receptor type is produced from a separate gene and differs in structure, tissue location, and function [10]. The similar activity of AF2 in ERα and ERβ shows their similar ability to bind coactivators [11]. To compare the activity of AF1 in ERα and ERβ, the two estrogen receptor were examined with different ligand in estrogen receptor element. The ability of ERE or SP-1 binding in ERα is better than ERβ, but ERβ has more affinity to bind to AP-1 [12]. Mitogen activated protein kinases (MAPKs) including extracellular signal regulated kinase (ERK), p38 MAPK and c-jun amino-terminal kinase (JNK) play important roles in cell function. In myocardial cells, ERK1/2 protects cell from apoptosis in ischemia and redox stress [13]. ERKs. 4.
(11) modulate transcription of inflammatory genes by up-regulation of AP-1 components [14]. p38 MAPK is referred to pro-apoptotic and anti-apoptotic actions. Chronic treatment with a p38 MAPK inhibitor, SB239063, prevents left ventricular (LV) hypertrophy and dysfunction in hypertensive rats [15]. Studies show that JNK signaling pathway contributes to the regulation of cell proliferation and apoptosis [16]. JNK reverse cell apoptosis and retard the cell death [17]. Protein expression may operate with multiple mechanisms, such as transcription, mRNA translation, and mRNA degradation. Cytokines and chemokine usually have low expression in non- stimuli cells [18], and increase mRNA level and translation of the mRNA during inflammatory responses. Unstable mRNAs often contain AU-rich elements (AREs) in their 3’untranslated region (UTR). The characteristic motif is AUUUA, but the number of copies of ARE and AU contents are very variable [19]. There are three classes of ARE. Class I contains one to three scattered AUUUA motif nearby U-rich sequence. Class II contains multiple cluster AUUUA copies. Class III ARE does not contain AUUUA motif but mediate mRNA degradation [20, 21]. AREs are functionally separated by their ability to confer instability to. 5.
(12) otherwise stable mRNAs. The regulatory function of AREs is apparently mediated through the RNA-binding proteins that recognize the ARE motif [22, 23], including human antigen R (HuR), AU-binding factor 1 (AUF 1), and the zinc finger protein tristetraprolin (TTP). HuR is one of embryonic lethal abnormal vision (ELAV) family of RNA-binding proteins which shuttle between the nucleus and cytoplasm and predominantly exist in nuclear proteins. HuR binds strongly to AREs and stabilizes mRNA [24]. HuR does not affect deadenylation but delay the commencement of decay of the RNA body and slow down its subsequent decay [25]. AUF1 is a member of the heteronuclear ribonucleoprotein (hnRNP) family which exists in four isoforms (37, 40, 42, and 45 kDa). AUF1 isoforms have different roles of mRNA turnover [25]. Overexpression of AUF1 may destabilize [26] or stabilize AREs [27]. TTP, a member of zinc finger proteins, is critically implicated in inflammation that binds to AREs and destabilizes TNFα mRNAs [28]. It hinders both the deadenylation and decays of the mRNA body [29].. 6.
(13) Aim Our previous results indicate that LPS induces myocardiac cell hypertrophy, apoptosis and fibrosis. We even found that 17β-estradiol and estrogen receptor alpha exhibit their cardioprotective effects by inhibiting JNK1/2-mediated LPS-induced TNF-α expression and cardiomyocyte apoptosis through activation of Akt [30]. Here, we would like to further investigate if the LPS induces the RNA binding protein, human antigen R (HuR), to regulate TLR4 mRNAs, which contain AU-rich element (ARE) in. their. 3’untranslation. regions(3’UTR). in. cardiomyocytes.. Additionally, using doxycycline (Dox)-induced Tet-On ERα H9c2 myocardic cell model, we want to identify whether the E2 and ERα manipulate the LPS-induced TLR4 mRNA stability.. 7.
(14) Materials and Methods Materials A、 、cell culture Dulbecco’s modified Eagle’s medium (no phenol red)(D2292 / Sigma / MO, USA) Sodium bicarbonate;NaHCO3(S-5761 / Sigma / MO, USA) Sodium pyruvate(11360-070 / GIBCO / Auckland, New Zealand) Antibiotic-Antimycotic(15240-062 / GIBCO / Auckland, New Zealand) Cosmic Calf Serum;CCS(SH30087.03 / Hyclone / USA) Trypsin-EDTA(25200-056 / GIBCO / Auckland, New Zealand) PBS(21600-010 / GIBCO / Auckland, New Zealand) DMSO(D2650 / Sigma / MO, USA) 17β-Estradiol(E8518 / Sigma / MO, USA) lipopolysacchride (LPS) (L4516 / Sigma / MO, USA) cyclosporine A (CsA) (C1832 / Sigma / MO, USA) PI3-K inhibitor, LY294002 (1130 /TOCRIS /Ellisville, MO, USA) MEK1/2 inhibitor, U0126 (1144 /TOCRIS /Ellisville, MO, USA) p38 MAPK inhibitor, SB203580 (1201 /TOCRIS /Ellisville, MO, USA) JNK inhibitor, SP600125 (1496 /TOCRIS /Ellisville, MO, USA) G418 (345810 / CALBIOCHEM / Darmstadt, Germany) Hygromycin B(400051 / CALBIOCHEM / Darmstadt, Germany) Doxycycline(8634-1 / Clontech/ CA, USA) B、 、measure of protein concentration BSA-Bovine serum albumin(A-7906 / Sigma / MO, USA) Coomassie billiant blue G-250 (Bio-Rad) 8.
(15) C、 、Western blotting/ Immunoblotting (1) )Cell lysis buffer Tris-base(T8600 / USB / OH, USA) Sodium chloride;NaCl(S7653 / Sigma / MO,USA) Ethylenediaminetetraacetic acid;EDTA(E5134 / Sigma / MO,USA) beta-mercaptoethanol(17-1317-01 / Pharmacia Biotech / Upsala, Sweden) NP40(I3021 / Sigma / MO, USA) Glycerol(0854 / Amresco / OH, USA) Protease inhibitor cocktail tablets(11836153001 / Roche / Mannheim, Germany) (2) )Western Blot buffer 40% Acrylamide/Bis solution 29:1(702159 / MD Bio / Taiwan) Sulution B: Tris-base(pH8.8)(75825 / USB / OH, USA) Sodium dodecyl sulfate;SDS(UN1325 / MERCK / Darmstadt, Germany) Sulution C: Tris-base(pH6.8)(75825 / USB / OH, USA) Sodium dodecyl sulfate;SDS(UN1325 / MERCK / Darmstadt, Germany) Ammonium persulfate;APS(17-1311-01 / Pharmacia Biotech / Upsala, Sweden) TEMED(T9281 / Sigma / MO, USA) Glycine(16407 / USB / OH, USA). 9.
(16) Glycerol(0854 / Amresco / OH, USA ) Mehanol 20L(M & J SCINTEK CO.,Taichung, Taiwan) Sodium chloride;NaCl(0241 / Amresco / OH, USA) Ponceau S solution(P7170 / Sigma / MO, USA) Western blotting luminal reagent Solution A、B(Sc-2048 / Santa Cruz Biotechnology / CA, USA) Blocking buffer(Anchor New Zealand Milk / New Zealand) (3) )5X loading dye: : Bromophenol Blue(B5525 / Sigma / MO, USA) beta-mercaptoethanol(17-1317-01 / Pharmacia Biotech / Upsala, Sweden) Sodium dodecyl sulfate;SDS(UN1325 / MERCK / Darmstadt, Germany) Glycerol(0854 / Amresco / OH, USA ) Tris-HCl(T8650 / USB / OH, USA) (4) )Antibody Antibody. Molecular. Company. Product number. weight α-Tubulin. 54. sc-5286. AUF-1. 37. ARP40230. HuR. 36. Sc-20694. Santa Cruz Biotechnology, Inc. Santa Cruz, California, USA. HDAC5. 140-150. Sc-11419. Santa Cruz Biotechnology, Inc. Santa Cruz, California, USA. IL-6. 25. Sc-1266. Santa Cruz Biotechnology, Inc. Santa Cruz, California, USA. TLR4. 89. sc-16240. Santa Cruz Biotechnology, Inc. Santa Cruz, California, USA. TNF-α. 26/17. sc-1350. Santa Cruz Biotechnology, Inc. Santa Cruz, California, USA. TTP. 44. sc-14030. Santa Cruz Biotechnology, Inc. Santa Cruz, California, USA. Santa Cruz Biotechnology, Inc. Santa Cruz, California, USA Aviva Systems Biology, San Diego,USA. 10.
(17) Goat anti-rabbit IgG antibody. Santa Cruz Biotechnology, Inc. Santa Cruz, California, USA. conjugated-HRP Goat anti-rabbit IgG antibody. Santa Cruz Biotechnology, Inc. Santa Cruz, California, USA. conjugated-HRP Goat anti-rabbit IgG antibody. Santa Cruz Biotechnology, Inc. Santa Cruz, California, USA. conjugated-HRP. (5) )TBS buffer: : Sodium Chloride;NaCl (0241 / Amresco / OH, USA) Tris(75825 / USB / OH, USA) Polyoxyethylenesorbitan monolaurate;Tween 20(63158 / Riedel-de Haen / Seeize, Germany) PVDF membrane pore size 0.45µm ( IPVH00010 / Millipore Corporation / MA, USA) D、 、Reverse Transcription-Polymerase Chain Reaction( (RT-PCR) ) (1) )R)A extract Ultraspec RNA reagent(BL-10500 / Biotecx / HO, USA) Chloroform(C2432 / Sigma / MO, USA) Isopropanol(I9516 / Sigma / MO, USA) Diethyl pyrocabonate;DEPC(D5758 / Sigma / MO, USA) (2) )RT-PCR and PCR MMLV Reverse transcriptase(M170B / Promega / WI, USA) MMLV Reverse 5×buffer(M531A / Promega / WI, USA) Recombinant RNasin Ribonuclease inhibitor(N251A / Promega / WI, 11.
(18) USA) dATP dCTP dTTP dGCP(U120A,U122A,U123A,U121A / Promega / WI, USA) Oligo-dT(MISSION BIOTECH / Taipei, Taiwan) DNA polymerase;Taq(611069 / MD Bio / Taiwan) 10×PCR buffer(708059 / MD Bio / Taiwan) (3) )D)A electrophoresis 5×TBE Buffer(MDTBE5 / MD Bio / Taiwan) Agarose(U3125 / Promega / WI, USA) Bio-100bp DNA ladder(M1-100T / PROTECH / Taipei, Taiwan) E、 、Immunofluorescence assay Paraformaldehyde(P6148 / Sigma / MO, USA) PBS(21600-010 / GIBCO / Auckland, New Zealand) Triton X-100(TR-1875 / TEDIA / Ohio, USA) Sodium citrate(S-4641 / Sigma / MO, USA) DAPI(D9564 / Sigma / MO, USA) F、 、)uclear Extraction HEPES(H3375 / Sigma / MO, USA) Sodium chloride;NaCl(S7653 / Sigma / MO,USA) Potassium chloride;KCL(P5405 / Sigma / MO,USA) Dithiothreitol;DTT(D9779 / Sigma / MO,USA) NP40(I3021 / Sigma / MO, USA) Glycerol(0854 / Amresco / OH, USA) Protease inhibitor cocktail tablets(11836153001 / Roche / Mannheim, Germany). 12.
(19) Methods Cell culture Heart-derived H9c2 myocardial cells were obtained from American Type Culture Collection (ATCC). H9c2 cells and Tet-On/ERα H9c2 cells were cultured in Dulbecco’s modified essential medium (DMEM) supplemented with 10% fetal bovine serum, 100 units/ml penicillin, 100 µg /ml streptomycin in 5% CO2 humidified air at 37◦C. Replace medium 2~3 times a week. H9c2 cells were cultured in serum-free medium with minimal essential medium for 12 h for drug treatment. The incubation was continued for 24 h, and then the cells were harvested and extracted for the analysis.. Total R)A Extraction Cells were lysed directly in a culture dish by adding the Ultraspec RNA and passing the cell lysate several times through a pipette. The cell lysate should be transferred immediately into centrifuge tubes. Store the cell for 5 minutes at 4◦C to permit the complete dissociation of nucleoprotein complexes. Next, add 0.2 ml of chloroform per 1 ml of Ultraspec RNA, cover the samples tightly, shake vigorously for 15 seconds and place on ice. 13.
(20) at 4◦C for 5 minutes. Centrifuge the homogenate at 12,000 g (4◦C) for 15 minutes. Carefully transfer the aqueous phase (40~50%) to a fresh tube while taking care not to disturb the interphase. Add equal volume of isopropanol and store samples for 10 minutes at 4◦ C. Centrifuge samples at 12,000g (4◦C) for 10 minutes. Remove the supernatant and wash RNA pellet twice with 75% ethanol by vortexing and subsequent centrifugation for 5 minutes at 7,500g (4◦C). Dissolve the RNA pellet in 50-100 ul of DEPC treated water or in an appropriate buffer by vortexing for 1 minute (An incubation for 10-15 minutes at 55-60◦C may be required to dissolve preparations of RNA). The RNA was quantified and checked for purity and condition by spectrophotometry at a wavelength of 260 nm.. RNA. concentration(µg/ml)=Dilution factor × RNA (OD260)×(40µg/ml/ OD260). Reverse Transcription (RT) and Polymerase Chain Reaction Amplification Reverse Transcription Reaction: : 59.5 µl DEPC-H2O containing 8 µg RNA add 0.5 µl RNase inhibitor (Promega;40 U/µl), 20 µl 5X RT buffer (Promega;50 mM Tris-HCl, 75 mM KCl, 3 mM MgCl2 and 10 mM DTT), 8 µl dNTP (Promega;2.5. 14.
(21) mM), and 10 µl Oligo dT(10 mM). reaction was initiated at 70◦C for 5 min, adding 2 µl RTase( Promega) after 5 min. The samples were then at 42◦C for 1 hr, 95◦C for 5 min, and store at 4◦C.. Polymerase Chain Reaction: : 5 µl RT product was diluted with the PCR buffer (50 mM KCl,10 mM Tris–HCl and 2 mM MgCl2), adding 0.5 µM dNTPs (final concentration, 0.8 mM) and 0.5 U of Taq DNA polymerase to a final volume of 50 µl. PCR was initiated with a hot start (5 min. at 95◦C); the samples were then subjected to 32 cycles at 95◦C for 1 min, annealing temperature for 1 min, and 72◦C for 2 min. The annealing temperature for the TLR4, and TNFα primers was 58◦C; GAPDH primers was 55◦C. This was followed by a final extension step at 72◦C for 20 min, and store at 4◦C. Primers were as follows: rat GAPDH forward primer:GGGTGTGAACCACGAGAAAT, reverse primer : CACAGTC TTCTGAGTGGCA; rat TNF-α forward primer:CCTCTTCTCATTCCTGCTCG, reverse primer:GGTATGAA ATGGCAAATCGG; TLR4 forward primer : CATGGCATTGTTCCTTTCCT, reverse primer:CATGGAGCCTAATTCCCTGA. DNA marker and 10 µl product adding 2 µl 6X loading dye was assessed. 15.
(22) by 1.5% agarose gel electrophoresis and DNA was visualized by ethidium bromide staining.. )uclear Extraction Cells were re-suspended with 200 µl ice-cold buffer-I (10 mM Hepes, pH 8.0; 1.5 mM MgCl2; 10 mM KCl; 1 mM dithiothreitol and proteinase inhibitor cocktail [Roche]) and incubated for 15 min on ice to allow cells to swell, followed by adding 20 µl IGEPAL-CA630. Centrifuge the homogenate at 14,000 g at 4◦C for 15 minutes. The cytoplasmic fraction was carefully aspirated. The pellet was re-suspended with ice-cold BUFFER-II (20 mM Hepes, pH 8.0; 1.5 mM MgCl2; 25% glycerol; 420 mM NaCl; 0.2 mM EDTA; 1 mM dithiothreitol and proteinase inhibitor cocktail [Roche]) and vigorously vortexed. Centrifuge the homogenate at 14,000 g at 4◦C for 15 minutes. Collect the supernatants (nuclear extracts). Cytoplasmic fraction and nuclear extracts were stored at -80◦C. The protein concentration was determined by the colorimetric assay (Bio-Rad).. Western blotting Cells were re-suspended in lysis buffer (50 mM Tris (pH 7.5); 0.5 M NaCl;. 16.
(23) 1.0 mM EDTA (pH 7.5); 10% glycerol; 1 mM BME; 1% IGEPAL-630 and a proteinase inhibitor cocktail [Roche])). Samples containing equal protein (40 µg) were separated by 12% SDS-PAGE and transferred onto polyvinylidene difluoride membranes (Millipore, Belford, MA). The membranes were incubated with the blocking buffer in a shaker for 1 h and then incubated overnight with first antibodies (dilution, 1:1,000) at 4◦C overnight. The membranes were washed with TBS three times and then incubated with Horseradish peroxidase conjugated secondary antibody (Santa Cruz). The membranes were washed with TBS three times. The proteins of interest were visualized by using the substrate buffer and were detected by exposure the autoradiograph to X-ray films.. Immunofluorescence H9c2 cells subjected to various treatments were subsequently fixed with 4% paraformaldehyde at room temperature for 30 min. Cells were permeabilized with 0.5% Triton X-100 for 10 min at 4◦C. The fixed cells was blocked with PBS containing 2% bovine serum albumin at 37◦C for 30 min, following incubation with DAPI and primary HuR antibody overnight at 4. ◦. C. After washing, cells were incubated with antirabbit. 17.
(24) FITC-conjugated antibody at 37 ◦ C for 1 hr. The fluorescence was visualized using a fluorescence microscope coupled with an image analysis system.. Statistical analysis All experiments were repeated at least three times using independent culture preparations. Values are shown as mean_SD. Student's t test was used to calculate the statistical significance of the experimental results for two groups; a P value of <0.05 was considered significant.. 18.
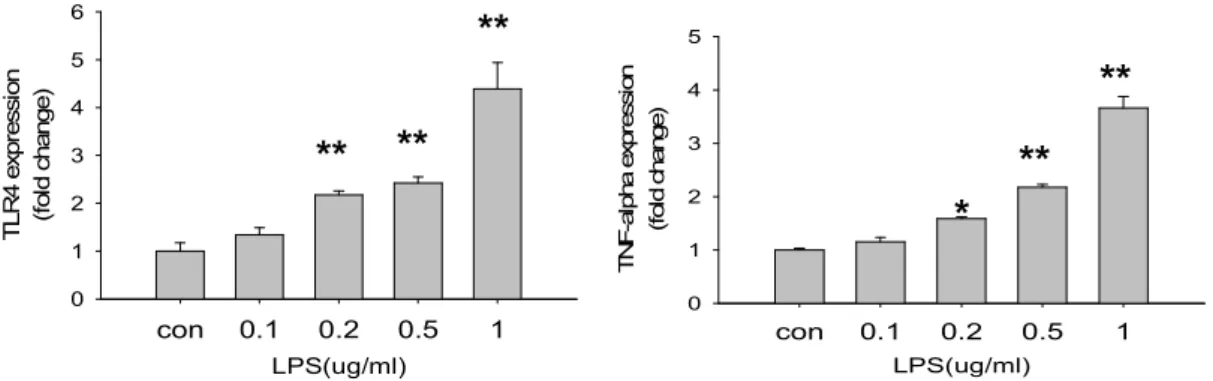
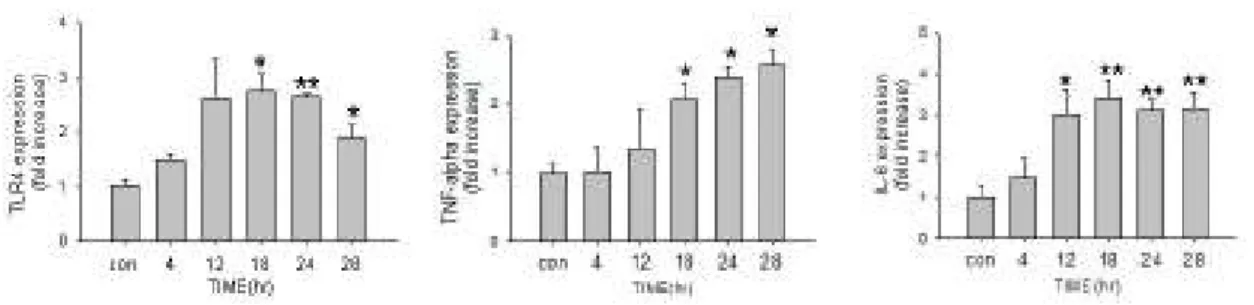
(25) Results LPS dose-dependently up-regulates TLR4 mR)A transcription and TLR4 protein expression. To investigate whether LPS can regulate TLR4 expression in myocardial cells, Tet-On/ERα H9c2 myocardial cells were pretreated for 24 h with 0–1.0 µg/ml of LPS, and TLR4 mRNA and protein were analyzed by semi-quantitative PCR and western blot analysis. Total RNA and protein were then extracted from the H9c2 cells. Figure 1A shows that LPS dose-dependently up-regulate TLR4 mRNA expression, elevated TLR4 mRNA at dose 1.0 µg/ml to a maximum. Similarly, LPS pretreatment significantly induced TLR4 protein expression in a dose dependent manner (Figure 1B).. LPS mediated TLR4 mR)A and protein expression in H9c2 cardiomyoblast cells in a time-depend manner. H9c2 cadiomyoblast cells were treated with vehicle or LPS (1 µg/ml) for 0, 4, 12, 24, and 28 hrs. Total RNA was extracted and analyzed by reverse transcription PCR. LPS significantly induced TLR4 and TNFα mRNA expressions. The expression of TLR4 and TNFα mRNA was reached a. 19.
(26) maximum at 24 hours after stimulation with LPS (Figure 2A). LPS also induced TLR4 and TNFα proteins after treatment for 12 hours (Figure 2B). The addition of actinomycin D (an RNA polymerase inhibitor) significantly reduced TLR4 expression in H9c2 cells treated with LPS (Figure 2C), suggesting that LPS affects TLR4 mRNA expression at transcription level.. J)K1/2 mediate LPS-induced TLR4 expression in Myocardiac cells We assessed the suppressive effects of inhibitors of U0126, SB203580, SP600125, CsA or Ly294002 on LPS-induced TLR4 expression in myocardial cells. H9c2 myocardiac cells were pretreated with vehicle, U0126 (ERK1/2 inhibitor, 1μM), SB203580 (p38 MAPK inhibitor, 1μ M), SP600125 (JNK1/2 inhibitor, 1μM), CsA (calcineurin inhibitor, 1μM) or Ly294002 (MEK inhibitor, 1μM) for 1 hour prior to the administration of LPS (1 μg/ml) for 24h, and subsequently subjected to immunoblotting assay. LPS significantly induced TLR4 expression. Pre-treatment of the JNK1/2 inhibitor SP600125 significantly inhibited LPS-induced TLR4 expression. The result suggests that JNK1/2 may mediate LPS-induced TLR4 expression in myocardial cells (Figure 3).. 20.
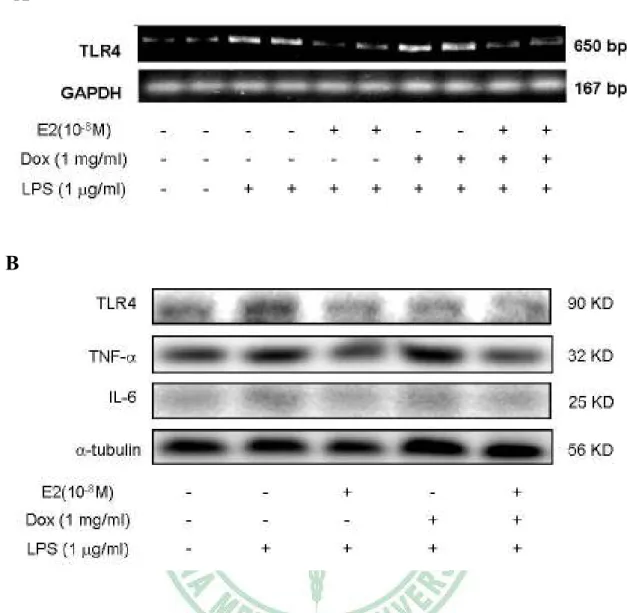
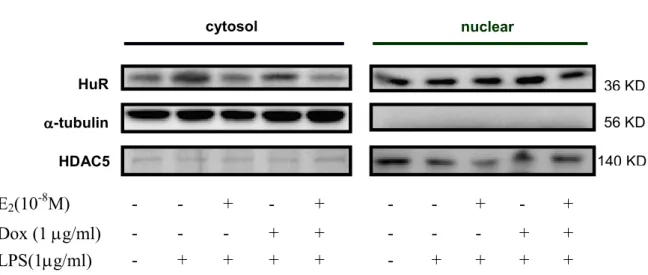
(27) 17β-estradiol and over-expressed ERα α inhibits TLR4 expression in LPS-treated H9c2 cadiomyoblast cells.. To examine whether 17β-estradiol and over-expressed ERα can inhibit the LPS-induced TLR4 expression, Tet-On/ERα H9c2 cells were treated with E2 (10-8 M), Dox (1µg/ml), which induces ERα over-expression, and E2 (10-8 M) plus Dox (1 µg/ml) for 1 hour before LPS treatment, E2 and E2 plus ERα over-expression but not ERα over-expression alone significantly inhibited LPS induced TLR4 mRNA expression (Figure 4A). Interestingly, pretreatment with E2, ERα over-expression, or both reduced TLR4 protein induced by LPS (Figure 4B).. LPS triggers a distinct increase in cytoplasmic HuR, which is significantly inhibited by E2, ERα α or both. H9c2 cadiomyoblast cells were treated with vehicle or LPS (1 µg/ml) for 0, 4, 12, 24, and 28 hrs. HuR was predominantly in the nucleus in un-treatment H9c2 cells. LPS time-dependently caused a significant accumulation of cytoplasmic HuR (Figure 5A). Pretreatment of Tet-On/ERα H9c2 cells with E2 (10-8 M), Dox (1µg/ml), and E2 (10-8 M). 21.
(28) plus Dox (1 µg/ml) for 1 hour before LPS treatment for 24 hours, E2, ERα over-expression, and E2 plus ERα over-expression reduce HuR protein expression. In contrast, the level of AUF1 and TTP protein was remained unchanged following LPS, E2 and/or Dox treatment (Figure 5B). In western blot analysis, LPS significantly increase the cytoplasmic level of HuR protein but E2, ERα over-expression, and E2 plus ERα over-expression significantly inhibited LPS enhanced cytoplasmic HuR expression, and the level of nuclear HuR did not decrease concomitantly with the increase in cytoplasmic HuR(Figure 5C).. 22.
(29) Discussion In this study, we found that MAPK signaling pathways play critical roles in LPS enhanced TLR4 expression and stimulates an inflammatory response in H9c2 cardiomyoblast cells. LPS enhance MAPK signaling transduction and activate HuR production to transcriptionally promote affect TLR4 gene expression. LPS from gram-negative bacteria is considered to be a strong stimulator of the pathogenesis of cardiac disease [2]. Evidence suggests that plasma concentrations of LPS raises in patients with chronic heart failure (CHF) and activated immune system [31]. The present study shows that cardiomyocytes exhibit lower expression of TLR4 under basal conditions. Low concentrations of LPS may contribute to the increased severity of ischemic heart disease [32]. Furthermore, studies have demonstrated that LPS directly decreases contractility [33] and dramatically induces TNF-α expression in cardiomyocytes through binding to TLR4 [34]. Clinical studies have shown that severe cardiac contractile dysfunction is the common symptoms in patients with sepsis [35]. The existence of TLR4 exists in myocardial cells may be a fundamentally significant contribution for the crucial pathological relationship between. 23.
(30) inflammation and cardiovascular disorders [36]. TLR4 expression under LPS stimulation is controlled by transcriptional and posttranscriptional mechanisms, which may be enhanced by MAPK signaling pathways. The Mitogen-Activated Protein Kinase (MAPK) signaling pathways play an important role in signal transduction in eukaryotic cells that transduce signals following growth or stress stimulation. MAPK protein including p38, JNK/SAPK, and ERK1/2 are associated with inflammatory stimuli and oxidative stress. The activation of MAPK has been showed to participate in cardiac pathologies [37]. Myocardiac were pretreated with vehicle, U0126 (ERK1/2 inhibitor, 1μM), SB203580 (p38 MAPK inhibitor, 1μM), SP600125 (JNK1/2 inhibitor, 1μM), CsA (calcineurin inhibitor, 1μM) or Ly294002 (MEK inhibitor, 1μM) for 1h and followed by LPS (1 μg/ml) administration for 24h, we found that incubation of LPS treated myocardial cells with JNK1/2 inhibitor SP600125 resulted in significant inhibition of LPS-induced TLR4 protein expression, suggesting that JNK1/2 may be the key mediator when LPS bind to TLR4. Studies have shown that women have lower mortality rate of sepsis or its related multi-organ disease than men [38]. Women have lower myocardial inflammatory responses, lower levels of cytokine production and better. 24.
(31) myocardial function after burn trauma injury [39]. In addition, clinical research shows that premenopausal women have lower TNFα product compared with men or postmenopausal women. In the present study, we observed that administration of E2, and/or Dox, which induces ERα over-expression, significantly provided cardioprotective effects by repressing. LPS-induced. TLR4. expression. and. down-regulated. proinflammatory TNFα production. Actinomycin D is a RNA polymerase inhibitor which prevents the transcription to form new mRNA. Our results show that LPS-induced TLR4 expression was blocked by actinomycin D, suggesting that the regulation of TLR4 expression by LPS might be mediated at transcriptional level. The basal expression of proteins associated with inflammatory responses is potentially unstable in normal cells, possibly because of the degradation of mRNA is facile. Unstable mRNAs often contain AU-rich elements (AREs) in their 3′ untranslated regions (UTR) [40]. HuR binds strongly to AREs and stabilized mRNA. LPS markedly increased the cytoplasmic level of HuR. Pretreatment of Tet-On/ERα H9c2 cells with E2 (10-8 M), Dox (1µg/ml), and E2 (10-8 M) plus Dox (1 µg/ml) for 1 hour before LPS, E2, ERα over-expression, and E2 plus ERα over-expression. 25.
(32) reduce HuR protein expression and reverse LPS-induced translocation of HuR to cytoplasm. However, the nuclear level of HuR was abundant and did not change at the same time. In conclusion, we found that LPS-enhanced TLR4 mRNA is mediated by HuR expression and up-regulates the expression of TLR4 through JNK1/2 pathway in myocardial cells. In addition, using Dox-induced Tet-On ERα H9c2 myocardial cells, E2 and ERα significantly abolish the LPS-induced cytoplasmic HuR expression to reduce TLR4 mRNA stability.. 26.
(33) Reference 1.. 2.. 3. 4.. 5.. 6.. 7. 8.. 9.. 10.. Bone, R.C., C.J. Grodzin, and R.A. Balk, Sepsis: a new hypothesis for pathogenesis of the disease process. Chest, 1997. 112(1): p. 235-243. Kiechl, S., G. Egger, M. Mayr, C.J. Wiedermann, E. Bonora, F. Oberhollenzer, M. Muggeo, Q. Xu, G. Wick, W. Poewe, and J. Willeit, Chronic infections and the risk of carotid atherosclerosis: prospective results from a large population study. Circulation, 2001. 103(8): p. 1064-1070. Raetz, C.R. and C. Whitfield, Lipopolysaccharide endotoxins. Annu Rev Biochem, 2002. 71: p. 635-700. Baumgarten, G., P. Knuefermann, G. Schuhmacher, V. Vervolgyi, J. von Rappard, U. Dreiner, K. Fink, C. Djoufack, A. Hoeft, C. Grohe, A.A. Knowlton, and R. Meyer, Toll-like receptor 4, nitric oxide, and myocardial depression in endotoxemia. Shock, 2006. 25(1): p. 43-49. Baker, L., K.K. Meldrum, M. Wang, R. Sankula, R. Vanam, A. Raiesdana, B. Tsai, K. Hile, J.W. Brown, and D.R. Meldrum, The role of estrogen in cardiovascular disease. J Surg Res, 2003. 115(2): p. 325-344. Mendelsohn, M.E. and R.H. Karas, The protective effects of estrogen on the cardiovascular system. N Engl J Med, 1999. 340(23): p. 1801-1811. Olefsky, J.M., !uclear receptor minireview series. J Biol Chem, 2001. 276(40): p. 36863-36864. Koide, A., C. Zhao, M. Naganuma, J. Abrams, S. Deighton-Collins, D.F. Skafar, and S. Koide, Identification of regions within the F domain of the human estrogen receptor alpha that are important for modulating transactivation and protein-protein interactions. Mol Endocrinol, 2007. 21(4): p. 829-842. Hall, J.M., J.F. Couse, and K.S. Korach, The multifaceted mechanisms of estradiol and estrogen receptor signaling. J Biol Chem, 2001. 276(40): p. 36869-36872. Couse, J.F., J. Lindzey, K. Grandien, J.A. Gustafsson, and K.S. Korach, Tissue distribution and quantitative analysis of estrogen receptor-alpha (ERalpha) and estrogen receptor-beta (ERbeta) messenger ribonucleic acid in the wild-type and ERalpha-knockout 27.
(34) 11.. 12.. 13.. 14.. 15.. 16. 17.. 18.. 19.. mouse. Endocrinology, 1997. 138(11): p. 4613-4621. Cowley, S.M. and M.G. Parker, A comparison of transcriptional activation by ER alpha and ER beta. J Steroid Biochem Mol Biol, 1999. 69(1-6): p. 165-175. Paech, K., P. Webb, G.G. Kuiper, S. Nilsson, J. Gustafsson, P.J. Kushner, and T.S. Scanlan, Differential ligand activation of estrogen receptors ERalpha and ERbeta at AP1 sites. Science, 1997. 277(5331): p. 1508-1510. Yue, T.L., C. Wang, J.L. Gu, X.L. Ma, S. Kumar, J.C. Lee, G.Z. Feuerstein, H. Thomas, B. Maleeff, and E.H. Ohlstein, Inhibition of extracellular signal-regulated kinase enhances Ischemia/Reoxygenation-induced apoptosis in cultured cardiac myocytes and exaggerates reperfusion injury in isolated perfused heart. Circ Res, 2000. 86(6): p. 692-699. Weber, N.C., J. Stursberg, N.M. Wirthle, O. Toma, W. Schlack, and B. Preckel, Xenon preconditioning differently regulates p44/42 MAPK (ERK 1/2) and p46/54 MAPK (J!K 1/2 and 3) in vivo. Br J Anaesth, 2006. 97(3): p. 298-306. Behr, T.M., S.S. Nerurkar, A.H. Nelson, R.W. Coatney, T.N. Woods, A. Sulpizio, S. Chandra, D.P. Brooks, S. Kumar, J.C. Lee, E.H. Ohlstein, C.E. Angermann, J.L. Adams, J. Sisko, J.D. Sackner-Bernstein, and R.N. Willette, Hypertensive end-organ damage and premature mortality are p38 mitogen-activated protein kinase-dependent in a rat model of cardiac hypertrophy and dysfunction. Circulation, 2001. 104(11): p. 1292-1298. Davis, R.J., Signal transduction by the J!K group of MAP kinases. Cell, 2000. 103(2): p. 239-252. Andreka, P., J. Zang, C. Dougherty, T.I. Slepak, K.A. Webster, and N.H. Bishopric, Cytoprotection by Jun kinase during nitric oxide-induced cardiac myocyte apoptosis. Circ Res, 2001. 88(3): p. 305-312. Bakheet, T., M. Frevel, B.R. Williams, W. Greer, and K.S. Khabar, ARED: human AU-rich element-containing mR!A database reveals an unexpectedly diverse functional repertoire of encoded proteins. Nucleic Acids Res, 2001. 29(1): p. 246-254. Kracht, M. and J. Saklatvala, Transcriptional and post-transcriptional control of gene expression in inflammation. Cytokine, 2002. 20(3): p. 91-106. 28.
(35) 20.. 21.. 22. 23.. 24.. 25.. 26.. 27.. 28.. 29.. Xu, N., C.Y. Chen, and A.B. Shyu, Modulation of the fate of cytoplasmic mR!A by AU-rich elements: key sequence features controlling mR!A deadenylation and decay. Mol Cell Biol, 1997. 17(8): p. 4611-4621. Shyu, A.B., J.G. Belasco, and M.E. Greenberg, Two distinct destabilizing elements in the c-fos message trigger deadenylation as a first step in rapid mR!A decay. Genes Dev, 1991. 5(2): p. 221-231. Brennan, C.M. and J.A. Steitz, HuR and mR!A stability. Cell Mol Life Sci, 2001. 58(2): p. 266-277. Carrick, D.M., W.S. Lai, and P.J. Blackshear, The tandem CCCH zinc finger protein tristetraprolin and its relevance to cytokine mR!A turnover and arthritis. Arthritis Res Ther, 2004. 6(6): p. 248-264. Lin, F.Y., Y.H. Chen, Y.W. Lin, J.S. Tsai, J.W. Chen, H.J. Wang, Y.L. Chen, C.Y. Li, and S.J. Lin, The role of human antigen R, an R!A-binding protein, in mediating the stabilization of toll-like receptor 4 mR!A induced by endotoxin: a novel mechanism involved in vascular inflammation. Arterioscler Thromb Vasc Biol, 2006. 26(12): p. 2622-2629. Peng, S.S., C.Y. Chen, N. Xu, and A.B. Shyu, R!A stabilization by the AU-rich element binding protein, HuR, an ELAV protein. Embo J, 1998. 17(12): p. 3461-3470. Loflin, P., C.Y. Chen, and A.B. Shyu, Unraveling a cytoplasmic role for hnR!P D in the in vivo mR!A destabilization directed by the AU-rich element. Genes Dev, 1999. 13(14): p. 1884-1897. Xu, N., C.Y. Chen, and A.B. Shyu, Versatile role for hnR!P D isoforms in the differential regulation of cytoplasmic mR!A turnover. Mol Cell Biol, 2001. 21(20): p. 6960-6971. Lai, W.S., E. Carballo, J.R. Strum, E.A. Kennington, R.S. Phillips, and P.J. Blackshear, Evidence that tristetraprolin binds to AU-rich elements and promotes the deadenylation and destabilization of tumor necrosis factor alpha mR!A. Mol Cell Biol, 1999. 19(6): p. 4311-4323. Mahtani, K.R., M. Brook, J.L. Dean, G. Sully, J. Saklatvala, and A.R. Clark, Mitogen-activated protein kinase p38 controls the expression and posttranslational modification of tristetraprolin, a regulator of tumor necrosis factor alpha mR!A stability. Mol Cell Biol, 2001. 29.
(36) 30.. 31.. 32.. 33. 34.. 35.. 36.. 37.. 38.. 39.. 21(19): p. 6461-6469. Liu, C.J., J.F. Lo, C.H. Kuo, C.H. Chu, L.M. Chen, F.J. Tsai, C.H. Tsai, B.S. Tzang, W.W. Kuo, and C.Y. Huang, Akt Mediates 17beta-Estradiol and/or Estrogen Receptor alpha Inhibition of LPS-Induced Tumor !ecrosis Factor-alpha Expression and Myocardial Cell Apoptosis by Suppressing the J!K1/2-!FkappaB Pathway. J Cell Mol Med, 2009. Niebauer, J., H.D. Volk, M. Kemp, M. Dominguez, R.R. Schumann, M. Rauchhaus, P.A. Poole-Wilson, A.J. Coats, and S.D. Anker, Endotoxin and immune activation in chronic heart failure: a prospective cohort study. Lancet, 1999. 353(9167): p. 1838-1842. Liu, S.F., R. Newton, T.W. Evans, and P.J. Barnes, Differential regulation of cyclo-oxygenase-1 and cyclo-oxygenase-2 gene expression by lipopolysaccharide treatment in vivo in the rat. Clin Sci (Lond), 1996. 90(4): p. 301-306. Astiz, M.E. and E.C. Rackow, Septic shock. Lancet, 1998. 351(9114): p. 1501-1505. Auphan, N., J.A. DiDonato, C. Rosette, A. Helmberg, and M. Karin, Immunosuppression by glucocorticoids: inhibition of !F-kappa B activity through induction of I kappa B synthesis. Science, 1995. 270(5234): p. 286-290. Barath, P., M.C. Fishbein, J. Cao, J. Berenson, R.H. Helfant, and J.S. Forrester, Detection and localization of tumor necrosis factor in human atheroma. Am J Cardiol, 1990. 65(5): p. 297-302. Yang, X., D. Coriolan, V. Murthy, K. Schultz, D.T. Golenbock, and D. Beasley, Proinflammatory phenotype of vascular smooth muscle cells: role of efficient Toll-like receptor 4 signaling. Am J Physiol Heart Circ Physiol, 2005. 289(3): p. H1069-1076. Baines, C.P. and J.D. Molkentin, STRESS signaling pathways that modulate cardiac myocyte apoptosis. J Mol Cell Cardiol, 2005. 38(1): p. 47-62. Schroder, J., V. Kahlke, M. Book, and F. Stuber, Gender differences in sepsis: genetically determined? Shock, 2000. 14(3): p. 307-310; discussion 310-303. Horton, J.W., D.J. White, and D.L. Maass, Gender-related differences in myocardial inflammatory and contractile responses to major burn trauma. Am J Physiol Heart Circ Physiol, 2004. 286(1): p. H202-213. 30.
(37) 40.. Wen, X. and G.D. Wu, Evidence for epigenetic mechanisms that silence both basal and immune-stimulated transcription of the IL-8 gene. J Immunol, 2001. 166(12): p. 7290-7299.. 31.
(38) Figures. ***. 8. *. 6 4. **. *. 2 0. con. 0.1. 0.2. 0.5. 1. 30 25. (fold change). 10. TNF-alpha mRNA expression. 12. (fold change). TLR4 mRNA expression. A. ***. 20 15. **. 10. *. 5 0. con. LPS(υg). 0.1. 0.2 LPS(ug). B. 32. ** 0.5. 1.
(39) **. 5. 5. TNF-alpha expression (fold change). TLR4 expression (fold change). 6. 4. **. 3. **. 2 1 0. **. 4 3. **. 2. *. 1 0. con. 0.1. 0.2. 0.5. 1. con. LPS(ug/ml). 0.1. 0.2. 0.5. 1. LPS(ug/ml). Figure 1. LPS up-regulates TLR4 and TNFa mRNA transcription and protein expression in a dose dependent manner. A. Tet-On/ERα H9c2 cells were incubated for 24 hrs with the indicated concentrations of LPS (0–1.0 µg/ml). TLR4 and TNFα mRNA transcription was analyzed by RT- PCR after normalization to GAPDH mRNA. B. Cell extracts were analyzed by western blotting with antibodies against proteins as indicated(relative to α-tubulin). Data represent the results of three independent experiments (means+SEM; *P<0.05, **P<0.01, ***P<0.001 compared with unstimulated group). 33.
(40) TLR4 mRNA expression (fold change) 3. 2. *. CON 4 12. *. 18. ** **. 1. 0. 24 TNF-alpha mRNA expression (fold change). A. B. 28. TIME(hr). 34 5. 4. *. 3. *. 2. 1. 0. con 4 12 18. TIME(hr). 24 28.
(41) Figure 2. LPS induces TLR4 mRNA and Protein Expression in H9c2 cadiomyoblast cells. Tet-On/ERα H9c2 cells were treated with vehicle or LPS (1 µg/ml) for 0–28 hrs. A. TLR4 mRNA expression was analyzed by RT- PCR after normalization to GAPDH mRNA. B. Cell extracts were analyzed by western blotting with antibodies against proteins as indicated (relative to α-tubulin).. Bar graphs show relative intensity of each band which was measured by densitometry. Data represent results from three independent experiments (mean ± SEM; *, P < 0.05, **P<0.01, compared with unstimulated. group).. 35.
(42) TLR4. 90KD. α-tubulin. 56KD SP600125. U0126. LPS (1µg/ml. CsA. LY294002. SB203580. ). Figure 3. LPS induced TLR4 protein expression is mediated through JNK1/2. H9c2 myocardiac cells were pretreated with vehicle, U0126 (ERK1/2 inhibitor, 1μM), SB203580 (p38 MAPK inhibitor, 1μM), SP600125 (JNK1/2 inhibitor, 1μM), CsA (calcineurin inhibitor, 1μM) or Ly294002 (MEK inhibitor, 1μM) for 1h and followed by LPS (1 μg/ml) administration for 24h. Total protein of cell extracts was separated by 12% SDS-PAGE, transferred to PVDF membranes, and immunoblotted with antibodies against TLR4 protein. Equal loading was assessed with an anti-α-tubulin antibody. Cells cultured without treatments were used as controls.. 36.
(43) A. B. Figure 4. Estrogen and over-expression ERα inhibits LPS-induced TLR4 expression. Tet-On/ERα H9c2 cells were incubated with E2 (10-8 M), Dox (1 µg/ml) in the present of LPS (1µg/ml) for 24 hrs. A. TLR4 mRNA expression was analyzed by RT- PCR after normalization to GAPDH mRNA. B. Cell extracts were analyzed by western blotting.. 37.
(44) A. B HuR. 36 KD. TTP. 44 KD. AUF-1. 39 KD. α-tubulin. 56 KD. E2(10-8M). -. -. -. -. +. +. -. -. +. +. Dox (1 µ g/ml). -. -. +. +. +. +. + +. + +. + +. + +. LPS(1µg/ml). 38.
(45) C cytosol. nuclear. HuR. 36 KD. α-tubulin. 56 KD. HDAC5. 140 KD. E2(10-8M). -. -. +. -. +. -. -. +. -. +. Dox (1 µg/ml). -. +. +. + +. + +. -. +. +. + +. + +. LPS(1µg/ml). Figure 5. E2 and ERα over-expression reverse the LPS-induced HuR cytoplasmic translocation. A. Tet-On/ERα H9c2 cells were incubated with LPS (1µg/ml) for 0-28 hrs. Cells were then fixed, and the immunoflurescence staining with antibody against HuR was performed and visualized with a fluorescence microscope coupled with an image analysis system. B. Tet-On/ERα H9c2 cells were incubated with E2 (10-8 M), Dox (1 µg/ml) in the present of LPS (1µg/ml) for 24 hrs. Cell lysates and nuclear extracts were prepared, and the levels of cytoplasmic HuR weredetermined by western blotting.. 39.
(46)
數據

+2
相關文件
臺大機構典藏NTUR (National Taiwan University 二 Repository, http://ntur.lib.ntu.edu.tw) 經驗與協助推 動臺灣學術機構典藏TAIR (Taiwan Academic Institutional Repository,
With regard to spending structure, visitors from Mainland China spent 60% of the per-capita spending on shopping, whereas those from Hong Kong and Taiwan, China spent 79% and 74% of
With regard to the spending structure, visitors from Mainland China spent 63% of their per- capita spending on shopping, whereas those from Hong Kong and Taiwan, China spent 79% and
With regard to spending structure, visitors from Mainland China spent 61% of their per- capita spending on shopping, whereas those from Hong Kong and Taiwan, China spent 78% and 71%
With regard to spending structure, visitors from Mainland China spent 66% of their per- capita spending on shopping, whereas those from Hong Kong and Taiwan, China spent 77% and 66%
- Teachers can use assessment data more efficiently to examine student performance and to share information about learning progress with individual students and their
By correcting for the speed of individual test takers, it is possible to reveal systematic differences between the items in a test, which were modeled by item discrimination and
After the SARS crisis in 2003, there is a huge and on-going reform in the post-graduate medical training program to improve the medical service in Taiwan. Our institute has
