Crassocolides A-F, Cembranoids with a trans-Fused Lactone from the Soft Coral Sarcophyton
crassocaule
Ho-Cheng Huang,†,‡Atallah F. Ahmed,†,§Jui-Hsin Su,†Chih-Hua Chao,†Yang-Chang Wu,|Michael Y. Chiang,⊥and Jyh-Horng Sheu*,†
Department of Marine Biotechnology and Resources, National Sun Yat-sen UniVersity, Kaohsiung 804, Taiwan, Republic of China, Department of Chemical and Materials Engineering, Cheng Shiu UniVersity, Kaohsiung 833, Taiwan, Republic of China, Department of Pharmacognosy, Faculty of Pharmacy, Mansoura UniVersity, Mansoura 35516, Egypt, Graduate Institute of Natural Products, Kaohsiung Medical UniVersity, Kaohsiung 807, Taiwan, Republic of China, and Department of Chemistry, National Sun Yat-sen UniVersity, Kaohsiung 804, Taiwan, Republic of China
ReceiVed April 25, 2006
Six new polyoxygenated cembrane-based diterpenoids possessing a trans-fused R-methylene-γ-lactone, crassocolides A-F (1-6), have been isolated along with the known compound 7 from the ethyl acetate extract of a Taiwanese soft coral, Sarcophyton crassocaule. The structures of 1-6 have been established by detailed spectroscopic analysis, including 2D NMR (1H-1H COSY, HMQC, HMBC, and NOESY) spectroscopy, while their absolute configurations were determined using a modified Mosher’s method for 1. The structure of 5 was further proven by X-ray diffraction analysis. The full assignment of NMR data of 7 is reported herein for the first time. The cytotoxicity of crassocolides 1-4, 6, and 7 against a limited panel of cancer cells was also determined.
Marine terpenoids are of considerable interest due to their unique structures and wide range of biological activities.1The macrocyclic cembrane-type compounds have been found to be the main terpenoidal constituents in marine coelenterates.1 Cembranoids possessing aγ-lactone2-17have been mostly isolated from octoc-orals (Alcyonaceae) belonging to the genera Sinularia,2-5Eunicea,6-12 and Lobophytum.13-17Some of these metabolites have been shown to exhibit cytotoxic activity against the growth of various cancer cell lines.2,6-8,14,15In our continuing search for bioactive metabolites from soft corals of Taiwanese waters,18-22 we have chemically investigated Sarcophyton crassocaule Moser and have succeeded in the isolation of six new polyoxygenated trans-fused cembrano-lides, crassocolides A-F (1-6), along with the known metabolite 723from the EtOAc extract of the organism. The structures of 1-6 have been established by extensive spectroscopic analysis, including 2D NMR (1H-1H COSY, HMQC, HMBC, and NOESY) spec-troscopy. Their absolute configurations were confirmed on the basis of the absolute structure of 1, determined by a modified Mosher’s method, and the absolute structure of 7, determined previously by X-ray crystallography.24 The relative structure of 5 was further proven by X-ray diffraction analysis. The full1H and13C NMR data of 7 are reported herein for the first time. Cytotoxicity of metabolites 1-4, 6, and 7 against Hep G2 (human liver carcinoma), MCF-7, MDA-MB-231 (human breast carcinoma), and A-549 (human lung carcinoma) is also discussed.
Results and Discussion
The EtOAc extract of the frozen animal was fractionated by silica gel column chromatography, and the eluted fractions were subjected to further separation and purification utilizing normal-phase HPLC to yield cembranolides 1-7 (see Experimental Section). All new metabolites were isolated as colorless oils (except 5) and showed positive optical rotations. Spectroscopic data revealed that all of these metabolites are cembranoids containing R-methylene- γ-lactones.
The HRFABMS (m/z 393.2275 [M + H]+) of the most polar metabolite, crassocolide A (1), established the molecular formula C22H32O6, requiring seven degrees of unsaturation. The IR spectrum of 1 revealed the presence of hydroxy (νmax3440 cm-1), R-meth-ylene-γ-lactone (νmax1753 and 1653 cm-1),2-17and ester (νmax1723 cm-1) moieties. The ion peaks at m/z 357 [M - 2 H2O + H]+, 297 [M - 2 H2O - AcOH + H]+in the FABMS indicated the presence of two hydroxyls and one acetoxyl in 1. The latter was indicated from the1H NMR data atδ 2.01 (3H, s) and the13C NMR data at δC20.8 (CH3) and 169.5 (qC). The 22 carbon signals appearing in the13C NMR spectrum of 1 (Table 1) were identified by DEPT to belong to four methyls, six methylenes (including one olefinic carbon), six methines (including two vinylic CH), and six quaternary carbons. These data suggested 1 was a diterpenoid containing an acetoxy function. The NMR signals observed atδC170.0 (qC), 139.0 (qC), 122.6 (CH2), 80.9 (CH), and 36.1 (CH) andδH6.27, 5.70 (each, 1H, d, J ) 2.5 Hz), 4.64 (1H, br s), and 3.31 (1H, m) revealed the presence of the R-methylene-γ-lactonic group as compared to those of similar metabolites.2-17 Two trisubstituted double bonds were also designated from the NMR signals atδC 136.7 (qC), 130.4 (qC), 127.4 (CH), and 125.4 (CH) and atδH 5.29 (1H, dd, J ) 7.0, 4.5 Hz) and 5.09 (1H, dd, J ) 7.0, 5.0 Hz). The three 3H singlets appearing in the1H NMR atδ 1.74, 1.68, and 1.12 were assigned to two olefinic methyls and one methyl bound to a quaternary oxycarbon, respectively. The remaining one
* Corresponding author. Tel: 5252000, ext. 5030. Fax: 886-7-5255020. E-mail: [email protected].
†Department of Marine Biotechnology and Resources, National Sun
Yat-sen University.
‡Cheng Shiu University. §Mansoura University. |Kaohsiung Medical University.
⊥Department of Chemistry, National Sun Yat-sen University.
10.1021/np060182w CCC: $33.50 © 2006 American Chemical Society and American Society of Pharmacognosy Published on Web 10/19/2006
degree of unsaturation was thus attributed to the 14-membered ring. Two oxymethines observed atδC76.4 (CH) and 74.9 (CH) andδH 5.40 (1H, s) and 3.52 (1H, d, J ) 9.5 Hz) and an oxycarbon observed atδC 74.3 (qC) indicated the presence of one acetoxy and two hydroxy substituents on the macrocyclic ring of 1. In the 1H-1H COSY spectrum, it was found that a ring-juncture methine proton (δ 3.31) exhibited allylic correlations with the exomethylene protons at C-17 (δH6.27 and 5.70, each d, J ) 2.5 Hz) and was assigned as H-1. Also, the 1H-1H COSY spectral analysis established three partial structures of consecutive proton spin systems (Figure 1). Moreover, it was found that the downfield shifted oxymethine proton at δ 5.40 showed 1H/13C long-range correlations in the HMBC spectrum with C-1 (δ 36.1, CH), C-14 (δ 80.9), the acetate carbonyl carbon (δ 169.5), and two olefinic carbons (δ 130.4, qC and 127.4, CH), implying the C-13 location of the acetoxy group and hence the presence of the C-11-C-12 olefin. Further analysis of the1H-1H COSY and HMBC correla-tions established the planar structure of 1, including the C-3 and C-4 positions of the remaining two hydroxy groups and the C-8 and C-9 location of the second olefinic moiety, as shown in Figure 1.
In order to resolve the absolute configuration of 1, we determined the absolute configuration at C-3 using a modified Mosher’s method.25,26 The (S)- and (R)-2-methoxy-2-(trifluoromethyl)-2-phenylacetic (MTPA) esters for 1 (1a and 1b, respectively) were prepared by using the corresponding (-)- and (+)-MTPA-chloride, respectively. The determination of ∆δ values (δS - δR) for the
protons neighboring C-3 led to the assignment of the S configuration at C-3 in 1 (Figure 2). An NOE correlation (Figure 3) was observed between H-3 (δ 3.52, d, J ) 9.5 Hz) and H3-18 (δ 1.12, s). Also, the1H NMR spectrum of 1 measured in C
5D5N showed a downfield
shift for H3-18 by the pyridine-induced deshielding effect (∆δ ) δCDCl3- δC5D5N ) - 0.32 ppm). This downfield shift value is equivalent only for a geminal deshielding resulting from 4-OH and not further deshielded by the vicinal 3-OH, revealing that 4-CH3 should be anti to the 3-OH.27By the above observations and NOE correlations between H-3 and H3-18, it was found that if H-3 is positioned on theβ-face, then the 4-CH3should be gauche to H-3 and located on the R-face (Figure 3). Furthermore, H-1 showed NOE correlation with H-3 but not with H-14. H-14 displayed NOE interactions with H-13, revealing theβ-, R-, and R-orientations of H-1, H-13, and H-14, respectively. Thus, 1 possesses a trans-fused γ-lactone ring at C-1 and C-14. The NOE correlations observed for the two olefinic methyls at C-8 and C-12 with H2-6 and H2-10, respectively, reflected the E geometry of the two trisubstituted double bonds in the molecule. On the basis of the above findings and other detailed NOE correlations (Figure 3), the structure of crassocolide A (1) was fully elucidated and established as (1R,3S,4R,- 13S,14R,7E,11E)-13-acetoxy-3,4-dihydroxycembra-7,11,15(17)-trien-16,14-olide.
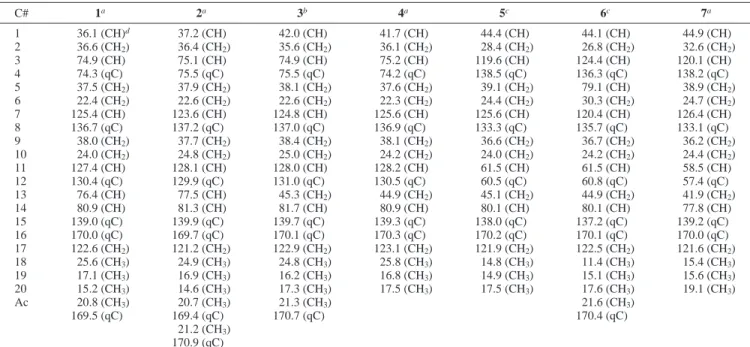
Crassocolide B (2) exhibited an ion peak at m/z 457.2204 [M + Na]+ in the HRESIMS, appropriate for a molecular formula of C24H34O7, with one more unsaturation degree relative to that of 1. The IR spectrum also revealed the presence of an R-methylene- γ-lactone (νmax1750 and 1653 cm-1). The presence of one hydroxyl and two acetoxyls was suggested from the IR absorptions (νmax 3447, 1734, and 1717 cm-1) and the ESIMS spectrum (m/z 315 [M - 2 AcOH + H]+and 297 [M - 2 AcOH - H2O + H]+). The NMR spectral data were found to be very similar to those of 1 (Tables 1 and 2), except for the presence of an additional acetoxyl (δC21.2, CH3and 170.9, qC;δH2.09, 3H, s) in 2, which replaces Table 1. 13C NMR Data for Compounds 1-7
C# 1a 2a 3b 4a 5c 6c 7a 1 36.1 (CH)d 37.2 (CH) 42.0 (CH) 41.7 (CH) 44.4 (CH) 44.1 (CH) 44.9 (CH) 2 36.6 (CH2) 36.4 (CH2) 35.6 (CH2) 36.1 (CH2) 28.4 (CH2) 26.8 (CH2) 32.6 (CH2) 3 74.9 (CH) 75.1 (CH) 74.9 (CH) 75.2 (CH) 119.6 (CH) 124.4 (CH) 120.1 (CH) 4 74.3 (qC) 75.5 (qC) 75.5 (qC) 74.2 (qC) 138.5 (qC) 136.3 (qC) 138.2 (qC) 5 37.5 (CH2) 37.9 (CH2) 38.1 (CH2) 37.6 (CH2) 39.1 (CH2) 79.1 (CH) 38.9 (CH2) 6 22.4 (CH2) 22.6 (CH2) 22.6 (CH2) 22.3 (CH2) 24.4 (CH2) 30.3 (CH2) 24.7 (CH2) 7 125.4 (CH) 123.6 (CH) 124.8 (CH) 125.6 (CH) 125.6 (CH) 120.4 (CH) 126.4 (CH) 8 136.7 (qC) 137.2 (qC) 137.0 (qC) 136.9 (qC) 133.3 (qC) 135.7 (qC) 133.1 (qC) 9 38.0 (CH2) 37.7 (CH2) 38.4 (CH2) 38.1 (CH2) 36.6 (CH2) 36.7 (CH2) 36.2 (CH2) 10 24.0 (CH2) 24.8 (CH2) 25.0 (CH2) 24.2 (CH2) 24.0 (CH2) 24.2 (CH2) 24.4 (CH2) 11 127.4 (CH) 128.1 (CH) 128.0 (CH) 128.2 (CH) 61.5 (CH) 61.5 (CH) 58.5 (CH) 12 130.4 (qC) 129.9 (qC) 131.0 (qC) 130.5 (qC) 60.5 (qC) 60.8 (qC) 57.4 (qC) 13 76.4 (CH) 77.5 (CH) 45.3 (CH2) 44.9 (CH2) 45.1 (CH2) 44.9 (CH2) 41.9 (CH2) 14 80.9 (CH) 81.3 (CH) 81.7 (CH) 80.9 (CH) 80.1 (CH) 80.1 (CH) 77.8 (CH) 15 139.0 (qC) 139.9 (qC) 139.7 (qC) 139.3 (qC) 138.0 (qC) 137.2 (qC) 139.2 (qC) 16 170.0 (qC) 169.7 (qC) 170.1 (qC) 170.3 (qC) 170.2 (qC) 170.1 (qC) 170.0 (qC) 17 122.6 (CH2) 121.2 (CH2) 122.9 (CH2) 123.1 (CH2) 121.9 (CH2) 122.5 (CH2) 121.6 (CH2) 18 25.6 (CH3) 24.9 (CH3) 24.8 (CH3) 25.8 (CH3) 14.8 (CH3) 11.4 (CH3) 15.4 (CH3) 19 17.1 (CH3) 16.9 (CH3) 16.2 (CH3) 16.8 (CH3) 14.9 (CH3) 15.1 (CH3) 15.6 (CH3) 20 15.2 (CH3) 14.6 (CH3) 17.3 (CH3) 17.5 (CH3) 17.5 (CH3) 17.6 (CH3) 19.1 (CH3) Ac 20.8 (CH3) 20.7 (CH3) 21.3 (CH3) 21.6 (CH3) 169.5 (qC) 169.4 (qC) 170.7 (qC) 170.4 (qC) 21.2 (CH3) 170.9 (qC)
aSpectra recorded at 125 MHz at 25°C. The values are in ppm downfield from TMS.bSpectra recorded at 100 MHz at 25°C.cSpectra recorded
at 75 MHz in CDCl3at 25°C.dAttached protons were determined by DEPT experiments.
Figure 1. 1H-1H COSY and HMBC correlations for 1 and 3. Figure 2. 1H NMR chemical shift differences∆δ (δS- δR) in
a hydroxyl in 1. The acetoxy group was located at C-3 due to the downfield shift observed for H-3 (δ 4.78, dd, J ) 6.0, 6.0 Hz) of 2 relative to that of 1 (δ 3.52, d, J ) 9.5 Hz). Moreover, acetylation of 1 with acetic anhydride in pyridine yielded a product that showed identical1H NMR data to that of 2, including the same splitting pattern and J value of H-3, and thus confirmed that 2 is the O-acetyl derivative of 1. On the basis of the above findings and the detailed analysis of the key NOESY correlations (Figure 3), the structure of crassocolide B (2) was established as (1R,3S,4R,13S,14R,7E,- 11E)-3,13-diacetoxy-4-hydroxycembra-7,11,15(17)-trien-16,14-olide.
Crassocolide C (3) was found to possess the molecular formula C22H32O5 from the HRFABMS (m/z 377.2325 [M + H]+). The presence of the R-methylene-γ-lactone, acetoxy, and hydroxy functionalities was indicated by IR (νmax1748 and 1660, 1715, and 3422 cm-1) and FABMS, which showed ion peaks at m/z 359 [M - H2O + H]+and 317 [M - AcOH + H]+. The13C NMR data of 3 were found to be very similar to those of 2 (Tables 1), except for the replacement of the three carbon signals of an acetoxymethine moiety in 2 by the signal of a methylene carbon in 3 (δ 45.3, CH2, C-13). The similar chemical shifts of C-2 and C-3 in both 2 and 3 positioned the acetate group at C-3 in 3. Furthermore, it was found that the splitting pattern and J value of the acetoxymethine proton H-3 (δ 4.84, dd, J ) 6.2, 6.2 Hz) of 3 were similar to those of 2 (δ 4.78, dd, J ) 6.0, 6.0 Hz), implying the very similar partial structure around C-3 in both 2 and 3. The overall planar structure of 3 was established by analyzing the1H-1H COSY and HMBC correlations (Figure 1). The above observations together with the very similar NOE correlations for both 2 and 3 revealed that they have the same configurations at C-1, C-3, C-4, and C-14, respectively. The structure of crassocolide C (3) was thus established as (1R,3S,4R,14S,7E,11E)-3-acetoxy-4-hydroxycembra-7,11,15(17)-trien-16,14-olide.
Crassocolide D (4) possessed the molecular formula C20H30O4 as revealed from its HRFABMS (m/z 335.2222 [M + H]+). The IR absorptions atνmax1750, 1653, and 3422 cm-1and the ion peaks at m/z 317 [M - H2O + H]+and 299 [M - 2 H2O + H]+indicated the presence of the R-methylene-γ-lactone and two hydroxyl groups.
Comparison of the1H and13C NMR spectral data of 4 with those of 3 revealed 4 as the deacetyl derivative of 3. It was found that H-3 of 4 resonated at higher field (δ 3.53, dd, J ) 9.5, 7.5 Hz) relative to that of 3 (δ 4.84, dd, J ) 6.2, 6.2 Hz). Thus, the acetoxy group at C-3 of 3 was replaced by a hydroxy group in 4. It was observed that the NOE correlations of 4 (Figure 3) are very similar to those of 1. Thus, the structure of crassocolide D (4) was deduced as (1R,3S,4R,14S,7E,11E)-3,4-dihydroxycembra-7,11,15(17)-trien-16,14-olide.
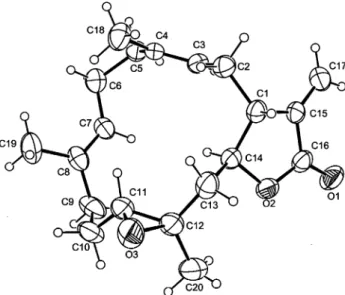
The least polar metabolite, crassocolide E (5), was isolated as colorless prisms. Its HRFABMS (m/z 317.2117, [M + H]+) and NMR data (Tables 1 and 2) established the molecular formula C20H28O3, implying seven degrees of unsaturation. The IR absorp-tions of 5 atνmax1765 and 1662 cm-1also revealed the presence of an R-methylene-γ-lactone moiety. This was further indicated from the1H NMR signals atδ 6.24 and 5.59 (each 1H, d, J ) 3.0 Hz) and13C NMR signals atδ 170.2 (qC, C-16), 138.0 (qC, C-15), 121.9 (CH2, C-17), 80.1 (CH, C-14), and 44.4 (CH, C-1). Moreover, the 13C NMR signals atδ 138.5 (qC), 133.3 (qC), 125.6 (CH), 119.6 (CH), 61.5 (CH), and 60.5 (qC) assigned two trisubstituted double bonds and one trisubstituted epoxide in the molecule, respectively. The 3H singlets appearing in the1H NMR spectrum atδ 1.66, 1.51, and 1.41 represented the methyl substituents at the two double bonds and the epoxide, respectively. By interpretation of 1H-1H COSY correlations, it was possible to establish three partial structures of consecutive proton systems extending from the olefinic proton H-3 to H2-13 through H-1 and H-14, from H2-5 to the olefinic H-7, and from H2-9 to H-11; the connectivities of these partial structures were further established by the HMBC correlations (Figure 4). The long-range1H/13C correlations observed from the methyl protons atδ 1.41 (H3-20) to C-11 (δ 61.5, CH), C-12 (60.5, qC), and C-13 (δ 45.1, CH2) indicated the C-11-C-12 position of the epoxide. On the basis of the above observations, and additional 2D NMR (1H-1H COSY, HMQC, and HMBC) experiments, the planar structure of 5 was established as illustrated in Figure 4. On comparison of chemical shifts and splitting patterns of the protons at C-1, C-3, C-7, C-11, C-14, and C-17-C-20 in 5 with the corresponding protons in lobophytolide (7), of which the structure was established by X-ray diffraction analysis,23,24it was found that the difference between these compounds was the configuration at the epoxide moiety. This was due to the variation in chemical shifts and splitting patterns for H-11 (δ 2.61, 1H, dd, J ) 7.5, 1.8 Hz) and H3-20 (δ 1.41, 3H, s) in 5 relative to those in 7 (δ 2.81, 1H, dd, J ) 6.0, 6.0 Hz and 1.28, 3H, s, respectively). Finally, the structure of 5 was unambiguously established by single-crystal X-ray diffraction analysis (Figure 5) as (1R,11R,12R,14S,3E,7E)-11,12-epoxycembra-3,7,15(17)-trien-16,14-olide.
The new metabolite crassocolide F (6) was found to have the molecular formula C22H30O5from its HRESIMS (m/z 397.1989, [M + Na]+) and NMR data (Table 1 and 2), implying eight degrees of unsaturation. Similar to 1-5, the IR spectrum of 6 indicated the presence of an R-methylene-γ-lactone group (νmax1767 and 1667 cm-1), and this was further supported from NMR shifts atδH6.40 and 5.62 (each 1H, d, J ) 3.0 Hz);δC170.1, 137.2, 122.5, 80.1, and 44.1. An acetoxy group was also revealed from the ESIMS (m/z 315 [M - AcOH + H]+) and IR (νmax 1734 cm-1) spectra and was supported by the1H NMR signals atδ 2.03 (3H, s). The NMR data of 6 showed signals of high similarity to those of 5 except for the replacement of a methylene group (δH2.27, 2H, m; δC39.1) in 5 with an acetoxymethine signal (δH5.24, 1H, dd, J ) 10.5, 4.8 Hz and 2.03, 3H, s;δC170.4, qC,δ 79.1, CH, and 21.6, CH3) in 6. The HMBC correlations observed from the methyl protons atδ 1.43 (H3-20) to C-11 (δ 61.5, CH), C-12 (60.8, qC), and C-13 (δ 44.9, CH2) and from the olefinic methyl protons atδ 1.54 to C-3 (δ 124.4, CH), C-4 (δ 136.3, qC), and C-5 (δ 79.1, CH) revealed that the epoxide and the acetoxy groups should be positioned at C-11-C-12 and C-5, respectively. On the basis of Figure 3. Selected NOESY correlations of 1-4.
the above observations and additional 2D NMR (1H-1H COSY, HMQC, and HMBC) experiments, the planar structure of 6 was established as shown in Figure 4.
The configurations of the five chiral centers at C-1, C-5, C-11, C-12, and C-14 in 6 were determined on the basis of NOE correlations observed from a NOESY experiment (Figure 6). It was found that H-11 (δ 2.53, m), but not H3-20, showed NOE interaction with the R-oriented H-14 (as deduced from the absolute
configu-ration of C-14 in the biogenetically related metabolite 1), implying the downward orientation of H-11. The methyl protons H3-20 showed NOE correlation with H2-10, but not with H-11 and thus assigned trans geometry to the epoxide ring. Theβ-orientation of the epoxide was established, since the NMR data of C-11-C-14 and C-20 and their attached protons in 6 were quite similar to those in 5. Moreover, H-14 exhibited an NOE interaction with H-3 (δ 5.35, dd, J ) 6.9, 6.9 Hz) and H-3 showed an NOE interaction with H-5 (δH 5.24, 1H, dd, J ) 10.5, 4.8 Hz), revealing the R-orientation of H-5, and thus the β-orientation of the acetoxy group at C-5 as shown in Figure 5. The two upfield shifted olefinic methyls that exhibited carbon signals below 20 ppm, together with the NOE interactions observed between H3-19 and H-6 and between H3-18 and H-2, confirmed the E geometry of the two trisubstituted double bonds. Further interpretation of NOESY data (Figure 6) again indicated the trans-fused lactone ring and hence established the stereochemistry of 6. On the basis of the above results, the structure of crassocolide F was elucidated as (1R,5R,11R,- 12R14S,7E,3E)-5-acetoxy-11,12-epoxycembra-3,7,15(17)-trien-16,-14-olide.
Compound 7 [(1R,11S,12S,14S,7E,3E)-11,12-epoxycembra-3,7,-15(17)-trien-16,14-olide] was also isolated from S. crassocaule and found to be identical to the previously reported metabolite lobo-phytolide (7), isolated from the soft coral Lobophytum cristagalli,26 by comparison of the physical (mp and [R]D) and spectroscopic (IR, MS,1H NMR) data. Moreover, by 2D NMR analysis, the full assignment of NMR data for 7 is reported here for the first time (Tables 1 and 2).
Table 2. 1H NMR Data for Crassocolides 1-7d
H# 1a 2a 3b 4a 5c 6c 7a 1 3.31 m (w1/2 12.0)c 3.32 m (w1/2 11.5) 3.10 m (w1/2 11.0) 2.99 m (w1/2 11.0) 2.77 m (w1/2 11.5) 2.80 m (w1/2 12.5) 2.64 m (w1/2 12.0) 2 1.54 m; 1.77 m 1.83 m; 2.01 m 1.88 m; 1.94 m 1.58 m; 1.74 m 2.30 2H, m 2.45 m; 2.49 m 2.27 2H, m 3 3.52 d (9.5) 4.78 dd (6.0, 6.0) 4.84 dd (6.2, 6.2) 3.53 dd (9.5, 7.5) 4.98 dd (6.6, 5.0) 5.35 dd (6.9, 6.9)c 5.06 dd (7.0, 7.0) 5 1.71 2H, m 1.74 m; 1.79 m 1.72 2H, m 1.73 2H, m 2.27 2H, m 5.24 dd (10.5, 4.8) 2.19 2H, m 6 2.06 m; 2.42 m 2.05 m; 2.27 m 2.09 m; 2.18 m 2.04 m; 2.46 m 2.10 m; 2.38 m 2.34, m; 2.55 m 2.21 m; 2.29 m 7 5.09 dd (7.0, 5.0) 5.23 dd (7.0, 7.0) 5.25 dd (7.0, 7.0) 5.10 dd (8.0, 4.5) 4.98 br dd (6.6, 5.0) 4.90 d (9.9) 5.01 br dd (6.0, 6.0) 9 2.16 m; 2.26 m 2.12 m; 2.21 m 2.18 2H, m 2.15 m; 2.21 m 2.06 m; 2.22 m 2.05 m; 2.32 m 2.09 m; 2.22 m 10 2.18; 2.39 m 2.20; 2.41 m 2.22 m; 2.34 m 2.14; 2.31 m 1.31 m; 2.06 m 1.34 m; 2.12 m 1.53 m; 1.78 m 11 5.29 d (7.0, 4.5) 5.31 dd (7.0, 7.0) 5.06 br dd (6.0, 6.0) 5.09 dd (6.5, 6.5) 2.61 dd (7.5, 1.8) 2.53 m 2.81 dd (6.0, 6.0) 13 5.40 s 5.38 s 2.08 m; 2.48 m 2.12 dd (14.0, 10.0) 1.31 m; 2.26 m 1.32 m; 2.29 m 2.04 m; 2.09 m 2.64 br d (14.0) 14 4.64 br s 4.63 br s 4.44 ddd (10.0, 5.0, 5.0) 4.52 ddd (10.5, 4.0, 3.0) 4.26 ddd (9.0, 6.6, 1.5) 4.12 ddd (9.9, 6.0, 1.5) 3.96 dd (11.0, 5.0) 17 5.70 d (2.5) 5.67 d (2.0) 5.68 d (1.8) 5.68 d (2.5) 5.59 d (3.0) 5.62 d (3.0) 5.59 d (2.0) 6.27 d (2.5) 6.23 d (2.0) 6.26 d (1.8) 6.27 d (2.5) 6.24 d (3.0) 6.40 d (3.0) 6.24 d (2.0) 18 1.12 3H, s 1.19 3H, s 1.19 3H, s 1.13 3H, s 1.51 3H, s 1.54 3H, s 1.58 3H, s 19 1.68 3H, s 1.68 3H, s 1.62 3H, s 1.66 3H, s 1.66 3H, s 1.69 3H, s 1.66 3H, s 20 1.74 3H, s 1.77 3H, s 1.74 3H, s 1.70 3H, s 1.41 3H, s 1.43 3H, s 1.28 3H, s Ac 2.01 3H, s 1.99 3H, s 2.07 3H, s 2.03 3H, s 2.09 3H, s
aSpectra recorded at 500 MHz at 25°C.bSpectra recorded at 400 MHz at 25°C.cSpectra recorded at 300 MHz in CDCl
3at 25°C.dThe J values
are in Hz in parentheses.
Figure 4. 1H-1H COSY and HMBC correlations for 5 and 6.
Figure 5. Molecular structure of 5 based on X-ray analysis.
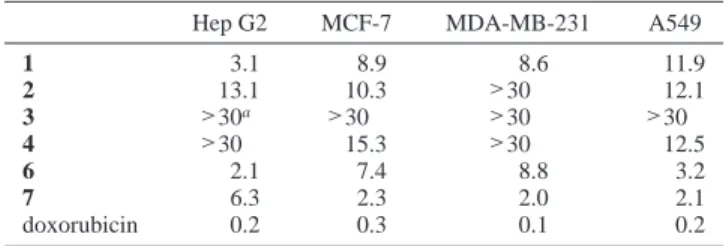
The cytotoxicity of the diterpenoids 1-4, 6, and 7 against HepG2, MCF7, MDA-MB-231, and A-549 cancer cells was studied. The results (Table 3) showed that the 13-acetoxy-3,4-dihydrox-ycembranolide (1) was cytotoxic against the four cancer cell lines, being significant (IC503.1µg/mL) to moderate (IC508.6-11.9µg/ mL) against Hep G2, and MCF-7, MDA-MB-231, and A549, respectively. The acetylation of 1 at the 4-OH position resulted in the formation of 2 and reduced cytotoxicity. The loss of the 13-acetoxy group from 2 to yield 3 was associated with a complete loss of cytotoxic activity against the four cell lines. Compound 4, which was not oxidized at C-13, but possessed two hydroxy groups at C-3 and C-4 like that of 1, retained partial cytotoxicity and exhibited moderate activity against two cell lines (MCF-7 and A549). The 5-O-acetylated epoxycembranolide 6 was strongly cytotoxic against Hep G2 and A549 but showed only moderate activity against MCF-7 and MDA-MB-231 cancer cell lines. While the known epoxycembranolide (7) showed only moderate cytotox-icity against Hep G2 cells (IC506.3µg/mL), it was found to exhibit significant cytotoxicity (IC50 ≈ 2.0 µg/mL) against other cancer cell lines.
Experimental Section
General Experimental Procedures. Melting points were determined using a Fisher-Johns melting point apparatus. Optical rotations were measured on a Jasco DIP-1000 digital polarimeter. IR spectra were recorded on a Hitachi I-2001 infrared spectrophotometer. FABMS were obtained with a VG Quattro GC/MS spectrometer. HRFABMS spectra were recorded on a JEOL-SX/SX 102A mass spectrometer. The NMR spectra were recorded on a Bruker AMX-400 FT-NMR at 400 MHz for1H and 100 MHz for13C or on a Varian Unity INOVA 500
FT-NMR at 500 MHz for1H and 125 MHz for13C, respectively, in CDCl 3
using TMS as internal standard. The crystallographic data were collected on a Rigaku AFC7S diffractometer using graphite-monochromated Mo KR radiation. Structure analysis was made by using the SHELXTL PLUS package. Si gel 60 (Merck, 230-400 mesh) was used for CC. Precoated Si gel plates (Merck, Kieselgel 60 F254, 0.2 mm) were used
for analytical TLC analyses.
Animal Material. S. crassocaule (Alcyoniidea) (1.05 kg, wet wt) was collected by hand via scuba at the coast of Kenting, in December 2001 and July 2002, at a depth of 10 to 15 m, and stored in a freezer until extraction. A voucher sample was deposited at the Department of Marine Resources, Sun Yat-sen University.
Extraction and Separation. The frozen bodies of S. crassocaule (1.05 kg, wet wt) were minced and exhaustively extracted with EtOAc (1 L× 5). The solvent-free EtOAc extract (15.0 g) was subjected to Si gel CC and eluted with EtOAc in n-hexane (0-100%, gradient) to yield 20 fractions. Fraction 10 eluted with EtOAc-n-hexane (1:10) and was purified by normal-phase HPLC using acetone-n-hexane (1: 20) to give 5 (27 mg). Fraction 12 eluted with EtOAc-n-hexane (1:5) and was further chromatographed by normal-phase HPLC using acetone-n-hexane (1:13) to give 7 (27.8 mg). Fraction 14 eluted with EtOAc-n-hexane (2:5) and was further separted by normal-phase HPLC using acetone-n-hexane-CH2Cl2(1:15:2) to yield 2 (3.8 mg)
and 6 (2.2 mg). Fraction 15 eluted with n-hexane-EtOAc (1:1) and was further purified by normal-phase HPLC using acetone-n-hexane (1:6) to afford 3 (5.5 mg). The next two successive fractions eluted with EtOAc-n-hexane (1:1 to 2:1) and were separately rechromato-graphed by normal-phase HPLC using acetone-n-hexane (1:4) to afford 4 (56.5 mg) and 1 (47.3 mg), respectively.
Crassocolide A (1): colorless oil; [R]25
D+7.0 (c 2.7, CHCl3); IR
(neat)νmax3440, 2958, 2928, 2878 1753, 1723,1653, 1437, 1373, 1273,
and 1232 cm-1;1H and13C NMR data (CDCl
3), see Tables 2 and 1,
respectively;1H NMR (C 5D5N)δ 6.29 (1H, s, H-17), 5.94 (1H, s, H-17), 5.63 (1H, s, H-13), 5.62 (1H, m, H-11), 5.49 (1H, m, H-7), 5.49 (1H, br s, H-14), 3.98 (1H, br d, J ) 9.3 Hz, H-3), 3.47 (1H, m, H-1), 2.66 (1H, m, H-6), 2.38 (1H, br d, J ) 14.0 Hz, H-2), 2.28 (2H, m, H-9, H-10), 2.18 (2H, m, H-9, 10), 2.13 (1H, m, H-5), 197 (3H, s, Ac), 1.93 (1H, m, H-2), 1.89 (3H, s, H3-20), 1.88 (2H, m, H-5, H-6), 1.63 (3H, s, H3-19), 1.44 (3H, s, H3-18); FABMS m/z 393 [0.6, (M + H)+], 357 [0.2, (M - 2 H2O + H)+], 297 [0.6, (M - 2 H2O - AcOH + H)+], 273 (1.9), 257 (2.0), 241 (1.6), and 229 (3.9); HRFABMS m/z 393.2275 (calcd for C22H33O6, 393.2277).
Crassocolide B (2): colorless oil; [R]25
D+31.6 (c 1.5, CHCl3); IR
(neat)νmax3447, 2956, 2932, 2872 1750, 1734, 1717, 1653, 1456, 1397,
1260, and 1238 cm-1;1H and13C NMR data (CDCl
3), see Tables 2
and 1, respectively; ESIMS m/z 457 [100, (M + Na)+], 375 [67, (M -AcOH + H)+], 315 [41, (M - 2 AcOH + H)+] and 297 [57, (M - 2 AcOH - H2O + H)+]; HRESIMS m/z 457.2204 (calcd for C24H34O7
-Na, 457.2202).
Crassocolide C (3): colorless oil; [R]25
D+19.1 (c 1.1, CHCl3); IR
(neat)νmax3422, 2958, 2932, 2880, 1748, 1715, 1660, 1456, 1375,
and 1242 cm-1;1H and13C NMR data (CDCl
3), see Tables 2 and 1,
respectively; FABMS m/z 399 [0.2, (M + Na)+], 377 [0.4, (M + H)+], 359 [0.4, (M - H2O + H)+], 317 [0.9, (M - AcOH + H)+], 299 [0.5,
(M - H2O - AcOH + H)+], 267 (0.8), and 253 (0.7); HRFABMS m/z 377.2325 (calcd for C22H33O5, 377.2328).
Crassocolide D (4): colorless oil; [R]25
D+21.9 (c 1.3, CHCl3); IR
(neat)νmax3422, 2958, 2932, 2880, 1750, 1653, 1435, 1387, and 1269
cm-1;1H and13C NMR data (CDCl
3), see Tables 2 and 1, respectively;
FABMS m/z 357 [2.1, (M + Na)+], 335 [1.7, (M + H)+], 317 [19.6, (M - H2O + H)+], 299 [1.2, (M - 2 H2O + H)+]; HRFABMS m/z
335.2222 (calcd for C20H31O4, 335.2222).
Crassocolide E (5): colorless needles (EtOAc) mp 95-96°C; [R]25D +108.9 (c 1.0, CHCl3); IR (neat)νmax2960, 2932, 2872, 1765, 1662,
1437, 1386, 1267, and 1240 cm-1;1H and13C NMR data (CDCl 3), see
Tables 2 and 1, respectively; FABMS m/z 317 [6.0, (M + H)+]; HRFABMS m/z 317.2117 (calcd for C20H29O3, 317.2117).
Crassocolide F (6): colorless oil; [R]25
D+138.3 (c 0.6, CHCl3); IR
(neat)νmax2956, 2926, 2870, 1767, 1734, 1667, 1437, 1372, and 1238
cm-1;1H and13C NMR data (CDCl
3), see Tables 3 and 1, respectively;
ESIMS m/z 397 [100, (M + Na)+], 381 [31, (M - O + Na)+], 315 [37, (M - AcOH + H)+]; HRESIMS m/z 397.1989 (calcd for C22H30O5
-Na, 397.1991).
Lobophytolide (7): colorless needles; mp 133-134°C (lit.,26
137-138°C); [R]25
D+23.1 (c 1.0, CHCl3) (lit.,26c 0.4, +7.0); IR (neat) νmax2928, 2851, 1752, 1655, 1543, 1385, 1338, and 1273 cm-1;1H
and13C NMR data (CDCl
3), see Tables 2 and 1, respectively; FABMS m/z 339 [16.9, (M + Na)+], 317 [12.6, (M + H)+].
Preparation of (S)- and (R)-MTPA Esters of 1. To a solution of 1 (4.7 mg, 12µM) in pyridine (50 µL) was added (-)-MTPA chloride (5µL, 26 µM), and the solution was allowed to stand overnight at room temperature. The reaction was stopped by the addition of 1.0 mL of water, followed by extraction with CH2Cl2(1.0 mL× 3). The
CH2Cl2-soluble layers were combined, dried over anhydrous MgSO4,
and evaporated. The residue was subjected to a short Si column using EOAC-n-hexane (2:5) to yield the (S)-MTPA ester 1a (1.1 mg, 18%) and an unreacted portion of 1 (3.0 mg). The same procedure was applied to obtain the (R)-MTPA ester 1b from the reaction of (+)-MTPA chloride with 1 in pyridine.1H NMR (CDCl
3) of 1a: δ 7.517 (2H, m, Ph), 7.436 (3H, m, Ph), 6.213 (1H, d, J ) 2.0 Hz, H-17), 5.677 (1H, d, J ) 2.0 Hz, H-17), 5.357 (1H, s, H-13), 5.280 (1H, m, H-7), 5.231 (1H, m, H-11), 4.781 (1H, m, H-3), 4.698 (1H, s, H-14), 3.473 (3H, s, OMe), 3.377 (1H, br d, J ) 9.3 Hz, H-1), 2.174 (1H, m, H-6), 1.970 (3H, s, OAc), 1.945 (1H, m, H-2), 1.812 (3H, H3-20), 1.794 (1H, m, H-6), 1.704 (1H, m, H-2), 1.644 (3H, H3-19), 1.614 (2H, m, H2-5), 1.162 (3H, H3-18). 1H NMR (CDCl3) of 1b: δ 7.517 (2H, m, Ph), 7.435 (3H, m, Ph), 6.207 (1H, d, J ) 2.0 Hz, H-17), 5.683 (1H, d, J ) 2.0 Hz, H-17), 5.346 (1H, s, H-13), 5.327 (1H, m, H-7), 5.254 (1H, m, H-11), 4.737 (1H, m, H-3), 4.652 (1H, s, H-14), 3.530 (3H, s, OMe), 3.465 (1H, br d, J ) 9.9 Hz, H-1), 2.133 (1H, m, H-6), 2.013 (1H, m, H-2), 1.961 (3H, s, OAc), 1.857 (3H, H3-20), 1.737 (1H, m, H-6), 1.715 (1H, m, H-2), 1.681 (3H, H3-19), 1.601 (2H, m, H2-5), 1.148 (3H, H3 -18).
Table 3. Cytotoxicity (IC50µg/mL) of Crassocolides 1-4, 6, and 7
Hep G2 MCF-7 MDA-MB-231 A549
1 3.1 8.9 8.6 11.9 2 13.1 10.3 >30 12.1 3 >30a >30 >30 >30 4 >30 15.3 >30 12.5 6 2.1 7.4 8.8 3.2 7 6.3 2.3 2.0 2.1 doxorubicin 0.2 0.3 0.1 0.2 aCompound is considered inactive when IC
Crystallographic Data and X-ray Structure Analysis of Crasso-colide E (5).28A suitable colorless crystal (0.65× 0.5 × 0.4 mm3) of
5 was grown by slow evaporation of the EtOAc solution. Crystal data: C20H28O3, monoclinic, Mr) 316.42 g/mol; a ) 6.2085(12) Å, b )
9.7196(19) Å, c ) 15.606(3) Å, V ) 937.5(3) Å,3space group P2 12121, Z ) 4, Dcalc) 1.121 g/cm3,λ ) 0.71073 Å, µ(Mo KR) ) 0.074 mm-1, F(000) ) 344, T ) 298(2) K. A total of 3691 reflections were collected
in the range 2.47°< θ < 26.01°, of which only 1961 unique reflections with I > 2σ(I) were used for the analysis. The structure was solved using direct methods and refined by full-matrix least-squares on F2
values. The non-hydrogen atoms were refined anisotropically. All hydrogen atoms were fixed at calculated positions. The final indices were R1) 0.0412, wR2) 0.1199 with goodness-of-fit ) 1.026.
Cytotoxicity Testing. Cell lines were purchased from the American Type Culture Collection (ATCC). Cytotoxicity assays were performed using the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] colorimetric method.29,30
Acknowledgment. This work was supported by grants from the Ministry of Education (C030313) and National Science Council of Taiwan (NSC 94-2323-B-110-002) awarded to J.-H.S.
Note Added after ASAP Publication: The version posted on October 19, 2006 had a misspelling in the title. The correct spelling of Crassocolides appears in the version posted on November 8, 2006.
Supporting Information Available: Description of X-ray crystal structure data of compound 5. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
(1) Blunt, J. W.; Copp, B. R.; Munro, M. H. G.; Northcote, P. T.; Prinsep, M. R. Nat. Prod. Rep. 2005, 22, 15-61, and previous reports in this series.
(2) Li, G.; Zhang, Y.; Deng, Z.; Ofwegen, L. V.; Proksch, P.; Lin, W.
J. Nat. Prod. 2005, 68, 649-652.
(3) Uchio, Y.; Eguchi, S.; Nakayama, M.; Hase, T. Chem. Lett. 1982, 277-278.
(4) Kobayashi, M.; Ishizaka, T.; Miura, N.; Mitsuhashi, H. Chem. Pharm.
Bull. 1987, 35, 2314-2318.
(5) Su, J.; Yang, R.; Kuang,Y.; Zeng, L. J. Nat. Prod. 2000, 63, 1543-1545.
(6) Shi, Y.-P.; Rodrı´guez, A. D.; Barnes, C. L.; Sa´nchez, J. A.; Raptis, R. G.; Baran, P. J. Nat. Prod. 2002, 65, 1232-1241.
(7) Rodrı´guez, A. D.; Dhasmana, H. J. Nat. Prod. 1993, 56, 564-570. (8) Rodrı´guez, A. D.; Soto, J. J.; Pin´a, I. C. J. Nat. Prod. 1995, 58,
1209-1216.
(9) Fontan, L. A.; Rodrı´guez, A. D. J. Nat. Prod. 1991, 54, 298-301. (10) Rodrı´guez, A. D.; Acosta, A. L. J. Nat. Prod. 1998, 61, 40-45. (11) Gopichand, Y.; Ciereszko, L. S.; Schmitz, F. J.; Switzner, A. R.;
Hossain, M. B.; van der Helm, D. J. Nat. Prod. 1984, 47, 607-614. (12) Rodrı´guez, A. D.; Li, Y.; Dhasmana, H. J. Nat. Prod. 1993, 56,
1101-1113.
(13) Rashid, N. A.; Custafson, K. R.; Boyd, M. R. J. Nat. Prod. 2000,
63, 531-533.
(14) Duh, C.-Y.; Wang, S. K.; Huang, B. T.; Dai, C.-F. J. Nat. Prod.
2000, 63, 884-885.
(15) Wang, S.-K.; Duh, C.-Y.; Wu, Y.-C.; Wang, Y.; Cheng, M.-C.; Soong, K.; Fang, L. S. J. Nat. Prod. 1992, 55, 1430-1435. (16) Coll, J. C.; Mitchell, S. J.; Stokie, G. J. Aust. J. Chem. 1977, 30,
1859-1863.
(17) Uchio, Y.; Eguchi, S.; Fukazawa, Y.; Kodama, M. Bull. Chem. Soc.
Jpn. 1992, 65, 1182-1184.
(18) Huang, H. C.; Chao, C.-H.; Liao, J.-H.; Chiang, M.-Y.; Dai, C.-F.; Wu, Y.-C.; Sheu, J.-H. Tetrahedron Lett 2005, 46, 7711-7714. (19) Ahmed, A. F.; Kuo, Y.-H.; Dai, C.-F.; Sheu, J.-H. J. Nat. Prod. 2005,
68, 1208-1212.
(20) Tseng, Y.-J.; Ahmed, A. F.; Dai, C.-F.; Chiang, M. Y.; Sheu, J.-H.
Org. Lett. 2005, 7, 3813-3816.
(21) Ahmed, A. F.; Su, J.-H.; Shiue, R.-T.; Pan, X.-J.; Dai, C.-F.; Kuo, Y.-H.; Sheu, J.-H. J. Nat. Prod. 2004, 67, 592-597.
(22) Ahmed, A. F.; Shiue, R.-T.; Wang, G.-H.; Dai, C.-F.; Kuo, Y.-H.; Sheu, J.-H. Tetrahedron 2003, 59, 7337-7344.
(23) Tursch, B.; Braekman, J. C.; Daloze, D.; Herin, M.; Karlsson, R.
Tetrahedron Lett. 1974, 15, 3769-3772.
(24) Karlsson, R. Acta Crystallogr. 1977, B33, 2032-2034.
(25) Ohtani, I.; Kusumi, T.; Kashman, Y.; Kakisawa, H. J. Am. Chem.
Soc. 1991, 113, 4092-4096.
(26) Randazzo, A.; Bifulco, G.; Giannini, C.; Bucci, M.; Debitus, C.; Cirino, G.; Gomez-Paloma, L. J. Am. Chem. Soc. 2001, 123, 10870-10876.
(27) Demarco, P. V.; Farkas, E.; Doddrell, D.; Mylari, B. L.; Wenkert, E. J. Am. Chem. Soc. 1968, 90, 5480-5486.
(28) Crystallographic data for compound 5 have been deposited with the Cambridge Crystallographic Data Centre (deposition number CCDC 604930). Copies of the data can be obtained, free of charge, on application to the Director, CCDC, 12 Union Road, Cambridge CB2 1EZ, UK (fax: +44-1223-336033 or e-mail: [email protected]). (29) Alley, M. C.; Scudiero, D. A.; Monks, A.; Hursey, M. L.; Czerwinski, M. J.; Fine, D. L.; Abbott, B. J.; Mayo, J. G.; Shoemaker, R. H.; Boyd, M. R. Cancer Res. 1988, 48, 589-601.
(30) Scudiero, D. A.; Shoemaker, R. H.; Paull, K. D.; Monks, A.; Tierney, S.; Nofziger, T. H.; Currens, M. J.; Seniff, D.; Boyd, M. R. Cancer
Res. 1988, 48, 4827-4833. NP060182W