1
國立臺灣大學醫學院臨床牙醫學研究所 博士論文
Graduate Institute of Clinical Dentistry College of Medicine
National Taiwan University Doctoral Dissertation
胎盤生長因子於口腔鱗狀細胞癌之表現
Expression of Placenta Growth Factor (PlGF) in Oral Squamous Cell Carcinomas
鄭世榮 Shih-Jung Cheng
指導教授:江俊斌教授、郭彥彬教授 Advisor: Professor Chun-Ping Chiang Professor Mark Yen-Ping Kuo
中華民國 101 年 6 月
June, 2012
2
謝誌
在繁忙的臨床工作壓力下,能順利完成博士學位,要感恩的人真是太多了。
首先感恩我的指導老師,江俊斌教授及郭彥彬教授,謝謝您們辛苦的指導,從 題目的找尋、實驗方法的確立、投稿文章的修改到最後博士論文的付梓,時時 刻刻無微不至的呵護,讓我的研究得以順利完成。也感謝口腔顎面外科的老師 們:韓良俊教授、楊博正教授、郭英雄教授、郭生興主任、李正喆醫師、陳信 銘醫師與章浩宏醫師的指導及協助,使我在臨床上無後顧之憂,得以全力衝刺。
再則感謝辛苦的口腔顎面外科住院醫師群:小馨、佳璇、纓絡、鴻穎、方瑜、
依青等都是我得力的助手,不論在工作或研究上都展現無窮的潛力,使得學位 歷程順利進行。也要感謝助理雅文的幫忙,能精確又快速的把實驗完成,獲得 有意義的結果。
更要感謝舍弟亞東醫院胸腔內科鄭世隆主任在研究上的幫忙,對我的博士 論文有著決定性的影響。特別感恩我在學佛路上的導師中台禪寺開山方丈
上惟
下覺大和尚及普眼精舍住持
上見
下聲法師多年的慈悲指導,讓我在遇到很多人生逆 境時有勇氣堅持下去,才能順利完成博士學位。最後感恩我敬愛的父母、岳父 母及最摯愛的妻子意芸、一對兒女穎澤、亦岑的默默支持,謝謝你們的鼓勵與 陪伴,讓我終於完成學位。
謹將我的博士論文獻給痛苦的口腔癌病人,希望自己更有能力為苦難的癌
症病人盡一份心力。
3
國立台灣大學醫學院臨床牙醫學研究所 博士學位論文
口試委員會審定書
論文題目
胎盤生長因子於口腔鱗狀細胞癌之表現
Expression of Placenta Growth Factor (PlGF) in Oral Squamous Cell Carcinomas
本論文係鄭世榮君(學號 D94422001)在國立臺灣大學醫學院臨床牙醫學研究
所完成之博士學位論文,於民國一 0 一年六月七日承下列考試委員審查通過及
口試及格,特此證明。
4
TABLE OF CONTENTS
中文摘要
….……….….………… 9
Abstract ..
………..………...11I. Introduction and literature review
………...….……….……. 141.1 Overview of oral squamous cell carcinoma …..………..….….………15
1.2 Angiogenesis ……….…………..….…... 23
1.3 VEGF and cancers ……….…………...………... 28
1.4 PlGF and cancers ………..…………... 40
II. Materials and methods
………... 48Part I: Tissue placenta growth factor mRNA level 2.1.1 Patients and oral cancer specimens ………... 48
2.1.2 Patients’ oral habits ………... 49
2.1.3 Quantitative real-time reverse transcription-polymerase chain reaction ... 50
2.1.4 Statistical analysis ………... 51
Part II: Serum placenta growth factor level 2.2.1 Patients and serum samples ……… 51
2.2.2 Patients’ oral habits ………. 53
2.2.3 Enzyme-linked immunosorbent assay (ELISA) ………. 53
2.2.4 Statistical analysis ………... 54
III. Results
……… 55Part I: Increased placenta growth factor mRNA level is significantly associated with progression, recurrence and poor prognosis of oral squamous cell carcinoma 3.1.1 PlGF mRNA levels in oral cancer tissues ... 55
5
3.1.2 Correlation between the PlGF mRNA levels in OSCC samples
and clinicopathological parameters of OSCC patients ... 55
3.1.3 Survival analysis ………... 56
Part II: Increased serum placenta growth factor level is significantly associated with progression, recurrence and poor prognosis of oral squamous cell carcinoma 3.2.1 Serum PlGF protein levels in normal controls and cancer patients …..….. 57
3.2.2 Correlation between the serum PlGF protein levels in pre-surgery OSCC patients and clinicopathological parameters of OSCC patients …..….….. 57
3.2.3 Survival analysis ……….……… 58
IV. Discussion
………..…... 60Part I: Increased placenta growth factor mRNA level is significantly associated with progression, recurrence and poor prognosis of oral squamous cell carcinoma ……… 60
Part II: Increased serum placenta growth factor level is significantly associated with progression, recurrence and poor prognosis of oral squamous cell carcinoma ……….……… 63
V. Conclusions
………..………..……….. 68VI. Tables
……… 70VII. Figures
………..………. 76VIII. References
………... 84IX. Appendix
……….………..…... 106A: Curriculum Vitae ………...…. 106
B: Publications ……….……. 108
6
Table Index
Table 1.1: Mean PlGF mRNA CT (threshold cycle) value in paired OSCC and non-OSCC tissue
samples ……….……… …….70
Table 1.2: Correlation between the mean PlGF mRNA -∆CT values and clinicopathological
parameters of 63 OSCC patients ………..……….. 71
Table 1.3: Univariate and multivariate recurrence-free survival analyses of the PlGF
mRNA expression and clinicopathological parameters of OSCC patients
by Cox proportional hazard regression model ………... .72
Table 2.1: Mean serum PlGF protein levels in normal control subjects and in OSCC patients before
and 3 months after total surgical excision of OSCCs ………. 73
Table 2.2: Correlation between the mean serum PlGF protein levels in pre-surgery OSCC patients
and clinicopathological parameters of 72 OSCC patients …………. ……….74
Table 2.3: Univariate and multivariate recurrence-free survival analyses of the serum
PlGF protein levels and clinicopathological parameters of OSCC patients by
Cox proportional hazard regression model ……….……… 75
7
Figure Index
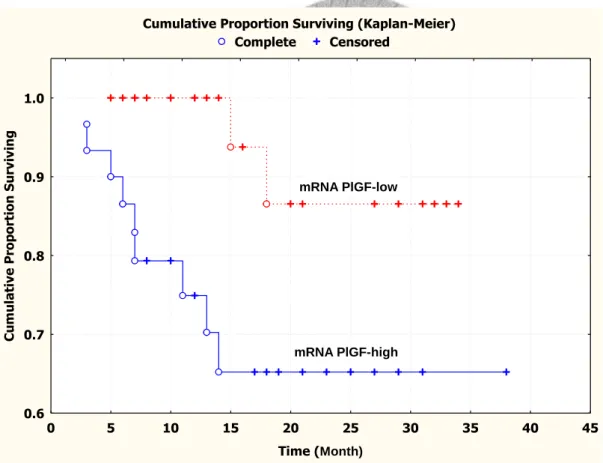
Figure 1.1: Kaplan-Meier survival curve showing that the cumulative recurrence-free
survival for patients with PlGF mRNA -∆CT value > 2 was significantly
poorer than that for patients with PlGF mRNA -∆CT value ≦ 2 ……….... 76
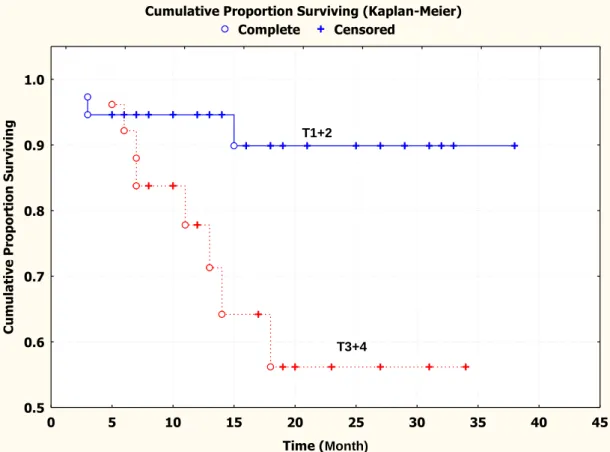
Figure 1.2: Kaplan-Meier survival curve showing that the cumulative recurrence-free
survival for patients with larger tumor size (T3+T4) was significantly
poorer than that for patients with smaller tumor size (T1+T2) ……….….. 77
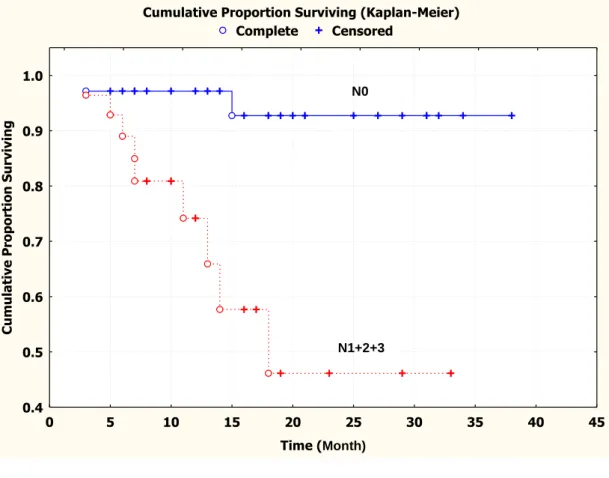
Figure 1.3: Kaplan-Meier survival curve showing that the cumulative recurrence-free
survival for patients with regional lymph node metastasis (N1+N2+N3)
was significantly poorer than that for patients without regional lymph
node metastasis (N0) ………..…………78
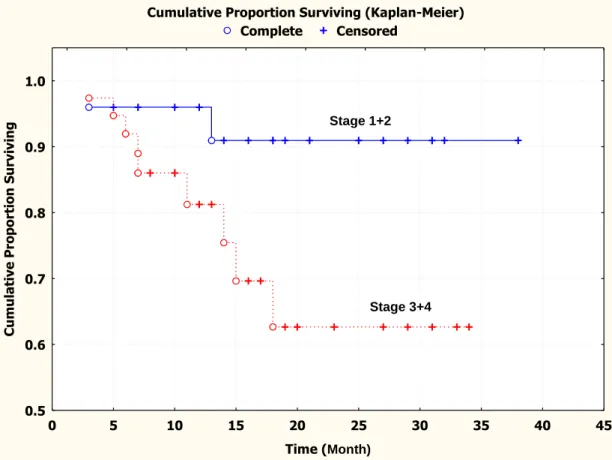
Figure1.4: Kaplan-Meier survival curve showing that the cumulative recurrence-free
survival for patients with higher clinical stages (stage 3+4) was
significantly poorer than that for patients with lower clinical stages
(stage 1+2) ……….……….……… 79
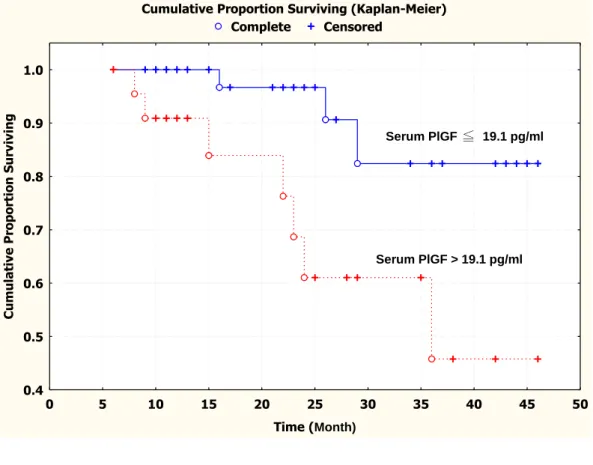
Figure 2.1: Kaplan-Meier survival curve showing that the cumulative recurrence-free
survival for patients with low serum PlGF protein level (≦ 19.1 pg/ml)
was significantly higher than that for patients with high serum PlGF
protein level (> 19.1 pg/ml) ……….……… 80
8
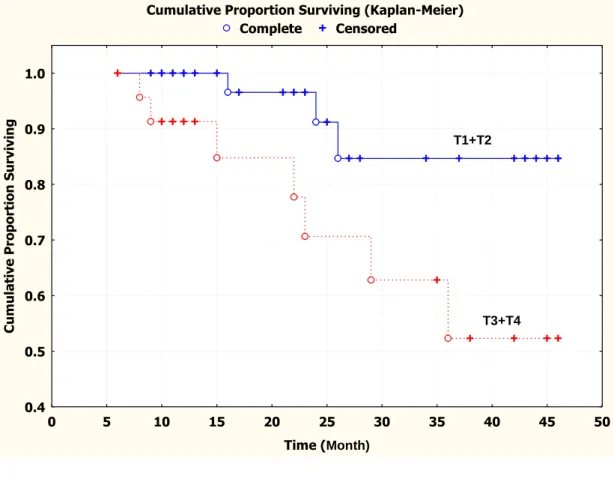
Figure 2.2: Kaplan-Meier survival curve showing that the cumulative recurrence-free
survival for patients with smaller tumor size (T1+T2) was significantly
higher than that for patients with larger tumor size (T3+T4) ……..…………. 81
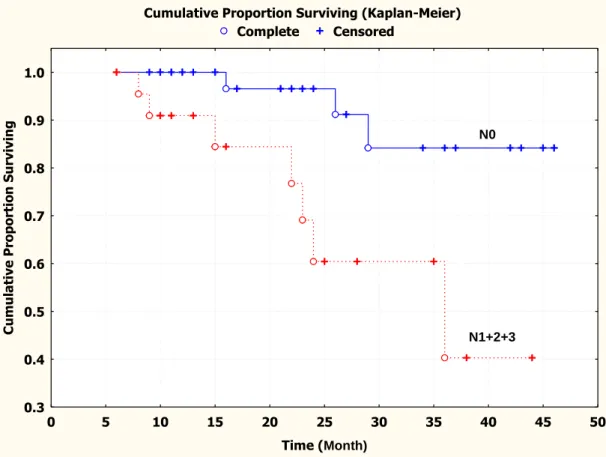
Figure 2.3: Kaplan-Meier survival curve showing that the cumulative recurrence-free survival for
patients without regional lymph node metastasis (N0) was
significantly higher than that for patients with regional lymph node
metastasis (N1+N2+N3) ……….……….. 82
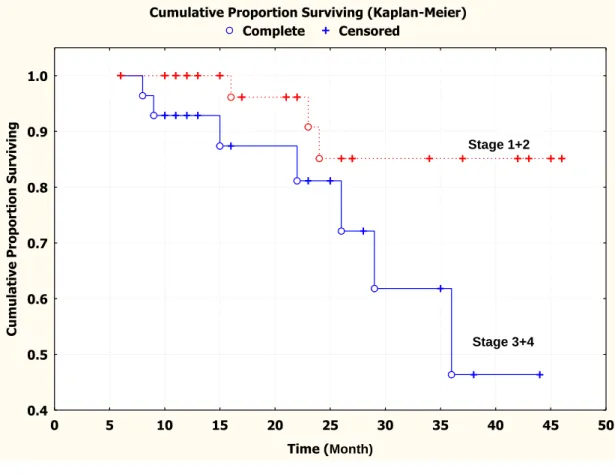
Figure 2.4: Kaplan-Meier survival curve showing that the cumulative recurrence-free
survival for patients with lower clinical stages (stage 1+2) was
significantly higher than that for patients with higher clinical stages
(stage 3+4) ……….……….………….. 83
9
中文摘要
背景: 胎盤生長因子(Placenta growth factor,PlGF)是屬於血管內皮生長因子家族(VEGF family)。PlGF 可藉由 VEGFR-1 引發腫瘤血管新生,PlGF 過度表現與全身很多癌症有關,亦 包括口腔癌。之前,我們利用免疫組織化學染色法研究顯示,口腔鱗狀細胞癌 (OSCC) 組織 中,有過度 PlGF 蛋白表現,則 OSCC 患者之預後較差。但並無文獻提到有關 OSCC 患者,
其癌組織中 PlGF mRNA 量及血清中 PlGF 蛋白量,與 OSCC 患者預後之相關性。因此,本研 究進一步檢測 OSCC 患者癌組織中 PlGF mRNA 量及血清中 PlGF 蛋白量,是否和 OSCC 患者 之腫瘤發展、復發及存活有關。
材料及方法
本 研 究 第 一 部 分 利 用 即 時 定 量 聚 合 酶 鏈 式 反 應(quantitative real-time reverse transcription-polymerase chain reaction; qRT-PCR)方法,測量 63 例 OSCC 癌組織及癌旁正常口 腔黏膜 (non-SCC) 中 PlGF mRNA 量。Threshold cycle (CT) 定義為 DNA 可被監測出之最少 PCR cycle 數目。CT數值大表示 PlGF mRNA copy number 少。OSCC 及 non-SCC 組織中,PlGF mRNA 的差異表現為 -∆CT = – (OSCC CT – non-OSCC CT) 。-∆CT數值愈大,表示 OSCC 組織 中 PlGF mRNA copy number 愈多。ANOVA、卡方檢定(Chi-square test)、Kaplan-Meier 存活 率方法及 Cox proportional hazard regression model,來分析癌組織中 PlGF mRNA 量與 OSCC 患 者 臨 床 病 理 參 數 及 存 活 率 之 相 關 性 。 本 研 究 第 二 部 分 利 用 酵 素 連 結 免 疫 吸 附 法 (enzyme-linked immunosorbent assay, ELISA),探討 72 位 OSCC 患者術前及術後三個月及 30 位具正常口腔黏膜者,其血清中 PlGF 蛋白量。再利用 ANOVA、卡方檢定、Kaplan-Meier 存 活率方法及 Cox proportional hazard regression model,來分析血清中 PlGF 蛋白量與 OSCC 患 者臨床病理參數及存活率之相關性,並試圖尋找預測存活時間的獨立預後因子。
結果
本研究第一部分發現,PlGF mRNA -∆CT的平均值較高和患者有較大腫瘤 (P = 0.03)、有
10
局部淋巴結轉移(P = 0.003)、有較高臨床分期 (P = 0.013) 及有局部復發 (P = 0.039) ,有統 計學上有意義之相關。以 Cox regression model 進行多變數分析發現,淋巴結轉移情形 (P = 0.019)、PlGF mRNA -∆CT value > 2 (P = 0.016)為影響 OSCC 患者存活時間的獨立預測因子。
Kaplan-Meier 存活分析發現, PlGF mRNA -∆CT value > 2 比 PlGF mRNA -∆CT value ≤ 2 之 OSCC 患者,有較差的無復發存活率 (log-rank test, P = 0.017)。
本研究第二部分發現,術前 OSCC 患者血清中 PlGF 蛋白量的平均值,比具正常口腔黏 膜者的平均值高,且具統計學上有意義之差別 (19.1 ± 10.7 pg/ml vs. 10.1 ± 4.5 pg/ml,P <
0.001)。術前 OSCC 患者血清中 PlGF 蛋白量平均值較高和患者有較大腫瘤 (P = 0.015)、有局 部淋巴結轉移 (P =0.001)、有較高臨床分期 (P = 0.002)及有局部復發(P = 0.037),有統計學 上有意義之相關。Cox regression model 進行多變數分析發現,血清中 PlGF 蛋白濃度為影響 存活時間的獨立預測因子(P = 0.014)。Kaplan-Meier 存活分析發現,血清中 PlGF 蛋白 > 19.1 pg/ml 比血清中 PlGF 蛋白 ≦ 19.1 pg/ml 者,會有較差的無復發存活率 (log-rank test,P = 0.009)。當以血清中 PlGF 蛋白為 19.1 pg/ml (正常平均值加 2 個標準差) 為切點時, 預測腫瘤 復發的敏感度 (sensitivity)、特異度 (specificity)及陽性預測值 (positive predictive value) 分別 為 80%、56% 和 78%。
結論
本研究發現,OSCC 患者組織中 PlGF mRNA 量及血清中 PlGF 蛋白量,可以預測 OSCC 之腫 瘤大小、有無局部淋巴結轉移、臨床分期、有無局部復發,及其無復發存活率,因此 OSCC 患者組織中 PlGF mRNA 量及血清中 PlGF 蛋白量,可以當作 OSCC 患者預後的指標。
關鍵詞: 胎盤生長因子、口腔癌、血管新生
11
Abstract
Background/Purpose
Placenta growth factor (PlGF) belongs to vascular endothelial growth factor (VEGF) family.
PlGF could induce tumor angiogenesis through binding to VEGFR-1, which was highly expressed
in a variety of human cancers, including oral cancer. Our previous study demonstrated a significant
association of higher expression of PlGF protein with poor prognosis of patients with oral squamous
cell carcinoma (OSCC). This study further examined whether the expression of PlGF mRNA in
OSCC tissues and the serum PlGF protein level in OSCC patients could be used to predict the
progression and prognosis of OSCCs in Taiwan.
Materials and methods
In the first part of this study, we used quantitative real-time reverse transcription-polymerase
chain reaction (quantitative RT-PCR) to detect the PlGF mRNA levels in 63 paired OSCC and
adjacent normal-looking oral mucosa (non-OSCC) tissues. Threshold cycle (CT) was defined as the
PCR cycle number needed to generate a pre-determined amount of DNA (threshold). For a chosen
threshold, a smaller starting copy number of mRNA results in a higher CT value. In this study, the
relative expression level of tissue PlGF mRNA in each OSCC patient was expressed as -∆CT = –
(OSCC CT – non-OSCC CT). Thus, the higher the -∆CT, the greater the copy number of PlGF
mRNA in tissues. In the second part of this study, serum samples were obtained from 72 OSCC
12
patients before and 3 months after surgical cancer excision and from 30 normal control subjects.
Serum PlGF protein levels were determined by enzyme-linked immunosorbent assay (ELISA).
Results
In the first part of this study, we found that the higher mean PlGF mRNA -∆CT value was
significantly associated with OSCCs with larger tumor size (P = 0.03), positive lymph node
metastasis (P = 0.003), more advanced clinical stages (P = 0.013) or the presence of loco-regional
recurrence (P = 0.039). Positive lymph node metastasis (P = 0.019) and PlGF mRNA -∆CT value >
2 (P = 0.016) were identified as two independent unfavorable prognosis factors by multivariate
analyses with Cox regression model. Moreover, Kaplan-Meier curve showed that OSCC patients
with a PlGF mRNA -∆CT value > 2 had a significantly poorer recurrence-free survival than those
with a PlGF mRNA -∆CT value ≦ 2 (log-rank test, P = 0.017). In the second part of this study, we
found that the mean serum PlGF protein levels were significantly higher in pre-surgery OSCC
patients than in normal controls (19.1 ± 10.7 pg/ml vs. 10.1 ± 4.5 pg/ml, P < 0.001). Serum PlGF
protein levels dropped to near the normal control levels after surgical cancer removal. Higher
pre-surgery serum PlGF protein levels were significantly associated with OSCCs with larger tumor
size (P = 0.015), positive lymph node metastasis (P = 0.001), more advanced clinical stages (P =
0.002), and loco-regional recurrence (P = 0.037). The serum PlGF protein level was identified as an
independent unfavorable prognosis factor by multivariate Cox regression analyses (P = 0.014).
13
Kaplan-Meier curve showed that OSCC patients with a higher serum PlGF protein level had a
significantly poorer cumulative recurrence-free survival than those with a lower serum PlGF protein
level (log-rank test, P = 0.009). When we used the serum PlGF protein level of 19.1 pg/ml (mean
normal control value plus 2 standard deviations) as a cutoff point, the sensitivity, specificity, and
positive predictive value for tumor recurrence was 80%, 56% and 78%, respectively.
Conclusion
This study found that PlGF mRNA level in OSCC tissues and serum PlGF protein level in
OSCC patients could be used to predict the tumor size, regional lymph node metastasis, clinical
stage, and recurrence of OSCCs and the prognosis of OSCC patients. Therefore, we conclude that
PlGF mRNA level in OSCC tissues and serum PlGF protein level in OSCC patients may be
valuable biomarkers for prediction of progression, recurrence and prognosis of OSCC in Taiwan.
Key words: Placenta growth factor、oral cancer、angiogenesis
14
I. Introduction and literature review
In Taiwan, oral squamous cell carcinomas (OSCC) rank as the sixth most prevalent cancer in
both sexes and account for the fourth most common cancers in males in 2010 (Cancer registry
annual report in Taiwan area, 2010). Despite significant efforts committed in recent years in
diagnosis and treatment of OSCC, the overall survival rate has remained approximately 50%
(Forastiere et al., 2001). Tumor recurrence is one of the major causes resulting in a poor survival of
OSCC patients. This suggests an urgent need for a novel biomarker to predict the progression,
recurrence and prognosis of OSCCs.
Placenta growth factor (PlGF) is a member of the vascular endothelial growth factor (VEGF)
family, which is related to angiogenesis and carcinogenesis (Fischer et al., 2008).PlGF was found
to promote proliferation, differentiation, and survival of endothelial cells by binding to VEGF
receptor 1 (VEGFR1, also known as FLT1) (Fischer et al., 2008). Clinically, overexpressed PlGF
mRNA or protein level correlates with pathological angiogenesis (Fischer et al., 2008), tumor cell
growth (Ikai et al., 2005; Lacal et al., 2000; Marcellini et al., 2006), positive lymph node metastasis
(Chen et al., 2004; Marcellini et al., 2006; Parr et al., 2005),advanced clinical stage (Chen et al.,
2004; Wei et al., 2005; Zhang et al., 2005), recurrence (Parr et al., 2005; Ho et al., 2007), and poor
prognosis (Chen et al., 2004; Parr et al., 2005; Wei et al., 2005)of a variety of cancers. Recently, we
reported that the higher mean PlGF labeling index assessed by immunohistochemical staining are
15
significantly associated with the more advanced progression and poorer prognosis of OSCC (Cheng
et al. 2010). Several previous studies have also reported increased serum PlGF protein levels in
patients with renal cell (Matsumoto et al., 2003), pancreatic (Chang et al., 2008; Sabbaghian et al.,
2010) or colorectal carcinomas (Rahbari et al., 2002; Wei et al., 2009).
In this study, we further evaluated whether the PlGF mRNA level in OSCC surgical specimen
measured by the quantitative real-time reverse transcription-polymerase chain reaction (quantitative
RT-PCR) and the serum PlGF protein levels in OSCC patients measured by the enzyme-linked
immunosorbent assay (ELISA) could be valuable biomarkers to predict the therapeutic effect,
progression, recurrence and prognosis of OSCC patients.
1.1 Overview of oral squamous cell carcinoma
1.1.1 Epidemiology of global oral cancers
Oral cancer is the sixth most common cancer worldwide, with a high prevalence in South Asia.
An estimated 263,900 new cases and 128,000 deaths from oral cavity cancer (including lip cancer)
were reported in 2008 worldwide. Generally, the highest oral cavity cancer rates are found in
Melanesia, South-Central Asia, and Central and Eastern Europe and the lowest in Africa, Central
America, and Eastern Asia for both males and females. Smoking, alcohol consumption, smokeless
tobacco use, and HPV infections are the major risk factors for oral cavity cancer, with smoking and
alcohol drinking having synergistic effects (Blot et al., 1988; Hashibe et al., 2009). Worldwide,
16
smoking accounts for 42% of deaths from cancers of the oral cavity (including the pharynx) and
heavy alcohol consumption for 16% of the deaths; the corresponding percentages in high-income
countries are about 70% and 30%, respectively (Danaei et al., 2005). The rise in the incidence rate
of oral cancer in Taiwan may have been in part due to the increased consumption of areca quid and
alcohol (Ho et al., 2002). Oral cavity cancer mortality rates among males decreased significantly in
most countries, including those of Europe and Asia, over the past decades (Garavello et al., 2010;
Mayne et al., 2006). However, rates continued to increase in several Eastern European countries,
including Hungary and Slovakia (Garavello et al., 2010). The mortality rate increase in females in
most European countries largely reflects the ongoing tobacco epidemic (Garavello et al., 2010).
This contrasts with the decreasing trends at all ages in both males and females in the United States
and United Kingdom, where the tobacco epidemic began and declined earlier (Garavello et al.,
2010). However, incidence rates for oral cancer sites related to HPV infections, such as the
oropharynx, tonsil, and base of the tongue, are increasing in young adults in the United States and in
some countries in Europe, which is hypothesized to be in part due to changes in oral sexual
behavior (D’Souza et al., 2009; Marur et al., 2010).
1.1.2 Epidemiology of oral cancers in Taiwan
With rapid aging of populations, cancer has become the first leading cause of death in Taiwan
since 1982 (Chen et al., 2002). In 1982, the incidence rate of head and neck cancer was 5.12 per
100,000 people in males and 1.54 per 100,000 people in females. In 1991, the incidence rate of head
17
and neck cancer had not much changed, with the incidence rate being 6.02 and 1.51 per 100,000
people in males and females, respectively. However, in 2003, the incidence rate of head and neck
cancer significantly increased to 35.08 and 3.56 per 100,000 people in males and females, an
alarming 5.82-fold increase in men and 2.35-fold increase in woman in a decade was found (Chen
et al., 2008). In 2008, the incidence rate of head and neck cancer significantly increased to 37.57
per 100,000 people in males. In 2010, oral cavity cancer had become the 5th most common cancer
causing mortality in Taiwanese men. Similarly, the mortality rate also increased significantly, from
4.25 per 100,000 in 1995 to 9.6 per 100,000 in 2006, a 2.26-fold increase in the past decade. There
has been a trend toward lower age at diagnosis of head and neck cancer over time. From 1989 to
1993, the peak of incidence rate was for people aged 50–59 years, but this shifted to ages 40–49
years between 1993 and 2000. A similar trend was also found in the mortality rate. During the
period between 1991 and 1994, mortality rate peaked at age 50–59 years, but shifted to age 40–49
years between 1999 and 2002. These data are consistent with other regional reports from northern
and southern Taiwan. In 2006, the median age at death from head and neck cancer was 54 years
compared with 69 years in other forms of cancer. Gender differences in head and neck cancer have
been described, with a marked male predominance. A study analyzing 703 OSCC patients between
1985 and 1996 in southern Taiwan found a 51:1 male-to-female ratio (Chen et al., 1999). The
overall 5-year survival rate in patients with head and neck cancer is one of the lowest among
common malignant neoplasms and has not significantly changed during the last two decades.
18
Cancer clinical stage is the major determinant of survival rate. The 5-year survival rates of oral
cavity cancer patients in stages I, II, III and IV are 72–90%, 39–85%, 27–70% and 12–50%,
respectively (Liao et al., 2008; Liao et al., 2007; Ko et al., 1995). Survival rates for head and neck
cancer are significantly influenced by tumor size, lymph node involvement, distant metastasis,
tumor differentiation and areca quid chewing (Liao et al., 2004; 2008). Areca quid chewing
independently contributes to the risk of head and neck cancer, and the estimated prevalence of areca
quid chewing in Taiwanese patients with head and neck cancer is approximately 85% (Liao et al.,
2004; 2007; 2008). Approximately 50% of patients who were areca quid chewers are also alcohol
drinkers and tobacco smokers (Liao et al., 2004; 2007; 2008). A cumulative effect from areca quid
chewing, alcohol drinking and tobacco smoking has been observed, with a 123-fold increased risk
of oral cancer when the three risk factors are present (Ko YC et al., 1995). With regard to the
anatomical location of oral cavity cancers, approximately 30–40% of all cases occur in the tongue
or in the buccal mucosa. Altogether, lesions at these sites account for approximately 70% of all oral
cavity malignancies ( Liao et al., 2008; Chen et al.,1999; Ko et al.,1995).
1.1.3 Risk factors related to oral cancers
Oral cancer was known to be associated with cigarette smoking, excessive alcohol consumption, areca quid chewing, viral or fungal infection (candida, human papilloma virus (HPV), Herpex simplex virus, etc.), nutrition deficiency (Plummer-Vinson Syndrome, Vitamin A deficiency, Vitamin C deficiency, etc.), family hereditary, immunodeficiency, gene mutation, and so on. Recent 2003 IARC monograph declared areca quid chewing, by itself, to be a Group 1 carcinogen. The
19
Taiwanese chewers commonly use fresh, unripe betel fruit with slaked lime as an essential ingredient. The composition of areca quid differs geographically; the areca quid used in Taiwan contains areca nut, lime and Piper betel inflorescence (Yang et al., 2001; 2005). Piper betel inflorescence contains high concentrations of hydroxychavicol and safrole, whereas arecoline, a major areca nut alkaloid, is considered to be the most important carcinogen in the areca nut.
Arecoline has been shown to induce structural chromosomal aberration, sister chromatid exchange and micronuclei formation in different cell types (Jeng et al., 2001; Shirname et al., 1983). Studies in human oral cancer cells have shown that exposure to arecoline or areca nut extract (ANE) results in growth arrest in the late S and G2/M phases (Lee et al., 2006). Piper betel inflorescence, which contains safrole, is a unique ingredient of areca quid in Taiwan. Safrole–DNA adducts have been suggested to play an important role in oral carcinogenesis (Chen et al., 1999). A further report has provided evidence that alkaline saliva generated by chewing areca quid may play a role in cigarette-related nicotine-induced DNA damage, and reactive oxygen species may be involved in generating this DNA damage (Wu et al., 2005). These findings provide a molecular explanation for the synergistic effect of areca quid chewing and tobacco smoking in the development of head and neck cancer in Taiwan.
1.1.4 Precancerous lesions and conditions
Many OSCCs develop from premalignant conditions of the oral cavity (Silverman et al., 1984;
1968). The World Health Organization classifies oral precancerous/potentially malignant disorders
into 2 general groups.A precancerous lesion is “a morphologically altered tissue in which oral cancer is more likely to occur than its apparently normal counterpart.” These precancerous lesions
include leukoplakia, erythroplakia, and the palatal lesions of reverse smokers. A precancerous
20
condition is “a generalized state associated with significantly increased risk of cancer.” The
precancerous conditions include submucous fibrosis, lichen planus, epidermolysis bullosa, and
discoid lupus erythematosus. Intervention should be based on histopathological features of a biopsy
of the lesions. In many cases, a biopsy is mandatory so that such lesions can be further evaluated.
Currently, histological criteria (presence and degree of dysplasia) represent the gold standard in
precancerous lesion risk evaluation (Blot et al., 1988). The latest WHO classification (Barnes et al.,
2005) recommends a more objective gradings. The criteria for grading of oral epithelial dysplasia
are summarized as follows:
Mild dysplasia (grade I) demonstrates proliferation or hyperplasia of cells of the basal and
parabasal layers which does not extend beyond the lower third of the epithelium. Cytological atypia
is generally slight with only mild pleomorphism of cells or nuclei. Mitoses are not prominent, and
when present are usually basally located and normal. Architectural changes are minimal.
Moderate dysplasia (grade II) demonstrates a proliferation of atypical cells extending into the
middle one-third of the epithelium. The cytological changes are more severe than in mild dysplasia
and changes such as hyperchromatism, and prominent cell and nuclear pleomorphism may be seen.
Increased and abnormal mitoses may be present, but these are usually located in the basal layers.
Architectural changes may be seen in the lower half of the epithelium where there may be loss of
basal polarity and hyperplasia leading to bulbous rete pegs. However stratification and maturation
are relatively normal, often with hyperkeratosis.
21
Severe dysplasia (grade III) shows abnormal cell proliferation from the basal layer into the
upper third of the epithelium. Cytological and architectural changes can be very prominent. All the
changes seen in mild and moderate dysplasia are seen but in addition there is marked pleomorphism
often with abnormally large nuclei with prominent or even multiple nucleoli. Prominent and
suprabasal mitoses are usually evident and abnormal tripolar or star-shaped forms may be seen.
Apoptotic bodies may also be prominent. Architectural changes are severe, often with complete loss
of stratification and with deep abnormal keratinization and even formation of keratin pearls.
Abnormal forms of rete pegs are usual and bulbous rete pegs are regarded as particularly significant
in the diagnosis of severe dysplasia. Abnormal shaped rete pegs may also be seen, with lateral
extensions or small branches. These are quite abnormal and may be the earliest signs of invasion.
Occasional lesions may show prominent acantholysis with severe disruption of the architecture.
Although the epithelium may be thickened, severe dysplasia is sometimes accompanied by marked
epithelial atrophy.
Carcinoma in situ is the most severe form of epithelial dysplasia and is characterized by full
thickness cytological and architectural changes. In the oral cavity, such changes are rare, and often,
even in the presence of the most severe atypia, there is still an intact keratinized surface layer.
Carcinoma in situ is thought by some to be a premalignancy but others regard it as evidence of
actual malignant change but without invasion. Severe epithelial dysplasia has an overall malignant
transformation rate of about 16% but studies show a wide range of 7% – 50% (Silverman et al.,
22
1984; Schepman et al., 1998; Gupta et al., 1990; Bouquot et al., 1988; JBanoczy et al., 1976;
Amagasa et al., 1985; Vedtofte et al., 1987; Mincer et al., 1972; Pindborg et al., 1977; Lumerman et
al., 1995; Jaber et al., 2003). Moderate dysplasia has a malignant transformation potential of 3% –
15%, whereas mild epithelial dysplasia shows a very low risk (less than 5%). It is always assumed,
however, that there is a temporal progression of disease, analogous to multistage carcinogenesis,
and that mild dysplasia will progress to severe dysplasia and then to carcinoma.
1.1.5 Clinicopathological classification and treatment modalities of oral squamous cell
carcinoma
The WHO grading system (Pindborg et al., 1997) recommends 3 categories: well-differentiated,
moderately-differentiated and poorly-differentiated. This usually depends on the subjective
assessment of the degree of keratinization, cellular and nuclear pleomorphism, and mitotic activity
(Woolgar, 2006). The influence of histologic grading as a prognostic factor in OSCC was assessed
in 215 patients and was found to be a significant predictor of locoregional failure and tumour
recurrence (Kademani et al., 2005).
Surgery is the most well-established mode of initial definitive treatment for a majority of oral
cancers, with a longstanding history of being the accepted method of treatment for oral cancers over
a century. Introduction of ionizing radiation, following the discovery of radium, became an
important means of non-surgical treatment of oral carcinoma. However, in the majority of patients
23
with advanced cancer, radiotherapy is employed in conjunction with surgery, most often offered as
post-operative treatment. Chemotherapy in the management of oral carcinoma was considered palliative in the 1950’s, 60’s and 70’s. However with the introduction of Cis-platinum, clinical trials
of induction chemotherapy demonstrated that response to chemotherapy is observed in a significant
number of patients (Shah et al., 2009). For patients with advanced-staged disease, the current
preference for the sequence of combined modality treatment program is surgical resection with
immediate appropriate reconstruction followed by post-operative radiation therapy or post-operative
concurrent chemoradiotherapy. The observations from two prospective randomized trials of
adjuvant chemoradiotherapy have shown that patients who have extracapsular extension of disease
in metastatic cervical lymph nodes and those who have positive margins have a significant
improvement in local regional control and disease free survival by addition of chemotherapy to
postoperative radiation therapy compared to post-operative radiation therapy alone (Bernier et al.,
2005). Targeted therapies with epidermal growth factor receptor (EGFR) inhibitors are an active
area of investigation at this time. Immunotherapy and gene therapy are also areas of research where
further work needs to be done (Shah et al., 2009).
1.2 Angiogenesis
The dependence of tumor growth on the development of a neovasculature is now a
well-established aspect of cancer biology (Folkman, 1971). Angiogenesis is important for supply of
24
oxygen, nutrients, growth factors, hormones, and proteolytic enzymes, influences on hemostatic
factors that control the coagulation and fibrinolytic system, and dissemination of tumor cells to
distal sites (Folkman, 1990; Fidler, 1994). The angiogenic process is a highly complex, dynamic
process regulated by a number of pro- and anti-angiogenic molecules. The so called ‘‘switch’’ to an
angiogenic phenotype is considered a hallmark of the malignant process whereby proangiogenic
mechanisms overwhelm or circumvent negative regulators of angiogenesis (Hanahan, 2000). In
general, increased tumor vascularization (eg, increased microvessel density) and tumor expression
of proangiogenic factors has been associated with advanced tumor stage and poor prognosis in a
variety of human cancers (Brown, 1997; Dvorak, 1995; Reinmuth , 2003).
1.2.1 Angiogenic growth factors
The known endothelial cell specific growth factors and their receptors can be classified into
vascular endothelial growth factor (VEGF) and angiopoietin (Ang) families. VEGF is a major
inducer of angiogenesis and it is structurally related to PlGF (placenta growth factor), VEGF-B,
VEGF-C, VEGF-D and Orf virus derived VEGF (also called VEGF-E) (Ferrara and Davis- Smyth,
1997; Erikson and Alitalo, 1999; Persico et al., 1999; Achen et al., 1998; Ogawa et al., 1998; Meyer
et al., 1999). Loss of even a single VEGF allele results in embryonic lethality, suggesting that
VEGF levels are critical during embryonic angiogenesis (Carmeliet et al., 1996). There are to date
three known receptor tyrosine kinase that bind VEGFs (VEGFR-1, VEGFR-2 and VEGFR-3)
25
(Neufeld et al., 1999; Veikkola et al., 2000; Aprelikova et al., 1992). In adults, VEGFR-1 and
VEGFR-2 are expressed mainly in the blood vascular endothelium, while VEGFR-3 is mostly
restricted to the lymphatic endothelium (Neufeld et al., 1999; Kaipainen et al., 1995). In addition to
endothelial cell specific growth factors, there is also a wide range of other less specific angiogenic
growth factors, including basic fibroblast growth factor (bFGF) and transforming growth factor- β
(TGF-β) (Friesel and Maciag, 1995). Recently, Eph receptor tyrosine kinases and their
cell-surface-bound ephrin ligands were found. Among nontyrosine kinase receptors, a recently
characterized member of the Delta family of Notch ligands, Dll4 is expressed specifically in
vascular endothelial cells, suggesting a role in the control of endothelial cell biology (Shutter et al.,
2000).
1.2.2 Angiogenic inhibitors
A large and structurally diverse group of endogenous angiogenesis inhibitors has also been
discovered, including thrombospondin-1 (Tolsma et al., 1993), interferon-α/β (Dvorak and Gresser,
1989), angiostatin (O'Reilly et al., 1994), endostatin (O'Reilly et al., 1997) and anti-thrombin III
(O'Reilly et al., 1999). Some of these are fragments of proteins, that are either components of the
extracellular matrix (ECM) or of enzymes of the blood clotting pathways.
1.2.3 Tumor angiogenesis
When a primary tumor first arises, proliferation of cancer cells may be balanced by apoptosis
26
and the tumor may remain undetectable for years (Hanahan and Folkman, 1996). Such a
phenomenon, called tumor dormancy, may depend on a rate-limiting role of neovascularization
(Holmgren, 1996). The onset of neovascularization in primary tumors can be relatively sudden,
described as the angiogenic switch (Hanahan and Folkman, 1996; Folkman et al., 1989). The
angiogenic switch in tumors is currently thought to be caused by a shift in the net balance of
positively and negatively acting angiogenic mediators, angiogenic growth factors and angiogenesis
inhibitors, respectively. The mechanisms underlying this shift of balance remain incompletely
understood. Thus, some oncogenes and tumor-suppressor genes contribute to the angiogenic switch
by causing up- or down-regulation of endogenous angiogenesis inhibitors or pro-angiogenic growth
factors (Kerbel et al., 1998; Hanahan and Folkman, 1996). On the other hand, the control of
bioavailability of angiogenic activators and inhibitors also has a role in the regulation of the
angiogenic switch. A variety of proteases can release TGF-β or bFGF stored in the extracellular
matrix (Taipale et al., 1998; Whitelock et al., 1996), whereas proteolytic cleavage of plasmin results
in the formation of the inhibitor, angiostatin (Gately et al., 1997). Some studies have suggested that
instead of growing as avascular masses, some tumors, especially metastases, may initially grow by
coopting existing host vessels (Holash et al., 1999). This coopted host vasculature would not
immediately undergo angiogenesis to support the tumor, but instead regresses, leading to secondary
hypoxia, massive tumor cell loss, and further to hypoxia-induced angiogenesis (Holash et al., 1999).
Furthermore, there is also evidence of mobilization of bone marrow-derived endothelial progenitor
27
cells (EPC) in various angiogenesis models, resulting inaugmented neovascularization (Asahara et
al., 1999; Peichev et al., 2000; Takahashi et al., 1999).
1.2.4 Angiogenesis and tumor metastasis
Several studies since Weidner's first report in 1991 reveal that the higher the microvessel count
is in areas of highest vessel density, the lower is the rate overall survival of the tumor patients
(Weidner et al., 1991). For a tumor cell to metastasize, it must pass through several barriers and
finally be able to survive and grow in the target tissue. First tumor cells must enter the vasculature
in the primary tumor. VEGF and bFGF, secreted by the tumor cells, induce the expression of
plasminogen activator and collagenases, contributing to the degradation of basement membranes
(Kalebic et al., 1983; Nagy et al., 1989). In addition, proliferating tumor capillaries with fragmented
and leaky basement membranes are easily penetrated by tumor cells. In fact, a considerable
percentage of tumor blood flow is indirect contact with tumor cells (Hashizume et al., 2000). After
overcoming the first vascular barrier, tumor cells have to survive the circulation, attach to the
micro-vasculature of the target organ, exit from this vasculature (usually through postcapillary
venular endothelium), and survive in the target tissue (Nicolson, 1988). Furthermore, in order to
form macrometastases, micrometastatic cells must also be able to induce angiogenesis in their new
target tissue. While angiogenesis is required for tumor growth, active lymphangiogenesis has not
been detected within tumors. functional lymphatic vessels are often absent from the interior of solid
28
tumors, possibly due to collapse of the vessels by the interstitial pressure induced by the growing
cancer cells and the leaky tumor blood vessels (Leu et al., 2000). Functional, enlarged lymphatics
are, however, detected in the tumor periphery. VEGF-C is a growth factor for both lymphatic and
blood vessels, and the expression of its specific receptor VEGFR-3 becomes restricted mainly to the
lymphatic endothelium during development (Kaipainen et al., 1995; Kukk et al., 1996). Several
recent reports have suggested a correlation between VEGF-C expression and lymphatic metastasis
in tumors (Ohta et al., 1999; Tsurusaki et al., 1999; Yonemura et al., 1999). Even though the tumor
itself lacks functional lymphatic vessels, the VEGF-C overexpression induced increase of
peritumoral lymphatics makes the lymphatic vessels more accessible to the metastases.
1.3 VEGF and cancers
1.3.1 Vascular endothelial growth factor family, VEGF family
VEGF-A (commonly referred to as VEGF) was first identified by Dvorak et al. (2002) as a
vascular permeabilitity-inducing factor secreted by tumor cells, and thus referred to as vascular
permeability factor (VPF). Ferrara et al. (1996) later isolated and cloned VEGF-A as an endothelial
specific mitogen. VEGF is a major inducer of angiogenesis and it is structurally related to PlGF
(placenta growth factor), VEGF-B, VEGF-C, VEGF-D and Orf virus derived VEGF (also called
VEGF-E) (Ferrara and Davis-Smyth, 1997; Erikson and Alitalo, 1999; Persico et al., 1999; Achen
et al., 1998; Ogawa et al., 1998; Meyer et al., 1999). The biological functions of the VEGF family
29
are mediated by activation of three structurally homologous tyrosine kinase receptors, VEGFR-1,
VEGFR-2, and VEGFR-3 (Neufeld et al., 1999; Veikkola et al., 2000; Aprelikova et al., 1992)
1.3.1.1 VEGF-A
VEGF-A is a 45-kDa homodimeric glycoprotein with a diverse range of angiogenic activities.
The VEGF-A gene undergoes alternative splicing to yield mature isoforms of 121, 165, 189, and
206 amino acids (Houck, 1991; Tischer et al., 1991). In addition, some less commonly expressed
variants have also been identified (VEGF145 and VEGF183) (Neufeld et al., 1999).
VEGF121 is freely secreted, whereas the largest isoforms (VEGF189 and VEGF206) are
sequestered in the extracellular matrix (ECM) and require cleavage by proteases for their activation
(Dvorak, 2002). VEGF165 exists in both a soluble and an ECM-bound form (Keyt et al., 1996). The
ECM-bound isoforms of VEGF-A, VEGF-C, and VEGF-D can be released in a diffusible form by
plasmin cleavage at the C-terminus, which generates a bioactive fragment (Park et al., 1993;
McColl et al., 2003). Alternatively, VEGF can be released from the ECM by MMP-9 to initiate the
angiogenic switch (Bergers et al., 2000). VEGF165 is the predominant isoform and is commonly
overexpressed in a variety of human solid tumors. In mice, homozygous or heterozygous deletion of
the VEGF gene is embryonically lethal, resulting in defects in vasculogenesis and cardiovascular
abnormalities, demonstrating that VEGF is essential for development (Carmeliet et al., 1996;
Ferrara, 1996). VEGF-A is also important to a number of postnatal angiogenic processes, including
30
wound healing, ovulation, menstruation, maintenance of blood pressure, and pregnancy (Brown,
1992). VEGF-A has also been linked to several pathologic conditions associated with increased
angiogenesis, including arthritis, psoriasis, macular degeneration, and diabetic retinopathy (Ferrara
et al., 2003). In healthy conditions, endothelial cells survive for prolonged periods owing to the low
level of VEGFA expression by the vasculature, which acts as a survival signal. Hence, when
quiescent endothelial cells are deprived of this trophic VEGFA signal, they become dysfunctional
(pro-thrombotic), lose their vasorelaxing activity (nitric oxide release) or even disappear altogether
(which leads to bleeding) (Baffert et al., 2006; Gerber et al., 1999; Lee et al., 2007). Furthermore,
large amounts of VEGFA are produced in healthy conditions, which suggest that VEGFA is needed
for endothelial-cell homeostasis (Rudge et al., 2007). VEGFA binds to FLT1 with an affinity
(dissociation constant; Kd ~2–10 pM) that is much higher than for FLK1 (Sawano et al., 2001). Yet,
VEGFA induces weaker tyrosine-kinase activity in FLT1, possibly because of an inhibitory
sequence in the juxtamembrane domain that represses FLT1 activity (Seetharam et al., 1995;
Waltenberger et al., 1994; Gille et al., 2000). This weak tyrosine-kinase activity of FLT1 and its
high affinity for VEGFA have led to the development of a model in which FLT1 acts as a decoy
receptor and modulates angiogenesis through its ability to sequester VEGFA, and thereby reduces
signalling through FLK1 (Park et al., 1994). Additional functional diversity may occur through the
formation of dimers between VEGF family members. For example, VEGF-A may form
heterodimers with either PlGF (Cao et al., 1996) or VEGF-B (Olofsson et al., 1996). In humans,
31
because of the alternative splice variants, this has the potential to create enormous diversity and
presents a challenge to assessing the functional consequences of heterodimerization (Birkenhager et
al., 1996). For example, the six human VEGF-A and four human PlGF isoforms could theoretically
form 24 VEGF-PlGF heterodimers, and VEGF-A could theoretically form 12 different
combinations with the two human VEGF-B variants.
1.3.1.2. VEGF-B and PlGF
VEGF-B and PlGF null mice display no defects in embryonic vasculogenesis or developmental
abnormalities, suggesting that the role of PlGF and VEGF-B may be redundant (Carmeliet, 2001).
However, loss of PlGF impairs angiogenesis, plasma extravasation, and collateral growth during
ischemia, inflammation, wound healing, and tumor growth, suggesting a role for PlGF in pathologic
states in the adult. VEGF-B exists as two isoforms (VEGF167 and VEGF 186) that bind to FLT1
and to neuropilin 1 (Neufeld, 2001). Neuropilin 1 is widely expressed in various tissues (Nash, 2006)
including endothelial and mural cells, brown fat, skin, lung, placenta, brain and retina, but it is
particularly abundant in the heart and skeletal muscle (Li, 2001). Despite some evidence for
VEGFB activity in vitro, genetic studies have revealed that VEGFB-deficient mice are healthy and
fertile, and do not display vascular defects, which indicates that VEGFB is redundant in
angiogenesis in the developing embryo and healthy adult in vivo (Li et al., 2008; Aase et al., 2001;
Bellomo et al., 2000). The role of VEGFB in tumor growth remains largely elusive. VEGFB has
32
been detected in a wide range of tumors, including meningioma, and colorectal, lung and breast
cancer (Andre et al., 2000; Donnini et al., 1999; Niki et al., 2000). Moreover, VEGFB expression
correlates with microvascular density in oral squamous cell carcinoma (Shintani et al., 2004).
1.3.1.3 VEGF-C and VEGF-D
The VEGF homologs VEGF-C and VEGF-D play key roles during embryonic and postnatal
lymphangiogenesis. Homozygous deletion of the VEGF-C gene in mice is embryonically lethal and
heterozygous deletion results in postnatal defects associated with defective lymphatic development
(Karkkainen et al., 2004). Both factors induce lymphangiogenesis in transgenic mouse models
(Jeltsch et al., 1997; Veikkola et al., 2001). VEGF-C and VEGF-D may also play a role in new
blood vessel growth as well, especially during pathological states such as tumor growth.
1.3.1.4. VEGF-E
VEGF-E is not a mammalian VEGF homolog, but rather a viral protein encoded by the
parapoxvirus Orf virus, which preferentially utilizes kinase-insert domain-receptor (KDR)/fetal
liver kinase-1 (Flk-1) receptor and carries a potent mitotic activity without heparin-binding domain
(Ogawa et al., 1998). VEGF-E shares 22% sequence identity to VEGF-A.
1.3.2 VEGF receptors
33
VEGF ligands mediate their angiogenic effects via several different receptors. Two receptors
were originally identified on endothelial cells and characterized as the specific tyrosine kinase
receptors VEGFR-1 (also referred to as fms-like tyrosine kinase 1 [Flt-1]) (Shibuya et al., 1990).
VEGFR-2 (also referred to KDR, (Terman, 1992) and the murine homologue, Flk-1) (Matthews et
al. 1991). These two receptors share 44% homology and possess a characteristic structure consisting
of seven extracellular immunoglobulin-like domains, a single transmembrane domain, and a
consensus tyrosine kinase domain interrupted by a kinase insert domain (Shibuya et al., 1990;
Terman, 1991). More recently, an additional tyrosine kinase receptor, VEGFR-3 (also referred to as
fms-like tyrosine kinase 4 [Flt-4]), was identified and has been found to be primarily associated
with lymphangiogenesis (Kaipainen et al., 1995; Paavonen et al., 2000). The various members of
the VEGF family have differing binding specificities for each of these receptors. All of the
VEGF-A isoforms bind to both VEGFR-1 and VEGFR-2, whereas PlGF-1, PlGF-2 and VEGF-B
are specific for VEGFR-1 binding and activation (Park et al., 1994; Olofsson et al., 1998; Silvestre
et al., 2003). Naturally occurring heterodimers of VEGF-A and PlGF have also been identified that
can bind to and activate VEGFR-2 (Cao et al., 1996; DiSalvo et al., 1995). VEGF-E specifically
interacts with VEGFR-2 whereas VEGF-C and VEGF-D interact with both VEGFR-3 and
VEGFR-2 (Shibuya et al., 2003; Achen et al., 1998; Joukov et al., 1996).
1.3.2.1 VEGF-R1
34
VEGFR-1 is a receptor for VEGF-A and has the unique ability to bind VEGF-B and PlGF.
VEGFR-1 is critical for physiologic and developmental angiogenesis. VEGFR-1 null mice die in
utero between days 8.5 to 9.5 due to excessive hemangioblast proliferation and poor organization of
vascular structures (Fong et al., 1995). VEGFR-1 was initially thought to be a negative regulator of
VEGF activity either by acting as a decoy receptor for VEGF (Hiratsuka et al., 1998) or by
downregulating VEGFR-2–mediated signaling (Dunk et al., 2001). VEGF-mediated stimulation of
VEGFR-1 autophosphorylation and signaling in endothelial cells is weak when compared to
signaling through VEGFR-2 (Waltenberger et al., 1994). A repressor motif has been identified in
the juxtamembrane region of VEGFR-1 that impairs phosphatidylinositol 3-kinase (PI3K) signaling
and endothelial cell migration in response to VEGF stimulation (Gille et al., 2000; Zeng et al.,
2001). However, other studies have indicated that VEGFR-1 has a positive, functional role in
certain cell types, participating in monocyte migration (Barleon et al., 1996; Clauss et al., 1996),
recruitment of endothelial cell progenitors (Lyden et al., 2001) increasing the adhesive properties of
natural killer cells (Chen et al., 2002) and inducing growth factors from liver sinusoidal endothelial
cells (LeCouter et al., 2003). A recent study (Autiero et al, 2003) showed that activation of
VEGFR-1 by PlGF resulted in transphosphorylation of VEGFR-2 in endothelial cells co-expressing
these receptors. Furthermore, VEGF/PlGF heterodimers are capable of activating intramolecular
VEGFR cross talk through formation of VEGFR-1/VEGFR-2 heterodimers. Other recent studies
have shown that during pathologic conditions such as tumorigenesis, VEGFR-1 is a potent, positive
35
regulator of angiogenesis (Hiratsuka et al., 2001). A naturally occurring, alternatively spliced
soluble form of VEGFR-1 (sVEGFR-1) also exists (Kendall et al., 1993). sVEGFR-1 may function
to reduce or modulate endogenous VEGF or PlGF activity.
1.3.2.2. VEGF-R2
VEGFR-2 mediates the majority of the downstream effects of VEGF-A in angiogenesis,
including microvascular permeability, endothelial cell proliferation, invasion, migration, and
survival (Zeng et al., 2001; Millauer et al., 1993). The importance of VEGFR-2 in vasculogenesis is
demonstrated by the fact that hetero and homozygous knockout mice die in utero of defects in blood
island formation and vascular development (Shalaby et al., 1995). VEGFR-2–mediated proliferation
of endothelial cells involves activation of a phospholipase C gamma-protein kinase C-Raf-MAP
kinase signaling pathway, whereas survival and migration is believed to involve PI3K and focal
adhesion kinase, respectively (Veikkola et al., 2000; Abedi et al., 1997).
1.3.2.3 VEGFR3
VEGFR-3 is a receptor tyrosine kinase originally cloned from a human leukemia cell line and
human placenta (Pajusola et al., 1993; Galland et al., 1993). VEGFR-3 preferentially binds
VEGF-C and VEGFD. VEGFR-3 is expressed throughout the embryonic vasculature, but during
development and in the adult, its expression is limited to lymphatic endothelial cells (Kaipainen et
al., 1995). Homozygous deletion of the VEGFR-3 gene in mice leads to embryonic death at day 10
36
to 12.5, with an underdeveloped yolk sac, poor perineural vasculature, and pericardial fluid
accumulation (Dumont et al., 1998). VEGFR-3 activation and upregulation of its ligands have been
observed in certain neoplastic conditions, including breast cancer and melanoma with elevated
levels of VEGF-C or VEGF-D associated with lymph node metastasis in patients (Achen et al.,
2001; Valtola et al., 1999; Pepper et al., 2003).
1.3.3 Functions of VEGF
1.3.3.1 Vessels Permeability
VEGF is also termed vascular permeability factor (VPF) (Dvorak et al., 1995). In fact, VEGF
is one of the most potent inducers of vascular permeability known - 50,000-fold more potent than
histamine (Dvorak, 2002). This ability to enhance microvascular permeability is one of the most
important properties of VEGF, especially with regards to the hyperpermeability of tumor vessels
that is thought to be largely attributable to tumor cell expression of VEGF. It has been suggested
that the increase in permeability results in the leakage of several plasma proteins, including
fibrinogen and other clotting proteins. This can lead to the deposition of fibrin in the extravascular
space, which subsequently retards the clearance of edema fluid and transforms the normally
antiangiogenic stroma of normal tissues into a proangiogenic environment (Dvorak et al., 1995;
2002). VEGF increases permeability in a variety of vascular beds, including the skin, peritoneal
wall, mesentery, and diaphragm, and can lead to pathologic conditions such as malignant ascites
37
and malignant pleural effusions (Yoshiji et al., 2001; Yuan et al., 1996).
1.3.3.2 Endothelial cell activation
VEGF exerts a number of different effects which include changes in endothelial cell
morphology, cytoskeleton alterations, and stimulation of endothelial cell migration and growth.
VEGF causes increased expression of a variety of different endothelial cell genes, including
procoagulant tissue factor and fibrinolytic pathway proteins, such as urokinase, tissue-type
plasminogen activator, type 1 plasminogen activator inhibitor, and urokinase inhibitor; and matrix
metalloproteases; GLUT-1 glucose transporter; nitric oxide synthase; and integrins (Dvorak et al.,
2002; Zachary, 2001; Eliceiri et al., 2001; 1999; Brooks et al., 1994).
1.3.3.3 Endothelial cell survival
VEGF was first shown to act as a survival factor for retinal endothelial cells (Alon et al., 1995).
In vitro, VEGF has been shown to inhibit apoptosis by activating the PI3K-Akt pathway (Gerber et
al., 1998; Gerber et al., 1998; Thakker et al., 1999) in addition to upregulating antiapoptotic
proteins such as bcl-2 and A1. VEGF has also been shown to activate focal adhesion kinase (FAK)
and associated proteins that have been shown to maintain survival signals in endothelial cells
(Abedi et al., 1997; Zachary et al., 2001).
1.3.3.4 Endothelial cell proliferation
38
VEGF is a mitogen for endothelial cells. This endothelial cell proliferation appears to involve
VEGFR-2–mediated activation of extracellular kinases Erk1/2 in addition to another member of the
MAP kinase family, JNK/SAPK (Zachary et al., 2001; Meadows et al., 2001).
1.3.3.5 Endothelial cell invasion and migration
Degradation of the basement membrane is necessary for endothelial cell migration and
invasion and is an important early step in the initiation of angiogenesis. VEGF induces a variety of
enzymes and proteins important in the degradation process, including matrix-degrading
metalloproteinases, metalloproteinase interstitial collagenase, and serine proteases such as
urokinase-type plasminogen activator (uPA) and tissue-type plasminogen activator (TTPA)
(Zachary et al., 2001; Choong et al., 2003). Activation of these various compounds leads to a
prodegradative environment that facilitates migration and sprouting of endothelial cells (Ferrara et
al., 1997). The intracellular mechanisms by which VEGF leads to increased endothelial cell
migration are not entirely clear, but appear to involve FAK-associated signaling leading to focal
adhesion turnover and actin filament organization as well as p38 MAPK-induced actin
reorganization (Abedi et al., 1997; Zachary et al., 2001)
1.3.4 Expression of VEGF on tumor cells
Several studies have reported the presence of VEGFRs on liquid and solid tumor cells,
39
including those of non–small cell lung carcinoma, melanoma, prostate carcinoma, leukemia,
mesothelioma, and breast carcinoma (Dias et al., 2001; Bellamy et al., 1999; Decaussin et al., 1999;
Dias et al., 2000; Ferrer et al., 1999; Hayashibara et al., 2001; Lacal et al., 2000; Price et al., 2001).
Various VEGF ligands support tumor growth, not only by inducing angiogenesis, but also by acting
directly through VEGFRs expressed by tumor cells. Moreover, since the vast majority of solid
tumors and a variety of hematologic malignancies have the capacity to express VEGF, expression
of VEGFRs by tumor cells implicates a potential role for VEGF/VEGFR autocrine loops in these
tumors.
1.3.5 Expression of VEGF on OSCCs
VEGF expression is up-regulated significantly during the transition from NOM, through
dysplasia to OSCC. For the dysplastic lesions, no correlation is found between VEGF expression
and grade of dysplasia (Johnstone et al., 2007). The head and neck squamous cell carcinoma
(HNSCC) tumor cells express VEGFR-1, VEGFR-2, and VEGFR-3 in all specimens evaluated.
Staining for all 3 receptors is also found on tumor associated macrophages and fibrobasts, except
that VEGFR-2 is not present on fibroblasts. Staining intensity for VEGFR-1 and VEGFR-2 is
significantly higher in tumor cells and macrophages than in vascular endothelial cells (VECs)
stained for the same receptor. In HNSCC, as well as other tumor systems, VEGF has been shown to
be expressed by tumor cells and to induce proliferation of adjacent VECs via a paracrine
40
mechanism (Benefield et al., 1996; Petruzzelli et al., 1997). Recent evidence has demonstrated that
VEGF may also have a direct effect on tumor cell activity (Masood et al., 2001). These data suggest
a possible autocrine mechanism for VEGF-regulated vasculogenesis and tumorigenesis.
1.4 PlGF and cancers
1.4.1. Placenta growth factor, PlGF
PlGF, shares 53% identity with the platelet-derived growth factor (PDGF)-like region of
VEGF (Nissen et al., 1998). The human PlGF gene has been mapped to chromosome 14q24 (Mattei
et al., 1996). PlGF is anticipated to have 149 amino acids and is encoded by 7 exons that span 800
kb (Roy et al., 2006). By alternative splicing, 4 forms of PlGF protein are generated: PlGF-1,
PlGF-2, PlGF-3, and PlGF-4 (Maglione et al., 2000). Only PlGF-2 is capable of binding heparin
(Hauser and Weich, 1993). Unlike VEGFA, which binds to both FLT1 and FLK1, PlGF binds to
FLT1 but not FLK1, and it also binds to neuropilin 1 and 2 (Persico et al., 1999; Migdal et al.,
1998)
1.4.2. Distribution of PlGF in tissues
PlGF is abundant in the trophoblasts of the placenta at the middle to late stages of pregnancy.
The amount of PlGF persistently increases toward the terminal development of the placenta, and its
41
biosynthesis seems to be limited to trophoblasts and stromal cells (Maglione et al., 1993; Hauser et
al., 1993). This temporal pattern of PlGF production may reflect a role in preventing excessive
neovascularization and overgrowth of the placenta tissue by downregulating angiogenesis. In
addition to the placenta, the gene is abundantly expressed in the thyroid under normal physiological
conditions (Viglietto et al., 1995).
1.4.3. Impact of PlGF gene knockout mice
Knockout of only one allele of the Vegfa gene, encoding VEGF-A, is lethal in the mouse
embryo (heterozygous lethality) with impaired angiogenesis and blood island formation leading to
developmental abnormalities (Ferrara et al., 1996; Carmeliet et al., 1996). In contrast, several
studies suggest that genetic deletion of either Vegfb or PlGF genes does not result in obvious
impairment of the vascular system (Luttun et al., 2002; Aase et al., 2001; Bellomo et al., 2001).
Although PlGF knockout mice under normal physiological conditions do not exhibit obvious
phenotypic changes, these animals show vascular defects under several pathological settings
(Luttun et al., 2002; Fischer et al., 2007; Carmeliet et al., 2001; Luttun et al., 2002). The response to
VEGF-A is also impaired, including reduced angiogenesis and vascular permeability under
ischemic insult in a hind limb model and recruitment of monocytes and macrophages in a skin
wound assay. Transplantation of wild-type bone marrow restores angiogenesis and collateral growth
in this knockout model under ischemic conditions, which suggests that PlGF may contribute to
42
vessel growth in adult animals through the mobilization of bone marrow–derived cells (Carmeliet et
al., 2001).
1.4.4 Mechanisms of PlGF
Several studies have analysed the molecular mechanisms of PlGF. This growth factor
stimulates angiogenesis, in part by enhancing VEGF signalling. It displaces VEGFA from FLT1,
which liberates VEGFA and allows it to activate FLK1 and enhance VEGF-driven angiogenesis
(Park et al., 1994). PlGF also upregulates the expression of VEGFA, fibroblast growth factor 2
(FGF2), platelet-derived growth factor-β (PDGFB), matrix metalloproteinases (MMPs), and other
angiogenic factors (Marcellini et al., 2006; Roy et al., 2005). Moreover, the binding of PlGF to
FLT1 leads to intermolecular crosstalk between FLT1 and FLK1, which amplifies FLK1 signalling
and consequently enhances VEGF-driven responses (Autiero et al., 2003). These effects of PlGF
suggest that endothelial cells enhance their own responsiveness to VEGFA by producing PlGF
(Autiero et al., 2003). However, PlGF is also capable of inducing its own signals through FLT1
independently of VEGFA. Indeed, gene expression profiling of endothelial cells has revealed that
PlGF signals directly through FLT1 and switches on a number of pro-angiogenic genes (Autiero et
al., 2003; Schoenfeld et al., 2004).
1.4.5. PlGF in primary human tumors