1
Hydrodeoxygenation of 4-methylguaiacol over silica-supported nickel phosphide catalysts:The particle size effect
Pei-Ju Hsu and Yu-Chuan Lin* Department of Chemical Engineering
National Cheng Kung University Tainan, Taiwan, 70101
* Corresponding author’s Tel.: (886) 6 275 7575 ext. 62668; Fax: (886) 6 234 4496; E-mail:
2
Abstract
Lignin is a natural-made heteroaromatic polymer, which is a potential feedstock for bio-based aromatics synthesis. Catalytic hydrodeoxygenation (HDO)
depolymerizes lignin through selectively oxygen extraction. This study investigated HDO of 4-methylguaiacol, a lignin model compound, via silica supported nickel phosphide catalysts with varying size of active phase. Physicochemical
characterizations, including N2 physisorption, XRD, TEM, XPS, H2-TPR, NH3-TPD, CO chemisorption, and CO-TPD were performed. HDO of 4-methylguaiacol was conducted in a H2-pressured batch reactor at 4 MPa. The reaction network of
4-methylguaiacol HDO was proposed based on the product distribution. Furthermore, nickel phosphide catalyst was found to be structure-sensitive in 4-methylguaiacol HDO, and is associated with Ni cations with different partial positive charges on the surface.
Keywords: nickel phosphide, hydrodeoxygenation, structure-sensitivity, 4-methylguaiacol
3
1. Introduction
Transforming abundant, renewable lignocellulosic biomass into fuels and chemicals alleviates our dependence on fossil resources. Amongst the three major components (cellulose, hemicellulose, and lignin) of lignocellulosics, lignin is a potential feedstock for aromatic synthesis. Developing technical and economical feasible lignin upgrading process is burgeoning, and catalytic hydrodeoxygenation (HDO) is promising in lignin and its fragments (e.g., pyrolytic lignin) conversion (1).
HDO is a process by which oxygen-containing feedstocks can be reduced
(oxygen removal) by gaseous hydrogen, yielding water as a major byproduct. High H2
partial pressures (2-20 MPa) and temperatures (200-300 °C) in a batch or a
continuous system are usually required for HDO. Lately, HDO at atmospheric (2-4) or low pressures (5-7) and using alternative hydrogen sources (such as formic acid and decalin) to generate H2 in situ (8-11) or to provide intramolecular H-transfer (12, 13) were investigated to minimize severity of reaction condition and to overcome
hydrogen deficiency in biorefineries. Detailed reaction conditions and exploited catalysts in lignin HDO can be found in earlier reviews (1, 14-16).
Using a model compound as a lignin representative is necessary to understand fundamental HDO chemistry and kinetics. Guaiacol (C7H8O2), anisole (C7H8O), and cresol (C7H8O) are the most used lignin representatives because these compounds possess functional groups of lignin, including methoxyl, hydroxyl, and methyl, and are frequently identified in pyrolytic oils (17). Applying these species in HDO allows us to understand how the functional groups evolved, making it possible to correlate physicochemical properties of catalysts and their HDO behaviors.
Transition metal phosphide catalysts, e.g., Ni2P, MoP, Fe2P, and CoP, were recently deployed in HDO of biocrude and lignin derivatives because of its effectiveness in analogue reactions like hydrodesulfurization (18) and
hydrodenitrogenation (19). The acidic/metallic nature of metal phosphide acts as a bifunctional center: the metal site mediates Caryl-O hydrogenolysis and hydrogenation, while acid site (e.g., Brønsted acids on phosphorous surface) promotes dehydration and transalkylation (3, 4, 20, 21). Ni2P seems to be the most effective in HDO among metal phosphides. Oyama et al. (2) surveyed a series of SiO2 supported transition metal phosphides (including Ni2P, Co2P, Fe2P, WP, and MoP), and found that Ni2P is the most active in guaiacol HDO. In addition, Ni2P/SiO2 displayed a greater durability than commercial hydrotreating catalysts such as Pd/Al2O3 and sulfide CoMo/Al2O3
due to its better resistance to coking. The Chen group (22, 23) investigated Ni2P crystalline size and Mo substituted Ni2P in HDO of methyl laurate and anisole. Small Ni2P size had negative effect in HDO activity due to strong interaction between Ni
4
and P (23). Additional Mo in Ni2P could hamper anisole conversion (22). This is in line with Whiffen and Smith, who showed that unsupported MoP is less active than Ni2P in cresol HDO (24). Our group investigated kinetics and mechanism of guaiacol HDO over Ni2P on supports with different acidities (i.e., SiO2, ZrO2, and Al2O3) (3).
Using differential kinetic analysis, small Ni2P particles are found to be intrinsic active in deoxygenation, while large Ni2P clusters are prone to demethylation.
Compared to guaiacol and anisole, little attention has been paid on HDO of alkylated guaiacols, particularly 4-alkylguaiacols. 4-alkylguaiacols are potential precursors of novel polymer monomers derived from lignin (25). For example, alkylated caprolactone and caprolactam can be synthesized through asymmetric Baeyer–Villiger oxidation of 4-alkylcyclohexanone (26, 27), which is derived from a series of demethoxylation, hydrogenation, and dehydrogenation of alkylated guaiacols (25). The Weckhuysen group compared HDO of guaiacol and 4-methylguaiacol (C8H10O2) over sulfide CoMo catalysts, and discovered that additional methyl species had steric effect in retarding oligomerization and hydrogenation (28). Since most lignin fragments carry alkyl functionalities, an in-depth investigation of HDO of alkylated guaiacols is necessary.
In this work, silica supported Ni2P with different dispersions were synthesized and tested in HDO of 4-methylguaiacol. By loading the same amount of nickel phosphide on silica with sequentially decreased surface area (i.e., SBA-15,
high-surface-area SiO2, and low-surface-area SiO2), the particle size of active center can be controlled in an increasing order. Changing size of active phase varies partial positive charges of Ni cations (Niδ+, known as the ensemble effect) (29, 30) on the surface, making Ni2P catalyst to be structure-sensitive (31) in 4-methylguaiacol HDO.
2. Experimental
2.1. Chemicals
Ni(NO3)2·6H2O (Alfa Aesar, 99%) and (NH4)2HPO4 (J.T. Baker, 99%) were used as the precursors of nickel phosphide. The precursors were dissolved in deionized water as aqueous nickel phosphate solution with Ni/P molar ratio equals to 0.5. A loading of 1.7 mmol Ni per gram of support (~12.6 wt% Ni2P) was impregnated on the support by incipient wetness.
Low-surface-area silica (SiO2-L, Cabosil L-90), high-surface-area silica (SiO2-H, Alfa Aesar), and SBA-15 were used as the supports. SBA-15 was synthesized
according to the method reported by the Ryoo group (32). In brief, 4.0 g of Pluronic P123 (Sigma-Aldrich) was dissolved in 76.0 g deionized water and 2.3 g of 37%
5
hydrochloric acid at room temperature. After vigorous stirring for 1 h, 8.6 g of tetraethoxysilane (TEOS, 99%) was added with continued stirring overnight. The mixture (TEOS : P123 : HCl : H2O = 1 : 0.016 : 0.54 : 100 in molar ratio) was then transferred to a Teflon liner, sealed in a bomb, and treated in a hydrothermal condition at 100 oC for 24 h. The collected paste was dried at 80 oC for 12 h and then calcined to 550 oC (3 oC/min) for 4 h in air.
After impregnation of aqueous nickel phosphate solution, the powder was air dried at 120 oC for 1 h and then calcined at 500 oC for 6 h. The particles were reduced in an 80% H2/N2 stream (150 mL/min) with a 2 oC/min heating rate from 30 to 670 oC and kept isotherm for 1 h. The reduction procedure transformed phosphate into a phosphide (19). After reduction, the sample was cooled to ambient temperature in a He stream (100 mL/min), and was passivated under a 2% O2/He (40 mL/min) stream for 1 h.
2.2. Characterization of catalysts
N2 physisorption was performed on a Micromeritics ASAP 2020 gas-adsorption analyzer at -196 °C. Approximately 0.1 g of a sample was dehydrated at 300 °C for 1 h prior to the measurement. Total surface area was calculated based on the
Brunauer-Emmett-Teller (BET) method. Powder X-ray diffraction (XRD) was conducted on a diffractometer (Rigaku D/Max-IIB) with Cu Kα radiation, excited at 40 kV and 40 mA. Transmission electron microscopy (TEM) images were acquired using a Hitachi H-7500 microscope at 100 kV. X-ray photoelectron spectroscopy (XPS) was performed using a Kratos Axis Ultra DLD equipped with a 180o
hemispherical sector analyzer and a focused monochromatic Al-Kα X-ray (1486.7 eV) source. Binding energy shift was corrected by using the C 1s signal of adventitious carbon at 284.6 eV. A Micromeritics Autochem II chemisorption analyzer with a thermal conductivity detector (TCD) was used to measure
H2-temperature-programmed reduction (TPR), H2-temperature-programmed
desorption (TPD), NH3-TPD, and CO pulse chemisorption with subsequent CO-TPD.
Before the test, each sample was dehydrated in a H2 stream (30 mL/min) for 1 h at 150 °C. Approximately 0.1 g of calcined precursors, i.e., NixPyOz/SiO2, was tested in H2-TPR using a 10% H2/Ar stream (100 mL/min) with a 5 °C/min from 100 to 1000
°C. For the remaining temperature-programmed techniques, approximately 0.1 g of passivated Ni2P catalyst was pre-reduced at 450 °C for 1 h then cooled to 100 °C before the test. For NH3-TPD, a 10% NH3/He stream with a 30 mL/min flow rate was used to flush the sample at 100 °C for 1 h. The NH3-treated sample was then purged with He for 40 min at 100 °C to remove weakly adsorbed NH3. NH3-TPD was
6
measured from 100 to 700 °C at a rate of 10 °C/min in a He stream. CO
chemisorption was performed at ambient temperature. Pulses of 10% CO/He were repeatedly injected into the system until achieving a breakthrough. The CO uptake was calculated based on the difference of the sum of pulse areas between a tested catalyst and a known volume. The CO-TPD was executed immediately after CO pulse chemisorption with a 15 °C/min heating rate in a He stream.
2.3. HDO activity evaluation
Reactivity tests were performed in a 300 mL stirred-batch autoclave (Parr Model 4561) with a system controller (Parr Model 4848). The temperature was set at 250 °C with an agitation rate of 500 rpm. Approximately 60 mL solvent (n-hexadecane), 1.5 mL reactant (4-methylguaicol or cresol), and 0.1 g passivated catalyst (200-120 mesh;
0.074-0.125 mm) were used in each trial. After transferring the mixture of solvent, reactant, and catalyst into the system, the autoclave was flushed with an Ar stream (60 mL/min) for 10 min and then purged with a H2 stream (60 mL/min) for 30 min to expel residual air and then sealed. Once the reaction temperature was achieved, the system was pressurized to 4 MPa with H2. Different reaction periods, ranging from 1 to 10 h, were used. Gaseous products, mostly methane and methanol, were analyzed by a parallel-dual-column gas chromatography (GC, SRI 8610C) with a 60-m MXT-1 capillary column and a HayeSep D packed column; liquid compounds including toluene (C7H8), methylcyclohexane (C7H14), cresol, 4-methylcyclohexanol (C7H14O), guaiacol, 2,4-dimethylphenol (C8H10O), 3-methylanisole (C8H10O), and
4-methylguaiacol were analyzed by a GC-MS (Agilent 5890, DB-5MS capillary column, 60 m x 0.25 mm). Internal and external mass transfer limitations of 4-methylguaiacol HDO were examined with different catalyst particle size and the stirring rate (see Figs. S1 and S2 of Supporting Information). The internal mass transfer limitation was negligible using catalyst particle size smaller than 0.177 mm (80 mesh), while the external mass transfer limitation could be avoided using the agitation rate higher than 500 rpm. Deposited coke was analyzed using the weight loss of spent catalyst by a thermogravimetric analysis (TA Q600). The sample was dried in a 10% O2/He stream at 150 °C for 30 min, followed by a 10 °C /min heating rate to 600 °C to burn coke out.
3. Results and discussion
Table 1 presents the surface area of each catalyst. After impregnating Ni2P, the surface area of each catalyst decreased compared to blank support. This implied pore
7
blockage and fouling by Ni2P and surplus phosphorus on catalyst surface. The surface area decreased followed the order as: Ni2P/SBA-15 (353 m2/g) > Ni2P/SiO2-H (156 m2/g) > Ni2P/SiO2-L (78 m2/g). The Ni loading for each catalyst was similar, ranging from 11.4 to 11.9 wt%. The Ni/P ratio displayed a decreasing trend as Ni2P/SiO2-L (1.5) > Ni2P/SiO2-H (1.4) > Ni2P/SBA-15 (1.2). In principle, a Ni/P ratio of 2 should be achieved when precursors of nickel phosphate solution were fully transformed into Ni2P. This suggests Ni-rich impurities such as Ni12P5 may be formed, or excess P may residue on the surface, particularly for Ni2P/SBA-15.
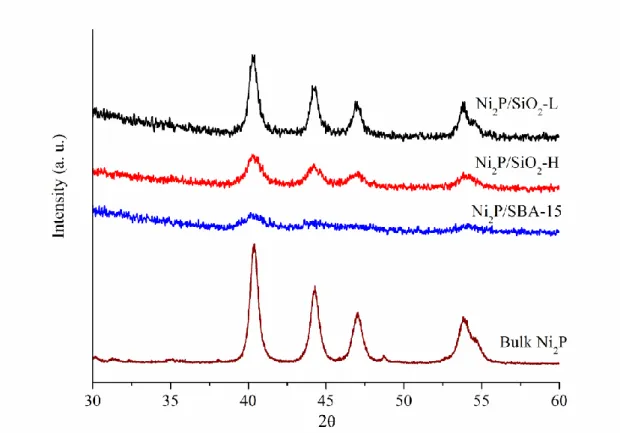
Figure 1 exhibits the XRD patterns and Table 1 lists the XRD-detected Ni2P crystalline sizes. The index responses of Ni2P (PDF no. 74-1385) at 2θ equal to 40.7o, 44.6o, 47.4o, and 54.2o, corresponding to (1 1 1), (2 0 1), (2 1 0), and (3 0 0) planes, were identified for each catalyst. This highlights that synthesized nickel phosphide was mainly Ni2P. The Scherrer equation-estimated crystalline size increased as Ni2P/SBA-15 (6.0 nm) < Ni2P/SiO2-H (8.6 nm) < Ni2P/SiO2-L (12.8 nm). Based on the crystalline size, theoretically calculated Ni2P concentration (nsite) and dispersion were presented in Table 1. Because similar Ni2P loading was used, the Ni2P dispersion decreased followed a decreasing trend of catalyst surface area as Ni2P/SBA-15
(17.6%) > Ni2P/SiO2-H (12.2%) > Ni2P/SiO2-L (8.2%).
Figure 2 displays the TEM images of tested catalysts. Since Ni2P has higher molecular weight than that of silica, it should have darker contrast in the bright-field micrographs against the support. Particle agglomeration was found on the external surface of SiO2, while the mesopores of SBA-15 could constrained the size of Ni2P.
The particle size ranged from 3 to 10 nm for Ni2P/SBA-15; 4 to 11 nm, Ni2P/SiO2-H;
6 to 16 nm, Ni2P/SiO2-L. The distribution is in line with the trend of XRD-estimated crystalline size: higher surface-area support could achieve higher dispersion (smaller particle size) of Ni2P.
Figure 3 exhibits the XPS patterns of Ni 2p3/2 region of tested catalysts. Three responses were identified at approximately 852, 856, and 861 eV, corresponding to Niδ+ in Ni2P (0 < δ < 2), Ni2+ ions, and the satellite response of Ni 2P3/2, respectively (33, 34). A shift to high energy level of Niδ+ response was observed, following the order: Ni2P/SiO2-L (852.2 eV) < Ni2P/SiO2-H (852.6 eV) < Ni2P/SBA-15 (852.8 eV).
This can be explained through the coordination between Ni and P in each Ni2P phase:
Ni cations coordinated with more numbers of P yield an additional electron transfer (higher δ) from Ni to P (4). Therefore, the δ value should be in the order as δ (Ni2P/SiO2-L) < δ (Ni2P/SiO2-H) < δ (Ni2P/SBA-15). Moreover, the increasing tendency of δ value matches the reverse sequence of Ni2P crystallite size. This
indicates that Ni cations in small Ni2P with smaller had higher number of coordinated P, while Ni cations in large Ni2P had lower number of coordinated P (30). The Ni2+
8
signal is higher than Ni in NiO (853.5-854.1 eV) (35), and is attributed to the interaction between Ni2+ and phosphate ions caused by superficial passivation (36).
Figure 4 shows the H2-TPR profiles of the calcined, silica supported Ni2P
precursors. A single reduction response was observed, and the maximal reduction rate (the apex of the peak, TM) elevated as Ni2P/SiO2-L (817 °C) < Ni2P/SiO2-H (828 °C)
< Ni2P/SBA-15 (833 °C). Oyama and Lee (30) reported that the trend of TM is
correlated to the interaction between Ni and P in Ni2P: the higher the TM, the stronger the Ni-P interaction can be. Moreover, small Ni2P particles had stronger Ni-P
interaction than large Ni2P particles. This indicates that the size of Ni2P particles should increase as Ni2P/SBA-15 < Ni2P/SiO2-H < Ni2P/SiO2-L, in agreement with the results observed by XRD and TEM.
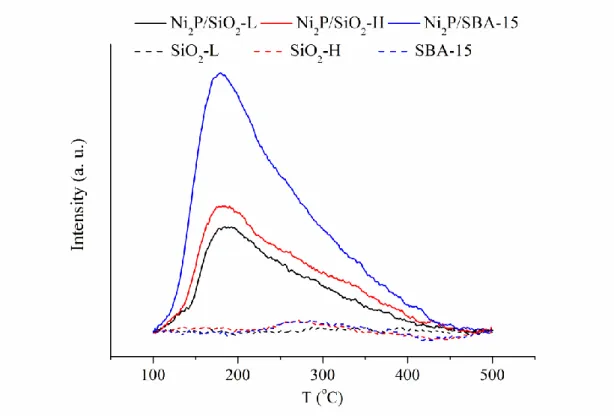
Figure 5 displays the NH3-TPD profiles. The NH3 desorption of each support is negligible. For Ni2P catalysts, the desorption profile was composed of a prominent peak at ~200 °C with a shoulder at ~300 °C. The response at ~ 200 °C is related to the Brønsted acid sites of PO-H species, while the shoulder contains the Lewis acid sites of Ni cations with partial positive charge in Ni2P at ~300 °C (37). Table 1 presents the NH3 uptakes over tested catalysts. The uptakes increased with increasing surface area of catalyst, particularly for Brønsted acid sites. This implied that higher surface area allow more PO-H species existing on catalyst surface.
Table 1 lists the CO uptake of each catalyst. The CO uptake declined as:
Ni2P/SBA-15 (102.9 μmol/g) > Ni2P/SiO2-H (78.2 μmol/g) > Ni2P/SiO2-L (69.0 μmol/g). The amount of chemisorbed CO reflects the number of Ni sites on the surface of catalyst according to a 1-to-1 ratio of CO on Ni cations of Ni2P (2, 37). The CO uptake was lower than theoretically estimated Ni site (nsite) for each sample. The overestimated nsite value can be explained by: 1) part of Ni cations was embedded inside Ni2P clusters (38), and 2) some Ni sites were blocked by surplus phosphorous.
The embedded/blocked Ni sites are inaccessible for CO chemisorption (33, 39, 40).
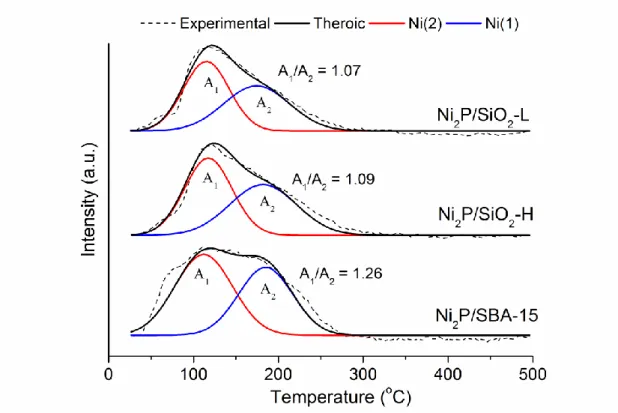
Figure 6 shows the CO-TPD profiles of Ni2P catalysts after CO pulse chemisorption. A combinational desorption signal was observed, and could be deconvoluted into two responses. The CO bonding strength depends upon the
availability of d electrons at the surface; therefore, stronger CO adsorption formed on lower oxidation state of Ni cations than higher positive charged Ni cations (41). de Silva and collaborators (42) attributed the low-temperature peak to the amounts of desorbed CO from higher positive charged Ni sites; high-temperature peak, lower positive charged Ni sites. The ratio of the areas of these two peaks (A1/A2) can then be interpreted as the relative composition of two Ni sites in Ni2P: tetrahedral Ni
surrounded by 4 nearest-neighbor P atoms (Ni(1), low δ) and square-pyramidal Ni surrounded by 5 nearest-neighbor P atoms (Ni(2), high δ) (30). The A1/A2 ratio
9
declined as Ni2P/SBA-15 (1.26) > Ni2P/SiO2-H (1.09) > Ni2P/ SiO2-L (1.07), indicating that Ni2P on SBA-15 had the highest concentration of Ni(2) sites while Ni2P/SiO2-L had the lowest.
Figure 7 shows H2-TPD profiles of tested catalysts. The profile can be correlated to H2 mobility on catalyst surface, and is usually categorized into two regions using 400 oC as the demarcation (22). The low-temperature region is related to the
desorption of H2 from Ni sites of Ni2P phase, while the high-temperature response is reverse spillover H2, from the interface of Ni2P and support (~530 °C) or from remote hydrogen species (such as P-OH) faraway of Ni2P particle (~850 °C) (23, 29). The low-temperature peak increased following the order as: Ni2P/SiO2-L (92 °C) <
Ni2P/SiO2-H (103 °C) < Ni2P/SBA-15 (157 °C), suggesting H2 bonded stronger on small Ni2P particles. A possible explanation is that hydrogen chemisorption forms Ni-hydrogen species via homolytic dissociation (H2 + Niδ+ + 2e- → 2NiH(δ-1)+) (41) on the Ni cations of Ni2P. Higher δ value requires higher thermal input in TPD to
decompose NiH(δ-1)+ to release H2. Therefore, higher partial positive charged Ni2P on SBA-15 had higher desorption temperature at low-temperature range; lower δ value of Ni2P on SiO2-L, vice versa. The desorption temperatures of spillover H2 were closely matched for all catalysts, indicating similar chemistry of phosphorous-coated support.
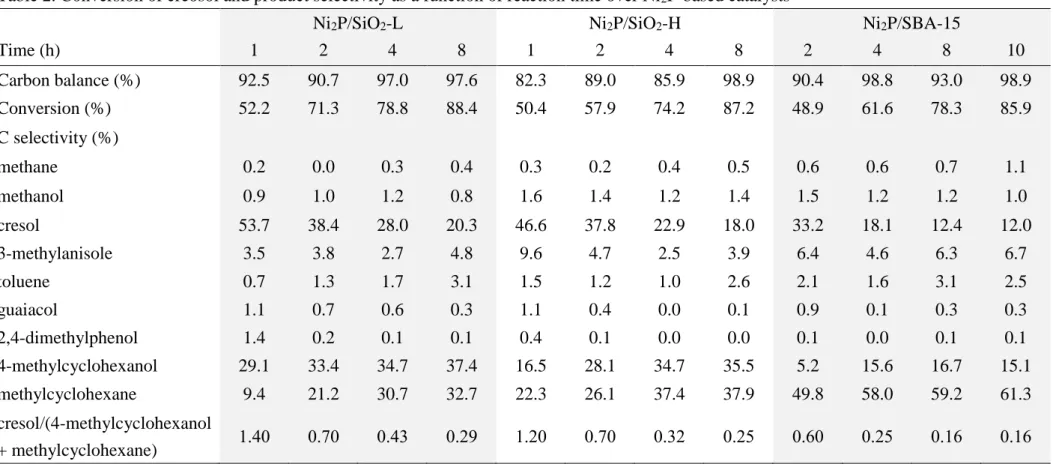
Table 2 presents the catalytic performances of Ni2P catalysts in 4-methylguaiacol HDO at 250 oC. Conversion increased with respect to the reaction time, ranging from 66.8% to 88.4% (Ni2P/SiO2-L), 57.1% to 87.2% (Ni2P/SiO2-H), and 47.4% to 78.3%
(Ni2P/SBA-15). At the same reaction time, the conversions followed the sequence as Ni2P/SiO2-L > Ni2P/SiO2-H > Ni2P/SBA-15. This implies that large Ni2P phases are more active than small Ni2P clusters. Cresol, 4-methylcycohexanol, and
4-methylcyclohexane were major products, accompanying with little amounts of methane, methanol, 3-methylanisole, toluene, guaiacol, and 2,4-dimethylphenol. Less than 0.5 wt% coke was detected on used catalyst for each test. Generally, selectivity of cresol decreased, while selectivities of 4-methylcycohexanol and
4-methylcyclohexane increased with increasing reaction time. Figure 8 shows the proposed reaction network of 4-methylguaiacol HDO, including direct deoxygenation (DDO), dehydrogenation (DH2), dehydration (DHY), demethylation (DME),
demethoxylation (DMO), and hydrogenation (HY), based on the product distributions (3-5, 13).
To gain insight into the sequence of formation of main products (i.e., cresol, 4-methylcycohexanol, and 4-methylcyclohexane), the ratio of cresol-to-the sum of 4-methylcyclohexanol and methylcyclohexane was included in Table 2. At the same conversion level, the ratio decreased as Ni2P/SiO2-L > Ni2P/SiO2-H > Ni2P/SBA-15.
This evidences that large Ni2P clusters have higher DMO activity than small Ni2P
10
particles. DMO of 4-methylguaiacol is a metal-catalyzed, Caryl-O bond cleavage (hydrogenolysis) route to form methanol and cresol (20, 21). The tendency of cresol selectivity under the same 4-methylguaiacol conversion can then be correlated to the chemisorbed H2 mobility of Ni sites of Ni2P particle as observed by H2-TPD. The great H2 mobility of lower partial positive charged Ni of large Ni2P particles is thereby responsible for its high DMO activity.
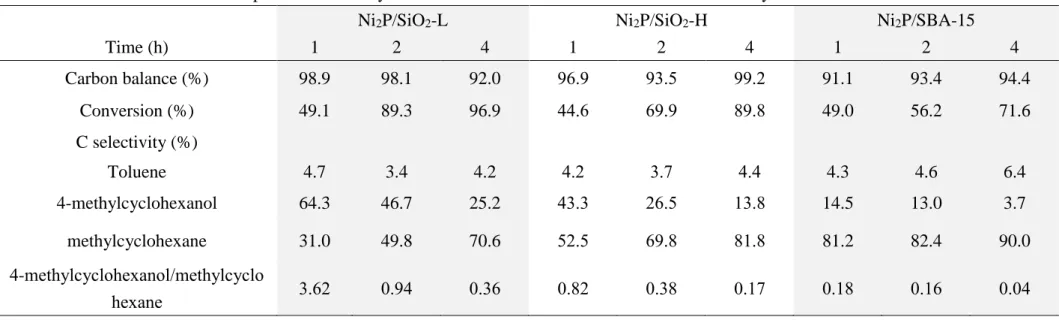
To further clarify the reaction pathways and the structure sensitivity effect of Ni2P, cresol was tested as the reactant under the same condition of 4-methylguaiacol HDO for 1, 2, and 4 h (Table 3). Again, conversion increased with extended reaction time, and Ni2P/SiO2-L was the most active among all catalysts. The comparatively high conversion of cresol over large, unsupported nickel phosphides was observed by Wang and coworkers (43). They attributed large nickel phosphide particle contains less impurities such as Ni5P4, and can lead to higher HDO activity. Similar to 4-methylguaiacol HDO, the high H2 mobility on Ni2P/SiO2-L (large Ni2P clusters) should be related to its high activity in cresol conversion.
Different groups mentioned that the 4-methylcyclohexanol DHY should first form 4-methylcyclohexene via dehydration (13, 44). 4-methylcycohexene is erratic under high H2 pressure, and can be hydrogenated to methylcyclohexane immediately (13, 44). The 4-methylcyclohexanol-to-methylcyclohexane ratio decreased as
Ni2P/SiO2-L > Ni2P/SiO2-H > Ni2P/SBA-15 at the same conversion level. Since a proton donor is decisive for dehydration and a metal site favors hydrogenation, the DHY activity of 4-methylcyclohexanol should rely on the Brønsted acidity of each catalyst (20) while the follow-up HY of 4-methylcyclohexene to methylcyclohexane is related to the number of Ni cations (42). Thus, the higher amount of Brønsted acid site and Ni dispersion of Ni2P/SBA-15 should promote the combinative step of 4-methylcyclohexanol DHY to 4-methylcyclohexene and 4-methylcyclohexene HY to methylcyclohexane.
4. Conclusions
Ni2P with varying particle size are structural sensitive in 4-methylguaiacol HDO.
Comparatively large Ni2P favors Caryl-O bond breakage, while small Ni2P clusters in collaboration with Brønsted acids promote a combinative dehydration-hydrogenation route. This is attributed to the different partial positive charged Ni cations in Ni2P:
large Ni2P contains a low Ni(2)/Ni(1) ratio, yielding Ni cations in Ni2P to have a low δ value; in contrast, small Ni2P had a high Ni(2)/Ni(1) ratio with a high δ value of Ni cations. Different Niδ+ results in varying H2 mobility on catalyst surface, by which extents of reaction routes such as DMO, DHY, and HY in 4-methylguaiacol HDO
11
could be manipulated.
Acknowledgement
This study was supported by Taiwan’s Ministry of Science and Technology (MOST projects 104-2628-E-006 -009 -MY2, 105-2221-E-006 -229, and
105-2218-E-155 -007).
References
1. Li C, Zhao X, Wang A, Huber GW, Zhang T. Catalytic Transformation of Lignin for the Production of Chemicals and Fuels. Chemical Reviews.
2015;115(21):11559-624.
2. Zhao HY, Li D, Bui P, Oyama ST. Hydrodeoxygenation of guaiacol as model compound for pyrolysis oil on transition metal phosphide hydroprocessing catalysts.
Appl Catal, A. 2011;391(1-2):305-10.
3. Wu S-K, Lai P-C, Lin Y-C, Wan H-P, Lee H-T, Chang Y-H. Atmospheric Hydrodeoxygenation of Guaiacol over Alumina-, Zirconia-, and Silica-Supported Nickel Phosphide Catalysts. ACS Sustainable Chemistry & Engineering. 2013.
4. Wu S-K, Lai P-C, Lin Y-C. Atmospheric Hydrodeoxygenation of Guaiacol over Nickel Phosphide Catalysts: Effect of Phosphorus Composition. Catalysis Letters.
2014;144(5):878-89.
5. Nimmanwudipong T, Runnebaum RC, Block DE, Gates BC. Catalytic Conversion of Guaiacol Catalyzed by Platinum Supported on Alumina: Reaction Network Including Hydrodeoxygenation Reactions. Energy Fuels.
2011;25(8):3417-27.
6. Runnebaum RC, Nimmanwudipong T, Block DE, Gates BC. Catalytic
conversion of compounds representative of lignin-derived bio-oils: a reaction network for guaiacol, anisole, 4-methylanisole, and cyclohexanone conversion catalysed by Pt/[gamma]-Al2O3. Catal Sci Technol. 2012;2(1):113-8.
7. Nimmanwudipong T, Aydin C, Lu J, Runnebaum R, Brodwater K, Browning N, et al. Selective Hydrodeoxygenation of Guaiacol Catalyzed by Platinum Supported on Magnesium Oxide. Catal Lett. 2012;142(10):1190-6.
8. Xu W, Miller SJ, Agrawal PK, Jones CW. Depolymerization and
Hydrodeoxygenation of Switchgrass Lignin with Formic Acid. ChemSusChem.
2012;5(4):667-75.
9. Holmelid B, Kleinert M, Barth T. Reactivity and reaction pathways in
thermochemical treatment of selected lignin-like model compounds under hydrogen rich conditions. Journal of Analytical and Applied Pyrolysis. 2012;98:37-44.
12
10. Grasemann M, Laurenczy G. Formic acid as a hydrogen source - recent developments and future trends. Energy & Environmental Science.
2012;5(8):8171-81.
11. Shafaghat H, Rezaei PS, Daud WMAW. Using decalin and tetralin as hydrogen source for transfer hydrogenation of renewable lignin-derived phenolics over
activated carbon supported Pd and Pt catalysts. Journal of the Taiwan Institute of Chemical Engineers. 2016;65:91-100.
12. Wang X, Rinaldi R. Exploiting H-transfer reactions with RANEY[registered sign]
Ni for upgrade of phenolic and aromatic biorefinery feeds under unusual, low-severity conditions. Energy & Environmental Science. 2012;5(8):8244-60.
13. Wang X, Rinaldi R. A Route for Lignin and Bio-Oil Conversion:
Dehydroxylation of Phenols into Arenes by Catalytic Tandem Reactions. Angewandte Chemie International Edition. 2013;52(44):11499-503.
14. Zakzeski J, Bruijnincx PCA, Jongerius AL, Weckhuysen BM. The Catalytic Valorization of Lignin for the Production of Renewable Chemicals. Chemical Reviews. 2010;110(6):3552-99.
15. Saidi M, Samimi F, Karimipourfard D, Nimmanwudipong T, Gates BC, Rahimpour MR. Upgrading of lignin-derived bio-oils by catalytic
hydrodeoxygenation. Energy & Environmental Science. 2014;7(1):103-29.
16. Ruddy DA, Schaidle JA, Ferrell Iii JR, Wang J, Moens L, Hensley JE. Recent advances in heterogeneous catalysts for bio-oil upgrading via "ex situ catalytic fast pyrolysis": catalyst development through the study of model compounds. Green Chemistry. 2014;16(2):454-90.
17. Dorrestijn E, Laarhoven LJJ, Arends IWCE, Mulder P. The occurrence and reactivity of phenoxyl linkages in lignin and low rank coal. J Anal Appl Pyrolysis.
2000;54(1-2):153-92.
18. Shu Y, Lee Y-K, Oyama ST. Structure-sensitivity of hydrodesulfurization of 4,6-dimethyldibenzothiophene over silica-supported nickel phosphide catalysts.
Journal of Catalysis. 2005;236(1):112-21.
19. Oyama ST. Novel catalysts for advanced hydroprocessing: transition metal phosphides. J Catal. 2003;216(1-2):343-52.
20. Sullivan MM, Chen C-J, Bhan A. Catalytic deoxygenation on transition metal carbide catalysts. Catalysis Science & Technology. 2016;6(3):602-16.
21. Jongerius AL, Gosselink RW, Dijkstra J, Bitter JH, Bruijnincx PCA,
Weckhuysen BM. Carbon Nanofiber Supported Transition-Metal Carbide Catalysts for the Hydrodeoxygenation of Guaiacol. ChemCatChem. 2013;5(10):2964-72.
22. Li K, Wang R, Chen J. Hydrodeoxygenation of Anisole over Silica-Supported Ni2P, MoP, and NiMoP Catalysts. Energy & Fuels. 2011;25(3):854-63.
13
23. Yang Y, Chen J, Shi H. Deoxygenation of Methyl Laurate as a Model Compound to Hydrocarbons on Ni2P/SiO2, Ni2P/MCM-41, and Ni2P/SBA-15 Catalysts with Different Dispersions. Energy & Fuels. 2013;27(6):3400-9.
24. Whiffen V, Smith K. A Comparative Study of 4-Methylphenol
Hydrodeoxygenation Over High Surface Area MoP and NiP. Topics in Catalysis.
2012;55(14/15):981-90.
25. Schutyser W, Van den Bosch S, Dijkmans J, Turner S, Meledina M, Van Tendeloo G, et al. Selective Nickel-Catalyzed Conversion of Model and
Lignin-Derived Phenolic Compounds to Cyclohexanone-Based Polymer Building Blocks. ChemSusChem. 2015;8(10):1805-18.
26. Peeters JW, van Leeuwen O, Palmans ARA, Meijer EW. Lipase-Catalyzed Ring-Opening Polymerizations of 4-Substituted ε-Caprolactones: Mechanistic Considerations. Macromolecules. 2005;38(13):5587-92.
27. Cavarzan A, Bianchini G, Sgarbossa P, Lefort L, Gladiali S, Scarso A, et al.
Catalytic Asymmetric Baeyer–Villiger Oxidation in Water by Using PtII Catalysts and Hydrogen Peroxide: Supramolecular Control of Enantioselectivity. Chemistry – A European Journal. 2009;15(32):7930-9.
28. Jongerius AL, Jastrzebski R, Bruijnincx PCA, Weckhuysen BM. CoMo sulfide-catalyzed hydrodeoxygenation of lignin model compounds: An extended reaction network for the conversion of monomeric and dimeric substrates. Journal of Catalysis. 2012;285(1):315-23.
29. Liu P, Rodriguez JA, Asakura T, Gomes J, Nakamura K. Desulfurization
Reactions on Ni2P(001) and α-Mo2C(001) Surfaces: Complex Role of P and C Sites.
The Journal of Physical Chemistry B. 2005;109(10):4575-83.
30. Oyama ST, Lee Y-K. The active site of nickel phosphide catalysts for the hydrodesulfurization of 4,6-DMDBT. Journal of Catalysis. 2008;258(2):393-400.
31. Boudart M. Catalysis by Supported Metals*. In: D.D. Eley HP, Paul BW, editors.
Advances in Catalysis. Volume 20: Academic Press; 1969. p. 153-66.
32. Choi M, Heo W, Kleitz F, Ryoo R. Facile synthesis of high quality mesoporous SBA-15 with enhanced control of the porous network connectivity and wall thickness.
Chemical Communications. 2003(12):1340-1.
33. Sawhill SJ, Phillips DC, Bussell ME. Thiophene hydrodesulfurization over supported nickel phosphide?catalysts. J Catal. 2003;215(2):208-19.
34. Zhao Y, Zhao Y, Feng H, Shen J. Synthesis of nickel phosphide nano-particles in a eutectic mixture for hydrotreating reactions. Journal of Materials Chemistry.
2011;21(22):8137-45.
35. Briggs D, Seah MP. Practical Structure Analysis. 2nd ed. Chichester: John Wiley;
1993.
14
36. Guan Q, Han F, Li W. Catalytic performance and deoxygenation path of methyl palmitate on Ni2P/SiO2 synthesized using the thermal decomposition of nickel hypophosphite. RSC Advances. 2016;6(37):31308-15.
37. Lee Y-K, Oyama ST. Bifunctional nature of a SiO2-supported Ni2P catalyst for hydrotreating: EXAFS and FTIR studies. Journal of Catalysis. 2006;239(2):376-89.
38. Cecilia JA, Infantes-Molina A, Rodriguez-Castellon E, Jimenez-Lopez A. The Influence of the Support on the Formation of Ni2P Based Catalysts by a New Synthetic Approach. Study of the Catalytic Activity in the Hydrodesulfurization of Dibenzothiophene. J Phys Chem C. 2009;113(39):17032-44.
39. Wang X, Clark P, Oyama ST. Synthesis, Characterization, and Hydrotreating Activity of Several Iron Group Transition Metal Phosphides. J Catal.
2002;208(2):321-31.
40. Koranyi TI, Vit Z, Poduval DG, Ryoo R, Kim HS, Hensen EJM.
SBA-15-supported nickel phosphide hydrotreating catalysts. J Catal.
2008;253(1):119-31.
41. Henrich VE, Cox PA. The Surface Science of Metal Oxides. Cambridge, UK:
Cambridge University Press; 1996.
42. Feitosa LF, Berhault G, Laurenti D, Davies TE, Teixeira da Silva V. Synthesis and hydrodeoxygenation activity of Ni2P/C – Effect of the palladium salt on lowering the nickel phosphide synthesis temperature. Journal of Catalysis. 2016;340:154-65.
43. Wang W, Zhang K, Liu H, Qiao Z, Yang Y, Ren K. Hydrodeoxygenation of p-cresol on unsupported Ni–P catalysts prepared by thermal decomposition method.
Catalysis Communications. 2013;41:41-6.
44. Huang Y-B, Yan L, Chen M-Y, Guo Q-X, Fu Y. Selective hydrogenolysis of phenols and phenyl ethers to arenes through direct C-O cleavage over
ruthenium-tungsten bifunctional catalysts. Green Chemistry. 2015;17(5):3010-7.
15
Table 1. Physicochemical properties of Ni2P-based catalysts.
Catalysts
SBET catalyst/support
(m2/g)
Ni loadinga
(wt%) Ni/Pa dXRDb
(nm)
nsitec
(μmol/g)
Dispersiond (%)
NH3 uptake catalyst/support
(μmol/g)
CO uptake (μmol/g)
Ni2P/SiO2-L 78/96 11.6 1.5 12.8 129 8.1 214/21 69.0
Ni2P/SiO2-H 156/205 11.4 1.4 8.6 192 12.0 263/59 78.2
Ni2P/SBA-15 353/687 11.9 1.2 6.0 275 17.2 484/70 102.9
aEstimated by ICP-AES. bEstimated by the (1 1 1) diffraction plane of Ni2P at 2θ = 40.7o. cEstimated from dXRD based on nsite = Seff * n * f. Seff is the effective surface area of Ni2P (Seff = 6/(ρ * dXRD): ρ is the density of Ni2P, 7.09 g/cm3); n is the mean surface metal atom density, 1.01 * 1015 atoms/cm2; f is the weight fraction of Ni2P. dEstimated by nsite/(1.6 mmol/g of Ni) * 100%.
16
Table 2. Conversion of creosol and product selectivity as a function of reaction time over Ni2P-based catalysts
Ni2P/SiO2-L Ni2P/SiO2-H Ni2P/SBA-15
Time (h) 1 2 4 8 1 2 4 8 2 4 8 10
Carbon balance (%) 92.5 90.7 97.0 97.6 82.3 89.0 85.9 98.9 90.4 98.8 93.0 98.9 Conversion (%) 52.2 71.3 78.8 88.4 50.4 57.9 74.2 87.2 48.9 61.6 78.3 85.9 C selectivity (%)
methane 0.2 0.0 0.3 0.4 0.3 0.2 0.4 0.5 0.6 0.6 0.7 1.1
methanol 0.9 1.0 1.2 0.8 1.6 1.4 1.2 1.4 1.5 1.2 1.2 1.0 cresol 53.7 38.4 28.0 20.3 46.6 37.8 22.9 18.0 33.2 18.1 12.4 12.0 3-methylanisole 3.5 3.8 2.7 4.8 9.6 4.7 2.5 3.9 6.4 4.6 6.3 6.7
toluene 0.7 1.3 1.7 3.1 1.5 1.2 1.0 2.6 2.1 1.6 3.1 2.5
guaiacol 1.1 0.7 0.6 0.3 1.1 0.4 0.0 0.1 0.9 0.1 0.3 0.3 2,4-dimethylphenol 1.4 0.2 0.1 0.1 0.4 0.1 0.0 0.0 0.1 0.0 0.1 0.1 4-methylcyclohexanol 29.1 33.4 34.7 37.4 16.5 28.1 34.7 35.5 5.2 15.6 16.7 15.1 methylcyclohexane 9.4 21.2 30.7 32.7 22.3 26.1 37.4 37.9 49.8 58.0 59.2 61.3 cresol/(4-methylcyclohexanol
+ methylcyclohexane) 1.40 0.70 0.43 0.29 1.20 0.70 0.32 0.25 0.60 0.25 0.16 0.16
17
Table 3. Conversion of cresol and product selectivity as a function of reaction time over Ni2P-based catalysts
Ni2P/SiO2-L Ni2P/SiO2-H Ni2P/SBA-15
Time (h) 1 2 4 1 2 4 1 2 4
Carbon balance (%) 98.9 98.1 92.0 96.9 93.5 99.2 91.1 93.4 94.4
Conversion (%) 49.1 89.3 96.9 44.6 69.9 89.8 49.0 56.2 71.6
C selectivity (%)
Toluene 4.7 3.4 4.2 4.2 3.7 4.4 4.3 4.6 6.4
4-methylcyclohexanol 64.3 46.7 25.2 43.3 26.5 13.8 14.5 13.0 3.7
methylcyclohexane 31.0 49.8 70.6 52.5 69.8 81.8 81.2 82.4 90.0
4-methylcyclohexanol/methylcyclo
hexane 3.62 0.94 0.36 0.82 0.38 0.17 0.18 0.16 0.04
18
Figure 1. XRD patterns of tested catalysts.
19
Figure 2. TEM micrographs of (a) Ni2P/SiO2-L, (b) Ni2P/SiO2-H, and (c) Ni2P/SBA-15.
20
Figure 3. XPS spectra in Ni 2P3/2 region for tested catalysts.
21
Figure 4. H2-TPR of calcined precursors of tested catalysts.
22
Figure 5. NH3-TPD of Ni2P-based catalysts and their corresponding supports.
23
Figure 6. CO-TPD of Ni2P-supported catalysts. The deconvolution was performed based on Gaussian function.
24
Figure 7. H2-TPD of Ni2P-supported catalysts.
25
Figure 8. Reaction network of 4-methylguaiacol HDO over Ni2P-based catalysts.
Unidentified products were bracketed. Abbreviations for the pathways are: direct deoxygenation (DDO), dehydrogenation (DH2), dehydration (DHY), demethylation (DME), demethoxylation (DMO), and hydrogenation (HY).