Targeting post-translational modifications of histones for cancer therapy
Y-C. Hsu
1,*, Y-H. Hsieh
2,3,*, C-C. Liao
4, L-W. Chong
5, C-Y. Lee
6, Y-L. Yu
7,8, and R-H. Chou
7,81 Institute of Biomedical Sciences, Mackay Medical College, New Taipei City, Taiwan
2 Department of Biochemistry, School of Medicine, Chung Shan Medical University, Taichung, Taiwan
3 Clinical Laboratory, Chung Shan Medical University Hospital, Taichung, Taiwan
4 Proteomics Research Center, National Yang-Ming University, Taipei, Taiwan
5 Division of Hepatology and Gastroenterology, Department of Internal Medicine, Shin Kong Wu Ho-Su Memorial Hospital, Taipei, Taiwan
6 School of Chinese Medicine for Post-Baccalaureate, I-Shou University, Kaohsiung, Taiwan
7 Graduate Institute of Cancer Biology and Center for Molecular Medicine, China Medical University, Taichung, Taiwan
8 Department of Biotechnology, Asia University, Taichung, Taiwan
Corresponding author: Dr. Ruey-Hwang Chou and Dr. Yung-Luen Yu, 9F, No.6, Hsueh-Shih Road, Taichung 404, Taiwan. E-mail:
rhchou@mail. cmu.edu.tw (Chou, RH), and [email protected] (Yu, YL)
* These authors contributed equally to this work.
Abstract
Post-translational modifications (PTMs) on histones including acetylation, methylation, phosphorylation, citrullination, ubiquitination, ADP ribosylation, and su- moylation, play important roles in different biological events including chromatin dynamics, DNA replication, and transcriptional regulation. Aberrant histones PTMs leads to abnormal gene expression and uncontrolled cell proliferation, followed by development of cancers. Therefore, targeting the enzymes required for specific histone PTMs holds a lot of potential for cancer treatment. In this review article, we retrospect the latest studies in the regulations of acetylation, methylation, and phosphorylation of histones. We also summarize inhibitors/drugs that target these modifications for cancer treatment.
Key words: Histone, post-translational modification, acetylation, methylation, phosphorylation, cancer.
Introduction
The term “epigenome” has been attracting a lot of attention recently. It represents various heritable post- translational modifications (PTMs) on histone proteins or DNA without altering DNA coding sequence (1).
These modifications transiently or permanently respond to internal or external stimuli to provide developmental cues. A well-known PTM on DNA is methylation while histones have been reported to undergo methylation, acetylation, phosphorylation, citrullination, ubiquitina- tion, ADP ribosylation and sumoylation (2). Different cues integrate together to direct stem cells to differen- tiate into various cell types. Dysregulation of this pro- cess in development can dictate defects that eventually lead to the susceptibility to diseases. Therefore, PTMs are potential pharmacological targets in many diseases including cancers (3,4). DNA methylation and the other PTMs on histones are major epigenetic features of chro- matin by altering nucleosomal organization, and result in gene activation or repression (5). In this review ar- ticle, we focus on the latest studies in the regulations of acetylation, methylation, and phosphorylation of his- tones, and also summarize inhibitors/drugs that target these modifications for cancer treatment.
Acetylation/de-acetylation of histones in cancers Currently, one of the most extensively studied PTMs is histone acetylation (2,6), which adds an acetyl group to the terminal of lysine residues within the tail domain of the core histones. This neutralizes the positive charge of the histones, and results in the relaxation of the chro-
matin. The relaxed chromatins are more accessible to the transcriptional machinery (6). In particular, acety- lated histones provide a binding platform for bromo- domain containing proteins to regulate transcriptional activity. Thus, histone acetylation facilitates the main- tenance of euchromatin structure and controls transcrip- tional activation (7). In contrast, histone deacetylation removes acetyl groups from the acetylated histones and leads to an inactive chromatin environment. In general, histone acetyl transferases (HATs) account for the func- tion of transcription co-activators, whereas histone dea- cetylases (HDACs) act as transcriptional co-repressors (2). The dysfunction of HATs and the increase of HDAC activity are suggested as epigenetic abnormalities in cancers. It has been shown that the dysfunction of tumor suppressor genes is a critical step for carcinogenesis and cancer progression. Notably, epigenetic modulators, such as HDACs, have been suggested to play important roles to suppress the expression of tumor suppressor genes. In addition, through the use of the high-affinity acetyl-lysine antibodies pull down method that couples to the mass spectrometry (MS) detection techniques, almost 2000 acetylated proteins that comprise around
3,600 acetylation sites was identified in the cell (8).
The term “acetylome” is therefore created to describe all acetylated sites on a set of acetylated proteins in a cell. These acetylated proteins not only include histone proteins, but also many transcription factors (9). The acetylation status in the cells is tightly regulated by the activities of HATs and HDACs. In normal physiological conditions, the acetylation of histones and transcription factors are precisely regulated. Such homeostasis is regulated by protein activity, concentration, and the re-
Copyright © 2015. All rights reserved.
1
cruitment of HATs and HDACs and is essential for gene regulation of normal cellular functions. In addition, it has been shown that differential recruitment of HATs and HDACs also regulates the acetylation homeostasis, and has been linked to various neurodegenerative disor- ders and cancerous diseases.
In the process of tumor for- mation, the acetylation machinery is impaired and the acetylation status in cells becomes deacetylation, thus resulting in dysregulation of proliferation, differentia- tion and apoptosis and, further transforming the cells into a malignant state and the initiation and progression of cancer (10).
The enzymes, HATs and HDACs, regulate the acetylation of histones to modulate gene expression.
It has been shown that the balance between HATs and HDACs is dysregulated in many types of cancer. Cur- rently, HDACs are classified into four groups namely class I (HDACs 1-3, and HDAC8), class II (HDACs 4-7, HDACs 9-10), class III (sirtuins) and class IV (HDAC11). HDAC inhibitors have been demonstrated to exert anticancer activity by promoting acetylation of both histones and non-histone protein substrates. Admi- nistration of HDAC inhibitors to cancerous cells results in cell cycle arrest, DNA repairs inhibition, and the in- duction of cell apoptosis.
Tumor suppressor genes negatively regulate the initiation and progression of cancers. HDACs, the his- tone acetylation erasers, have been shown to silence the tumor suppressor genes. During the initiation phase of cancer, HDACs play roles in silencing the expression of genes that control cell cycle and differentiation. The ele- vated HDAC activity has been observed in promyelocy- tic leukemia (11), non-Hodgkin lymphoma (12), colon cancer and gastric cancer (13).
HDACs and cancer cell proliferation
Dysregulation of genes that negatively regulate cell proliferation is a hallmark of carcinogenesis. HDACs participate in the regulation of cell proliferation through repressing the expression of selective inhibitors, such as p21 and p27, of cyclin dependent kinase (CDK).
Notably, HDAC1-null mouse embryos and ES cells showed increased levels of p21 and p27 that are cor- related with a decreased level of cell proliferation (14).
It has been demonstrated that HDACs cooperate with SP1 to repress p21 and enhances proliferation of mouse embryonic fibroblasts (14,15). In addition, HDAC4 also cooperates with Sp1/Sp3 to repress p21 expression in colon cancer cells (16) and in glioblastoma cells (17).
Moreover, silenced HDAC4 upregulates the expression of p21 and thus inhibits human glioblastoma cell growth in vitro and in vivo (17). Furthermore, the expression le- vels of HDACs, such as HDAC1 (18- 20), HDAC2 and HDAC3 (21) are highly upregulated in prostate cancer.
HDACs and cancer cell differentiation
Dysregulated cancer cell proliferation is usually accompanied by the loss of differentiation capacity.
HDACs have been demonstrated to play important roles in the regulation of histone acetylation, thus affecting
the expression of differentiation-related genes. In addi- tion, Mucins are a family of proteins that are highly gly-
Y-C. Hsu et al. / Histone PTMs as cancer therapeutic targets.
cosylated. Of note, Mucin2 (MUC2) is the most abun- dant gastrointestinal secreted form that is involved in gastrointestinal cell differentiation. Decreased MUC2 expression is observed in both pancreatic and colorectal cancers. MUC2-null mice have been demonstrated to develop adenocarcinoma (22). In particular, H3-K9 and H3-K27 acetylation has been reported to be enhanced in MUC2 promoter region to activate the expression of MUC2. Trichostatin A (TSA), a HDAC inhibitor, has been shown to induce MUC2 mRNA and protein ex- pression in the pancreatic cancer cells that lack MUC2 expression (23). Furthermore, treatment of colon cancer cells with sodium butyrate, another HDAC inhibitor, has also been demonstrated to induce MUC2 mRNA and protein expression through enhancing the acetyla- tion of H3 (24). In addition, the increased acetylation of histone H3 and H4 at the MUC2 promoter and up- regulation of MUC2 mRNA were also observed upon sodium butyrate treatment (25).
HDACs and the regulation of cancer cell apoptosis In addition to cell proliferation and differentiation, HDACs also play a role to suppress apoptosis in cancer cells. For example, HDAC2 reduce apoptosis of pan- creatic cancer cells (26). Furthermore, knockdown of HDAC2 in pancreatic cancer cells accelerates the pro- cessing of caspase 8 and increases apoptosis. Additio- nally, co-treatment of valproic acid (VPA) in HDAC2 suppressing pancreatic cells further enhances apopto- sis of them (26). Interestingly, knockdown of HDAC2 upregulates the expression of NOXA gene, which is pro-apoptotic and can sensitize pancreatic cancer cells to etoposide-induced apoptosis, suggesting that HDAC2 can silence NOXA in pancreatic cancer cells (27). Simi- larly, knockdown of HDAC1 also enhances apoptosis in osteosarcoma cells (28), whereas knockdown of HDAC2 induces apoptosis in HeLa cells (12). In par- ticular, synergistic treatment of two HDAC inhibitors, VPA and CI-994, can lead to the stabilization of histone acetylation and the restoration of caspase-8 in small cell lung carcinomas cells (29). Notably, treatment of medulloblastoma cells with HDAC inhibitor, MS-275, induces caspase-8 activity and increases apoptosis.
This coincides with the increased acetylation levels of histone H3 and H4 at the promoter of TRAIL receptor 1 and increased levels of TRAIL receptor 1 gene and protein (30). In lung cancer cells, silence of HDAC2 induces cellular apoptosis through the activation of p53 and Bax activation and the suppression of Bcl2.
In contrast, overexpression of HDAC2 inhibits p53 ex- pression and Bax, as well as increases the expression of Bcl2, thus let the transformed cells overcoming the apoptotic signal (31). Moreover, treatment with another HDAC inhibitor, suberanilohydroxamic acid (SAHA;
also named as vorinostat), also sensitizes breast cancer cells and induce the activation of caspase 8, caspase 3, Bid and PARP cleavage concomitantly, as well as the enhanced expression of Bax and TRAIL receptor 1 (32).
In vivo, treatment of the breast cancer cells in nude mice with SAHA induce the expression of Bax, Bim, NOXA, p21, DR4, DR5, tissue inhibitor of metalloproteinase-1 (TIMP-1) and TIMP-2 (33). HDAC5 is required for the maintenance of heterochromatin and the refractory
Y-C. Hsu et al. / Histone PTMs as cancer therapeutic targets.
to apoptosis of human cancer cells. In contrast, loss of HDAC5 induced heterochromatin de-condensation and enhanced sensitivity of DNA mutation agents, thus resulting in cancer cell apoptosis (34). Currently, it is clear that HDAC1, HDAC2 and HDAC5 are involved in the attenuated apoptosis during carcinogenesis.
Cancer progression and the roles of HDACs
In addition to the initiation of cancer, HDACs also regulate the genes implicated in the progression of can- cer. Decreased histone acetylation has been suggested to be involved in tumorigenesis, tumor invasion and me- tastasis (35). Furthermore, the status of acetylation and deacetylation of the transcription factor HIF1α regulate angiogenesis and cellular metabolism in carcinogenesis. The acetylation and deacetylation at different lysine re- sidues in HIF1α protein result in different biological ef- fects. Acetylation at the N- terminus (Lys-10, 11, 12, 19,
21) and at the oxygen-dependent degradation domain (Lys-532) can promote HIF1α protein degradation and inhibition of downstream HIF1 activity (36). In particu- lar, HDAC4 can deacetylate the N-terminal of HIF1α and increases the stability, transcriptional activity of HIF1α and thus the expression of a subset of HIF1α downstream target genes, including VEGF-α, lactate dehydrogenase A, and GLUT1 (36). Furthermore, such deacetylation directs the cells towards transformation.
Notably, glycolytic genes, such as phosphoglycerate ki- nase 1 and pyruvate kinase M2 and glucose transporters GLUT1 and GLUT3, are also the downstream targets of HIF1α and are upregulated in cancer cells (37-39). The- refore, HDACs regulate the metabolic changes in can- cer cells through the deacetylation machinery to meet the demand of extra energy requirement of cancer cells.
HDAC inhibitors
HDACs have attracted a lot of attention as anti- cancer therapeutic targets. It has been suggested that HDACs are involved in tumorigenesis (40,41). HDACs negatively regulate the gene expression of cell cycle inhibitors, differentiation factors, and pro-apoptotic fac- tors. Notably, HDACs also up-regulate the expression of genes associated with angiogenesis, cell invasion and migration. In the previous sections, we have shown that HDACs are involved in the regulation of cancer initia- tion and progression. Therefore, HDAC inhibition may show promising antitumor effects on various types of cancer. Specifically, HDAC inhibitors could exert their anti-cancer effects through inducing cell cycle arrest, apoptosis, and differentiation (40,41). In addition, the specific HDAC inhibitors should be designed to avoid in targeting all of the HDACs. Due to different chemical structures and mechanisms of inhibition, seven catego- ries of HDAC inhibitors have been reported, including short chain fatty acids, cyclic peptides, benzamides, hydroxamine-acid-derived compounds, electrophilic ketones, miscellaneous compounds and sirtuin inhi- bitors. Inhibitors for class I, II, IV of HDACs have a common metal binding domain that functions to block Zn
2+chelation at the active site (42). In particular, zinc- dependent HDAC
inhibitors are ineffective against class III HDACs
(sirtuin) (43). The mechanisms of action
and the biological consequences of Class III HDAC inhibitors remain unclear (44-46). Furthermore, sirtuin inhibitors were shown to be ineffective in human cells (47). Nevertheless, specific inhibitors against sirtuin I (SEN96) (48) and sirtuin 2 (compound 6J) (49) are cur- rently in clinical trials. Zinc-dependent HDAC inhibi- tors have been developed as anticancer drugs, and two sirtuin inhibitors have been approved for cancer treat- ment (50). Specifically, inhibition of HDAC results in cell cycle arrest at G1 phase and upregulation of p21 in a p53-independent manner (51) through increase of histones H3 and H4 acetylation at the p21 promoter (52). In addition, HDAC inhibitors, such as butyrate and TSA, exert the anti-cancer effects by stabilizing p21 mRNA (52), repressing cyclins A, D, and activa- ting p16 and p27 in cell cycle arrest (53,54). Interestin- gly, TSA has also been reported to induce microRNA-7 expression, leading to suppression of epidermal growth factor receptor (EGFR) expression in an HDAC-inde- pendent manner in lapatinib-treated breast cancer cells (55). Furthermore, treatments of HDAC inhibitors have been shown to activate the expression of pro-apoptotic genes and/or to reduce the expression of anti-apopto- tic genes (56). Comparing with other epigenetic drugs, the potency is the major advantage of HDAC inhibitors, due to that most of their effective doses are in nano- and micro-molar range (57,58). It has also been shown that HDAC inhibitors also inhibit angiogenesis and increase the host immune system in cancer patients (56,59,60).
Therefore, HDAC inhibitors can be used with other different drugs in cancer therapies. Notably, HDAC inhibitors can function synergistically with different chemotherapeutic agents and biologic polypeptides. For example, pretreatment with the HDAC inhibitor, vori- nostat, enhances the drug effects of topoisomerase II inhibitors (61). In addition, HDAC inhibitors are also combined with DNA demethylating agents to reacti- vate the silenced tumor suppression genes. As a proof of principle, decitabine was combined with HDAC in- hibitors (such as phenyl butyrate or VPA) to treat the patients with acute myeloid leukemia (AML) or mye- lodysplastic syndrome (MDS). The results have been shown more tolerable and promising efficacy in three Phase I/II trials (62-64). Notably, another promising strategy is the use of HDAC inhibitors with tyrosine kinase inhibitors in cancers that are characterized with overexpressed anti-apoptotic genes. For example, vori- nostat, LBH-589, LAQ-824 and romidepsin have been demonstrated there synergistic effects with imatinib for pro-apoptotic activity in the treatment of both imatinib- sensitive and imatinib-resistant leukemic cells (65-67).
HAT inhibitors
HAT is responsible for the reaction of histone acety- lation, which transfers the acetyl group of acetyl coen- zyme A (acetyl-CoA) to the ε-amino group of lysine (68,69). HATs have been classified into two groups, type-A and B, based on sequence divergence of the HAT domain and intracellular localization. Type A HATs are nuclear localized HATs that acetylate histones and other chromatin-associated proteins.
Interestingly, although Type B HATs are localized in
both nuclear and cytoplasm, it is responsible that
acetylating the newly
synthesized histones in the cytoplasm promotes their nuclear localization (70,71). Type A HATs consist of three families: GNATs, P300/CBP, and MYST.
Type B HAT consist only the histone acetyltransferase- 1 (HAT1/KAT1) (69,72). Little sequence similarity and no homology domain have been shown in the type A HAT families, but the acetyl-CoA binding domain is highly conserved in all HATs (73).
Due to the specific catalytic activity of HATs relying on association, HAT inhibitors are therefore mainly the acetyl-CoA-related derivatives, conjugates.
Currently, HAT inhibitors are classified into synthetic peptide CoA-based, natural product or small molecule.
Notably, the synthetic HAT inhibitors were identified from the finding that polyamine-CoA conjugates can inhibit HAT activity in vitro (74). Most of the synthetic bi-substrate HAT inhibitors mimic the complex of acetyl CoA-ly- sine intermediate and exert the inhibitory effects. The major disadvantage for this class of HAT inhibitors is their high degree of cellular impermeability. Further- more, most of the HAT inhibitors from natural products also have the same disadvantage. The best characterized of HAT inhibitors from natural products is Curcumin, which is isolated from the rhizome. It has been demons- trated that Curcumin is effective in the prevention and treatment of many types of cancer, such as colorectal, breast, cervical kidney, lung, prostate, ovarian and liver cancers (75,76).
The third class of HAT inhibitors is a number of small molecules to deal with permeability. These inhi- bitors include butyrolactone 3 (MB-3), quinoline and isothiazolone and their derivatives. Isothiazolone has been shown to inhibit the cell proliferation of human ovarian and colon cancer cell lines (77). The derivatives of isothiazolone also showed inhibitory effects on the enzyme activities of HAT. Using HATs as drug targets is complicated due to the fact that most HATs are the component of large protein complexes (78), which may increase the variability of HATs. It has been shown that the protein complexes have been shown to reduce the HAT activity from certain loci (79). In addition, the pro- tein complexes facilitate the different enzymatic activity required for different cellular functions. Based on these findings, making HAT inhibitors with cell permeability will facilitate the drug development for cancer treat- ment.
Methylation/de-methylation of histones in cancers The dynamical activities of lysine methyltransfe- rases (KMTs) and demethylases (KDMs) control the methylation status of lysine residues on histone pro- teins, which modulate chromatin structure to regulate different cellular functions, including transcription, re- plication and repair (80). The specificity and degree of methylation at lysine residues rely on particular KMTs and KDMs. The first site-specific histone KMT, SU- V39H1 (KMT1A), which contains a conserved enzy- matic SET domain, has been identified in 2000 (81).The SET domain, a 130 amino acid with catalytic activity towards lysine residues of histones, was initially found to be conserved in Su(var)3-9, E(z) (enhancer of zeste)
and trithorax (82). By homology alignments with the
SET domain, dozens of KMTs have further identified
(83). In addition to SET domain-containing KMTs, ano- ther class of KMTs without SET domain, such as KMT4 (also known as Dot1p in yeast and Dot1L in human), has also been clarified (84,85). Although these two classes of KMTs do not possess conserved catalytic domains, both of them utilize S-adenosyl-L- methionine (SAM) as the methyl group donor for lysine methylation on his- tones (83,86). The presence of histone KDMs was first identified from a part of the C- terminal binding protein 1 (CtBP1) corepressor complex as LSD1/KDM1A, which contains a flavin adenine dinucleotide (FAD)-dependent amine oxidase domain that demethylates H3K4me2 and H3K4me1 and modulates gene expression (87,88). Additionally, another type of histone KDMs, such as KDM2A/B, which utilizes the Jumonji (JmjC) domain to catalyze demethylation through the oxidation of methyl groups, is dependent on a-ketoglutarate, mole- cular oxygen, and Fe(II) (89-91). However, the afore- mentioned KDM1 and KDM2, as well as KDM3 can’t demethylate trimethylated lysine until the discovery of KDM4A-KDM4D (also known as JMJD2A-JMJD2D), which are capable of demethylating histone H3K9me3/
H3K9me2, H3K36me3/H3K36me2, and H1.4K26me3/
H1.4K26me2, but are unable to remove H3K9me1 or H3K36me1 (92-96). These studies demonstrate the spe- cificity of KMTs and KDMs for both the site and degree of methylation.
The site and the degree of methylation on lysine resi- dues are important in generation of open or closed chro- matin. For instance, histones H3K4me3, H3K36me2/3, H3K79me2/3, and H4K20me1 correlate with active states of open chromatin; in contrast, H3K9me3, H3K27me3, and H4K20me3 marks are associated with closed heterochromatin (97,98). The above histone
‘marks’ provide a particular surface for recognition by ‘reader’ proteins. For example, the basal transcrip- tion factor TFIID directly recognizes and binds to the H3K4me3 mark via the plant homeodomain (PHD) fin- ger of TAF3, leading to activation of transcription (99).
In contrast, a heterochromatic adaptor HP1 recognizes
H3K9me3 mark through its chromo domain, thereby ge-
nerating the closed supra-nucleosomal chromatin struc-
ture and silencing transcription (100). Accumulated evi-
dences demonstrate that lysine methylation of histones
play important roles in cancer progression. Alterations
of histone KMTs and KDMs, such as overexpression,
downregulation, mistargeting, mutations and gene
fusions caused by chromosomal translocations, result
in aberrant modification marks and structures of chro-
matin, leading to deregulation of cancer-specific gene
expressions, uncontrolled cell proliferation, and ge-
nome instability, in different types of cancers (101). For
example, KMT5A (also known as PR-SET7 and SET8)
directly interacts with PCNA and mono-methylates his-
tone H4K20 to regulate S phase progression during the
cell cycle (102,103). The amounts of KMT5A and his-
tone H4K20me1 increase with cell cycle progression,
reach to the highest levels during G2/M and early G1
phase (104), subsequently, followed by marked degra-
dation of KMT5A via PCNA-coupled CRL4 (CDT2)
ubiquitination during S-phase (105). KMT5A and
H4K20 methylation are tightly regulated in replication
fork to prevent aberrant re-replication and maintain
fork stability during DNA replication (106,107). His-
tone H4K20me1 can be further di- and tri-methylated by Suv4-20h1/2(107). Histone H4K20me2 is required for recruiting 53BP1 at sites of DNA damage to initiate DNA repair process (108). We also find a crosstalk of histone H4K20 methylation and H4Y72 phosphoryla- tion in regulation of the above processes (109). Histone H4K20me3 and Suv4-20h enzyme coupled with HP1 are focally enriched at pericentric heterochromatin in higher order chromatin structure (110). In addition, KMT5A interacts with TWIST, a master regulator of epithelial–mesenchymal transition (EMT), to activate the transcription of N-cadherin gene and repress that of E-cadherin gene via its mono-methylation activity to- wards H4K20, thereby promoting EMT and enhancing the invasive potential of breast cancer cells both in vitro and in vivo. Importantly, the expression of KMT5A is clinically positive-correlated with expressions of N- cadherin and TWIST expression, as well as metastasis, but negative-correlated with E- cadherin expression in human breast carcinoma, suggesting KMT5A as a po- tential therapeutic target of breast cancer via suppres- sing metastatic properties (111).
DOT1L (as known as KMT4) specifically methy- lates histone H3K79 at various degree including mono- , di- and tri-methylation (85,112). Although DOT1L does not possess a common SET domain, its catalytic domain shows structural similarity with that of classic non-histone methyltransferases (113), such as catechol O-methyltransferase COMPT (114). Mistargeting of DOT1L by association with different MLL fusion pro- teins, such as MLL-AF4, MLL-AF9, MLL-AF10, and MLL-ENL caused by translocation, leading to their abnormal expressions have been demonstrated in leuke- mias (115,116). Additionally, the activity of DOT1L is required for maintaining the MLL-AF6-driven oncoge- nic gene-expression program in accordance with high levels of histone H3K79me2 in hematologic malignan- cy. Conditional knockout of Dot1l results in suppressing the MLL-AF6-mediated leukemogenesis in a mouse model (117).
Polycomb group (PcG) proteins serve as the regu- lators to control the process through histone modifica- tions (118,119). PcG proteins comprise of two poly- comb repressor complexes (PRCs), PRC1 and PRC2.
The PRC2 complex consists of EZH2, SUZ12, EED, and RBAP48, in which EZH2 is a catalytic subunit with histone methyltransferase activity that tri-methylates lysine 27 of histone H3 (H3K27me3). Previous studies have demonstrated that EZH2 is overexpressed in a wide variety of cancerous tissue types, such as prostate, breast (119-121), and brain (122-125) cancers. Its roles in maintenance of stemness and neuron regeneration have been illustrated (126-129). In addition, accumu- lating evidences have demonstrated that EZH2 is criti- cal in drug resistance and overexpressed in cancer stem cells (also named as tumor initiating cells, TICs). For instance, high expression of EZH2 is associated with tumor aggressiveness and poor prognosis in patients with esophageal squamous cell carcinoma treated with definitive chemoradiotherapy (130). Overexpression of EZH2 contributes to the acquired cisplatin resistance in ovarian cancer cells (131). Overexpression of EZH2 and chemotherapy have been shown to enrich stem cell-
like side populations in ovarian cancer (132), breast
cancer
(133), and prostate cancer (134). In contrast, decreased expression of EZH2 shows favorable outcome to ta- moxifen, an antagonist of the estrogen receptor in ad- vanced breast cancer (135,136). Knockdown of EZH2 by RNAi re-sensitizes drug-resistant ovarian cancer cells to cisplatin (131), reverses the drug resistance to 5-Fu in human hepatic multidrug-resistant cancer cells (137), and decreases MDR1 expression and sensitizes multidrug-resistant hepatocellular carcinoma cells to chemotherapy (138).
Histone KMT inhibitors
Due to the roles of KMTs/KDMs in cancers, several inhibitors against some of these molecules have been developed. The aminonucleoside inhibitors, such as EPZ004777, have been used for targeting DOT1L cata- lytic activity by competition with SAM for binding to its active site. The compound EPZ004777 selectively decreases the global levels of H3K79 methylation and kills mixed lineage leukemia cells bearing the MLL gene translocation(139,140). Although it has been re- ported that activation of Wnt-targeted genes also de- pends on H3K79 methylation, recently, treating human colon adenocarcinoma-derived cell lines by inhibiting DOT1L with EPZ004777 does not affect the canonical Wnt signaling pathway and H3K79 methylation is not increased in clinical human colon carcinoma samples comparing with that in normal colon tissue, suggesting that DOT1L might be not a favorable therapeutic tar- get in colon cancer (141). Another advanced DOT1L inhibitor, EPZ-5676, has been designed and synthesized as a SAM-competitor, which induces conformational changes in the active site, and possesses attractive se- lectivity and a slow off-rate with the most potent effects on inhibition of H3K79 methylation and MLL-fusion target gene expression, leading to effective cell killing for acute leukemia (142). It demonstrates biexponen- tial pharmacokinetics following intravenous adminis- tration, but has low oral bioavailability in animal mo- dels including mouse, rat and dog (143). EPZ-5676 is also effective in AML with partial tandem duplication (PTD), MLL-PTD (144). Moreover, the synergistic anti-proliferative activity of EPZ-5676 has been obser- ved in combination with cytarabine and daunorubicinin MLL-rearranged leukemia cells (145). Recently, EPZ- 5676 has been applied to a phase I clinical trial in the adult and pediatric patients with MLL-rearranged AML (http://clinicaltrials.go v ).
A series of small molecules, such as GSK126 (146),
EI1 (147), UNC-1999 (148), and E7438 (EPZ-6438)
(149), have been developed for inhibition of the cata-
lytic activity of EZH2 and thereby decreasing global
tri-methylation at histone H3K27. These inhibitors are
selectively against EZH2 over other KMTs and func-
tion by competition with SAM. The turnover rates of
H3K27me3 are low, thus prolong treatment of EZH2
inhibitors is required for its efficacy in non-Hodgkin’s
lymphoma without altering H3K27me1 (150). EZH2 is
normally expressed in the germinal center (GC) B cells
and decreases to turn on genes required for B cells
diffe- rentiation. Around 1800 EZH2-targeted genes in
GC B cells are identified. Accordingly, knockdown of
EZH2 in diffuse large B-cell lymphoma (DLBCL) cells
leads
to acute cell cycle arrest at the G1/S transition and up- regulation of its tumor suppressor target genes. These results suggest the critical roles of EZH2 involved in regulation of a specific epigenetic program in normal GC B cells, and aberration of the epigenetic program may contribute to their malignant transformation into DLBCLs (151). Somatic mutations of EZH2 frequently occur in B cell lymphomas. Conditional expression of mutant EZH2 in mice promotes hyperplasia and lym- phomagenesis via abnormal suppression of genes requi- red in B-cell differentiation. The discovery provides the potential therapeutic strategy by targeting EZH2 in DLBCLs (152). EZH2 inhibitors have effective an- ti-tumor activity in different animal models including GCB-DLBCL xenograft models (146,149,150,152) and SMARCB1-deletion driven rhabdoid tumor models (153). The clinical trials of EZH2 inhibitors, including E7438 (EPZ-6438), CPI-1205, and GSK2816126, are in progress for different types of lymphomas (http://clini- caltrials.gov). The anti-tumors activity of other histone KMT inhibitors, such as A-366 against EHMT1 (154), BIX-01294 against EHMT2 (155-157), AZ505 and LLY-507 against SMYD2 (158,159), have also docu- mented in preclinical stage.
Histone KDM inhibitors
Human KDMs for histones consist of two distinct enzyme classes: FAD-dependent amine oxidase type demethylase, such as LSD1/KDM1A (87,88), and the JmjC-domain containing demethylases, such as KDM2A/B (89-91). Tranylcypromine (TCP) is an irre- versible inhibitor of KDM1A via covalently binding to the FAD co-factor that resides at the base of the active site (160). The cellular effects of the KDM1A inhibitors on induction of histone H3K4 methylation and anti-pro- liferative activity are demonstrated as well (161). Afte- rward, a variety of TCP analogs have been generated by substituting the phenyl group of TCP with functio- nal groups ranging from fluoro- and bromo-additions (162,163). TCP analogs, trans-N-[1-(2,3-dihydro-1,4- benzodioxin-6-yl)ethyl]-2-phenylcyclopropan-1-amine (Compound A) and trans-N-[(2-methoxypyridin-3-yl) methyl]-2-phenylcyclopropan-1-amine (Compound B, also known as Oryzon), effectively inhibit KDM1A- mediated responses in the nanomolar range and show si- milar phenotypes of Kdm1a knockdown in both murine and primary human AML cells bearing the MLL gene translocations exhibiting MLL translocations(164). Two irreversible, TCP-derived KDM1A inhibitors, ORY-
1001 (165) and GSK2879552 (165,166), are currently being applied to clinical trials in patients with AML or small cell lung cancer (http://clinicaltrials.go v ).
Another type of KDM1A inhibitors is reversible, such as GSK690 and N’-(1-phenylethylidene)-benzohy- drazides (167,168). The most potent inhibitor, SP2509, attenuates the interaction between LSD1 and the core- pressor CoREST, enhances the permissive H3K4me3 mark on the target gene promoters, and increases the le- vels of p21, p27 and CCAAT/enhancer binding protein alpha, leading to suppressing cell growth in AML cells.
Combinational treatment with SP2509 and panobinos-
tat, a pan-HDAC inhibitor, synergistically kills AML
cells and also improves the survival in a human AML
cell-inoculated mouse model (169). In addition, low molecular weight amidoximes, such as bis-guanidines, bis-biguanides, and their urea- and thiourea isosteres, have been identified as KDM1A inhibitors by structure- based virtual screening. These amidoximes potently inhibit LSD1/ KDM1A and induce the re-expression of aberrantly silenced tumor suppressor genes in tumor cells (170). However, the potentials of these novel re- versible KDM1A inhibitors in pre-clinical development need to be further explored and evaluated.
The other KDM members containing the JmjC do- main utilize 2-oxoglutarate (2-OG; α-ketoglutarate) as a co-factor. Several inhibitors of JmjC domain-containing KDMs have been designed by interfering with the asso- ciation between KDMs and 2-OG (171). For instance, GSK-J1 is the first selective and potent H3K27-specific demethylase inhibitor from high throughput screening.
GSK-J1 mimics 2-OG binding by a propanoic acid moiety, and also chelates the Fe
2+on active site by a py- ridyl-pyrimidine biaryl, thereby inducing conformatio- nal change of this catalytic divalent cation in the active site to suppress the activity of KDMs. A cell-permeable derivative of GSK-J1 is synthesized by esterification of its polar carboxylate group and named as GSK-J4 (172).
The effects of GSK-J4 on tumor suppression have been observed in T-ALL leukemia cells (173).
Phosphorylation of histones in cancers
Mitogen- and stress-activated kinase 1/2 (MSK1/2) elicits histone H3 phosphorylation at S10 (H3S10p) and S28 (H3S28p) (174), in which H3S10p contributes to EGF-stimulated neoplastic cell transformation (175).
The MAP kinase cascades are key regulators in the phos- phorylation of H3S10 and activation of immediate early (IE) response genes upon different stimuli, including growth factors, cytokines, and stress.
Increased H3S10p induces the expression of the IE
response genes, inclu- ding proto-oncogenes c-fos and
c-jun, in cancer pro- gression (176). Activating
mutations in K-Ras elevates the Ras-MAPK pathway
and results in the MSK1-me- diated H3S10p in
neoplastic transformation of pancrea- tic cancer cells
(177). The transcription byproducts, R loops, constitute
a threat to genome integrity and are tightly linked to
H3S10p for chromatin condensation (178,179). In
addition to acetylation and methylation of histones,
gene transcription is also regulated through
phosphorylation of histone H2B at S33 by TAF1 (180)
and histone H3 at T11 by PRK1 (181). Moreover, tyro-
sine phosphorylation of histones has also been identi-
fied in recent years. WSTF, a transcription factor, has
intrinsic tyrosine kinase activity toward Y142 of his-
tone H2AX to maintain S139 phosphorylation and IR-
induced foci formation, which is crucial for regulation
of the DNA damage response (182). Rad53-associated
Y99 phosphorylation of histone H3 is also critical for
efficient ubiquitination and degradation in the regula-
tion of histone levels (183). JAK2 phosphorylates his-
tone H3-Y41 and prevents HP1α binding to chromatin
(184). WEE1 has been identified as the tyrosine kinase
towards Y37 on histone H2B, and histone H2B-Y37
phosphorylation suppresses expression of replication-
dependent core histone genes via excluding binding of
the transcriptional coactivator NPAT and RNA poly-
merase II, and recruiting the histone chaperone HIRA (185). Recently, we have demonstrated that EGFR can translocate into the nucleus to phosphorylate histone H4 at Y72 and results in enhancing the recruitment of histone KMTs to facilitating its K20 methylation and consequently promotes DNA synthesis and repair (109). The evidence supports that non-canonical nuclear EGFR works as a modifier of nuclear proteins, such as PCNA (186), RNA helicase A (187), PNPase (188), and histone H4 (109), and thus regulates the relative cellular functions. Disruption of the interaction between EGFR and the associated nuclear proteins leads to suppress cell growth of prostate cancer (189) and breast cancer (109,190).
Kinase inhibitors
Based on the previous documents, several kinases have been known to possess kinase activity toward his- tones, such as MSK1 (174,177), JAK2 (184), WEE1 (185), and EGFR (109). Inhibition of MSK1 activity by addition of its inhibitor, H89, or by knockdown of MSK1 with specific short hairpin RNAs (shRNAs) specifical- ly abrogates cell proliferation in response to estrogens or progestins in breast cancer cells (191).
Recently, to Inhibition of MSK1 by the same approach significantly reduces latent membrane protein 1 (LMP1)-promoted cell proliferation, and induces cell cycle arrest at G0/ G1 phase. Knockdown of MSK1 attenuates the LMP1- promoted anchorage-independent cell growth in naso- pharyngeal carcinoma cells (192).
Because of the off- target effects of MSK inhibitor H89, another more spe- cific MSK1 inhibitor, SB- 747651A, has been developed with an IC50 value of 11 nM. Cellular study reveals that SB-747651A inhibits production of the anti-inflamma- tory cytokine IL-10 (193). Treatment with SB-747651A dramatically diminishes invasiveness of aggressive oral squamous cell carcinoma (194).
Somatic mutations of JAK2 such as JAK2-V617F, a recurring gain-of-function mutation, are correlative to development of myeloproliferative disorders (MPD).
Small-molecule inhibitors of JAK2 kinase can be clas- sified into two types: JAK2-selective (Class I) and non- JAK2 selective (Class II). The JAK2-selective inhibi- tors, including INCB018424, XL019, and G101348, and non-JAK2 selective inhibitors, such as MK-0457 (VX680), CEP-701 (Lestaurtinib), and AT9283, have been employed in clinical trials in patients with MPD, acute lymphoblastic leukemia (ALL), and CML (195).
The type II inhibitor of JAK2, CHZ868, stabilizes JAK2 protein in an inactive conformation. CHZ868 potently inhibits the growth of CRLF2-rearranged human B- ALL cells, disrupts JAK2 signaling, and improves sur- vival in B-ALL cells-inoculated mice.
Combinational treatment with CHZ868 and dexamethasone synergis- tically induces apoptotic response in JAK2-dependent B-ALLs comparing to CHZ868 alone (196). These fin- dings support the therapeutic strategy for the patients with JAK2- dependent leukemias and other disorders by targeting JAK2 kinase.
WEE1 kinase is a key G2-M checkpoint regulator, which phosphorylates CDC2 at Y15, leading to inac-
tivation of the CDC2/cyclin B complex and induction
of cell cycle arrest at G2/M. Inhibition of WEE1 by
either small molecule inhibitors or shRNAs results in premature entry into mitosis and consequently inducing cell death via mitotic catastrophe or apoptosis (197). A series of WEE1 inhibitors have been synthesized, inclu- ding PD0166285 (198,199) and MK-1775 (also named as AZD1775) (200-208). In addition to the small com- pound inhibitors, a microRNA, miR-381, has been iden- tified as a novel intrinsic WEE1 inhibitor, which up-re- gulates CDC2 activity to sensitize renal cancer cells to 5-FU by induction of mitotic catastrophe and apoptosis (209). MK-1775 (AZD1775) has been employed in cli- nical trials in patients with wide types of cancers inclu- ding lung cancer, ovarian cancer, fallopian tube cancer, peritoneal cancer, cervical cancer, glioblastoma, pan- creatic cancer, head and neck cancer, and AML (http://
clinicaltrials.gov).
Overexpression of EGFR have been observed in different types of cancers, including ovarian cancer (210), breast cancer (211-215), lung cancer (216-219), colorectal cancer (220). Several tyrosine kinase inhibi- tors (TKIs) against EGFR are currently used in clinical treatment of cancers, such as Erlotinib (Tarceva) (221), Gefitinib (Iressa) (222), Erbitux (Cetuximab) (223) and Tykerb (Lapatinib), which is a dual TKI to EGFR and HER2 (224). Lapatinib promotes the sensitivity to pro- teasome inhibitors via inducing NF-kappaB activation in triple-negative breast cancer cells (225). However, drug resistance to these clinical used TKIs sometimes occurs. The worse event-free survival rate of lapatinib in breast cancer may partly attribute to the elevation of EGFR through the downregulation of microRNA-7, subsequently leading to overexpression of cyclooxy- genase-2 independent of EGFR kinase activity (226).
Moreover, the specific monoclonal antibody against EGFR, such as Panitumumab, has also been developed for target therapy in cancer (227).
Conclusion
We described the machinery to maintain the homeos- tasis of PTMs on histones by specific enzymes, such as HATs/HDACs, KMTs/KDMs, and kinases, within cells. Abnormal activation of these enzymes frequently occurs in different types of cancers.
Several inhibitors/ drugs have been developed for specifically suppressing the overexpressed enzymatic activity in pre-clinical or clinical treatment of cancers (summarized in Table 1). Targets validation for personalized medication, and the specificity, biostability, and reactivity of the inhibitors/ drugs are critical for the outcome of cancer patients af- ter treatments. Importantly, drug resistance is another difficult issue needed to be solved. Compensation with activating alternative signal pathways has been uncove- red in cells with drug resistance. Therefore, combina- tional treatments with different inhibitors/drugs might overcome the difficult problems. To sum up, understan- ding the detail molecular mechanisms of specific inhi- bitors/drugs and their resistance will benefit to improve the therapeutic strategy for better clinical outcome of cancer patients.
Acknowledgements
We appreciate the financial support by grants from
Ministry of Science and Technology (MOST103-
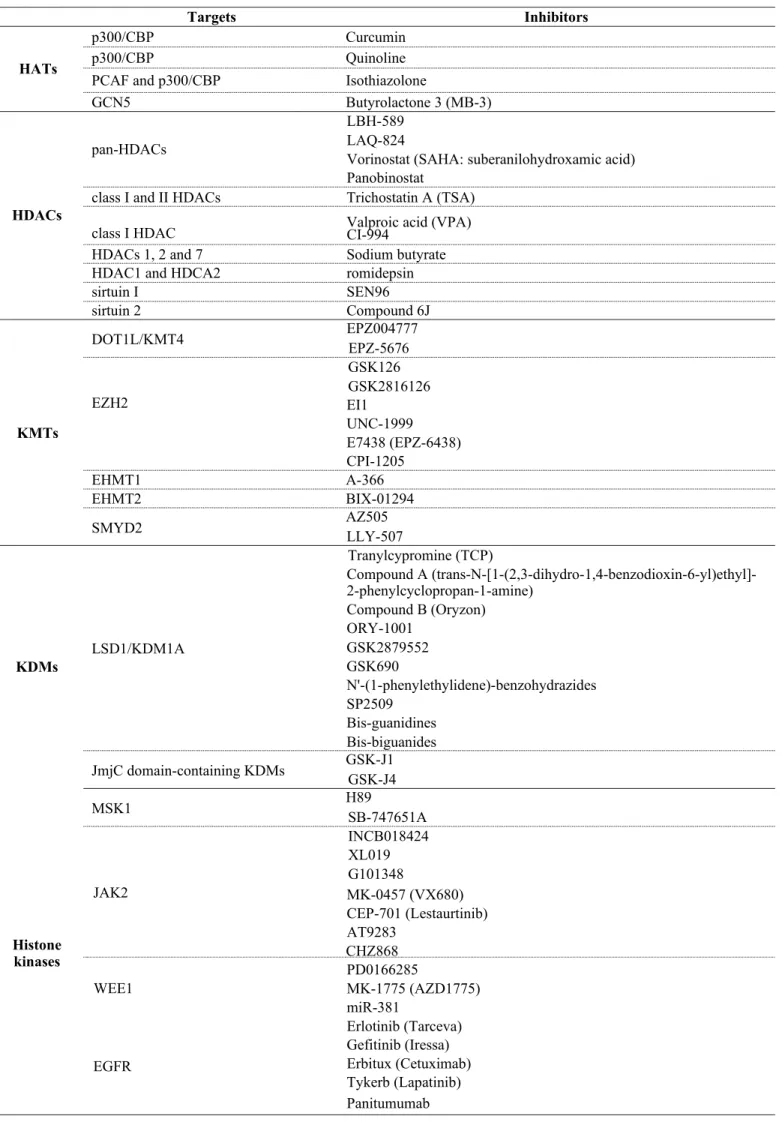
Table 1. The candidate targets and inhibitors of histone PTMs in oncology.
Targets Inhibitors
p300/CBP Curcumin
p300/CBP Quinoline
HATs
PCAF and p300/CBP Isothiazolone
GCN5 Butyrolactone 3 (MB-3)
LBH-589
pan-HDACs LAQ-824
Vorinostat (SAHA: suberanilohydroxamic acid) Panobinostat
HDACs
class I and II HDACs Trichostatin A (TSA)
class I HDAC Valproic acid (VPA)
CI-994
HDACs 1, 2 and 7 Sodium butyrate
HDAC1 and HDCA2 romidepsin
sirtuin I SEN96
sirtuin 2 Compound 6J
DOT1L/KMT4 EPZ004777
EPZ-5676 GSK126 GSK2816126
KMTs
EZH2 EI1
UNC-1999
E7438 (EPZ-6438) CPI-1205
EHMT1 A-366
EHMT2 BIX-01294
SMYD2 AZ505
LLY-507
Tranylcypromine (TCP)
Compound A (trans-N-[1-(2,3-dihydro-1,4-benzodioxin-6-yl)ethyl]- 2-phenylcyclopropan-1-amine)
Compound B (Oryzon) ORY-1001
KDMs
LSD1/KDM1A GSK2879552
GSK690
N'-(1-phenylethylidene)-benzohydrazides SP2509
Bis-guanidines Bis-biguanides JmjC domain-containing KDMs GSK-J1
GSK-J4
MSK1 H89
SB-747651A INCB018424 XL019 G101348
JAK2 MK-0457 (VX680)
CEP-701 (Lestaurtinib) AT9283
Histone
kinases
CHZ868 PD0166285
WEE1
EGFR
MK-1775 (AZD1775) miR-381
Erlotinib (Tarceva)
Gefitinib (Iressa)
Erbitux (Cetuximab)
Tykerb (Lapatinib)
Panitumumab
2320-B-039-052-MY3, MOST104-2321-B-039-005, MOST104-2320-B-039-031, MOST103-2314-B-715- 001-MY2, and MOST104-2314-B-715-003-MY3), National Health Research Institutes (NHRI-EX102- 10245BI), China Medical University (CMU104-S-02), Taiwan.
Other articles in this theme issue include references (228-239).
References
1. Goldberg, A. D., Allis, C. D., Bernstein, E. Epigenetics: a landscape takes shape. Cell, 2007, 128:635-638, doi:10.1016/j.
cell.2007.02.006
2. Parbin, S., Kar, S., Shilpi, A., Sengupta, D., Deb, M., Rath, S. K., et al. Histone deacetylases: a saga of perturbed acetylation homeostasis in cancer. J Histochem Cytochem, 2014, 62:11-33, doi:10.1369/0022155413506582
3. Alhazzazi, T. Y., Kamarajan, P., Verdin, E., Kapila, Y. L.
SIRT3 and cancer: tumor promoter or suppressor? Biochim Biophys Acta,
2011, 1816:80-88, doi:10.1016/j.bbcan.2011.04.004
4. Alhazzazi, T. Y., Kamarajan, P., Joo, N., Huang, J. Y., Verdin, E., D’Silva, N. J., et al. Sirtuin-3 (SIRT3), a novel potential therapeutic target for oral cancer. Cancer, 2011, 117:1670-1678, doi:10.1002/ cncr.25676
5. Vaissiere, T., Sawan, C., Herceg, Z. Epigenetic interplay be- tween histone modifications and DNA methylation in gene silenc- ing. Mutat Res, 2008, 659:40-48, doi:10.1016/j.mrrev.2008.02.004 6. Grunstein, M. Histone acetylation in chromatin structure and transcription. Nature, 1997, 389:349-352, doi:10.1038/38664 7. Shogren-Knaak, M., Ishii, H., Sun, J. M., Pazin, M. J., Davie, J. R., Peterson, C. L. Histone H4-K16 acetylation controls chroma- tin structure and protein interactions. Science, 2006, 311:844-847, doi:10.1126/science.1124000
8. Choudhary, C., Kumar, C., Gnad, F., Nielsen, M. L., Rehman, M., Walther, T. C., et al. Lysine acetylation targets protein com- plexes and co-regulates major cellular functions. Science, 2009, 325:834-840, doi:10.1126/science.1175371
9. Yang, X. J., Gregoire, S. Metabolism, cytoskeleton and cellular signalling in the grip of protein Nepsilon - and O-acetylation.
EMBO Rep, 2007, 8:556-562, doi:10.1038/sj.embor.7400977 10. Joanna, F., van Grunsven, L. A., Mathieu, V., Sarah, S., Sarah, D., Karin, V., et al. Histone deacetylase inhibition and the regula- tion of cell growth with particular reference to liver pathobiol- ogy. J Cell Mol Med, 2009, 13:2990-3005, doi:10.1111/j.1582- 4934.2009.00831.x
11. Chevallier, N., Corcoran, C. M., Lennon, C., Hyjek, E., Chad- burn, A., Bardwell, V. J., et al. ETO protein of t(8;21) AML is a corepressor for Bcl-6 B-cell lymphoma oncoprotein. Blood, 2004, 103:1454-1463, doi:10.1182/blood-2003-06-2081
12. Huang, B. H., Laban, M., Leung, C. H., Lee, L., Lee, C. K., Salto-Tellez, M., et al. Inhibition of histone deacetylase 2 increases apoptosis and p21Cip1/WAF1 expression, independent of histone deacetylase 1. Cell Death Differ, 2005, 12:395-404, doi:10.1038/
sj.cdd.4401567
13. Song, J., Noh, J. H., Lee, J. H., Eun, J. W., Ahn, Y. M., Kim, S. Y., et al. Increased expression of histone deacetylase 2 is found in human gastric cancer. APMIS, 2005, 113:264-268, doi:10.1111/j.1600-0463.2005.apm_04.x
14. Lagger, G., Doetzlhofer, A., Schuettengruber, B., Haidweger, E., Simboeck, E., Tischler, J., et al. The tumor suppressor p53 and histone deacetylase 1 are antagonistic regulators of the cyclin-de-
pendent kinase inhibitor p21/WAF1/CIP1 gene. Mol Cell Biol, 2003,
23:2669-2679,
15. Zupkovitz, G., Grausenburger, R., Brunmeir, R., Senese, S., Tischler, J., Jurkin, J., et al. The cyclin-dependent kinase inhibitor p21 is a crucial target for histone deacetylase 1 as a regulator of cel- lular proliferation. Mol Cell Biol, 2010, 30:1171-1181, doi:10.1128/
MCB.01500-09
16. Wilson, A. J., Byun, D. S., Nasser, S., Murray, L. B., Ayyanar, K., Arango, D., et al. HDAC4 promotes growth of colon cancer cells via repression of p21. Mol Biol Cell, 2008, 19:4062-4075, doi:10.1091/ mbc.E08-02-0139
17. Mottet, D., Pirotte, S., Lamour, V., Hagedorn, M., Javerzat, S., Bikfalvi, A., et al. HDAC4 represses p21(WAF1/Cip1) expres- sion in human cancer cells through a Sp1-dependent, p53-inde- pendent mechanism. Oncogene, 2009, 28:243-256, doi:10.1038/
onc.2008.371
18. Patra, S. K., Patra, A., Dahiya, R. Histone deacetylase and DNA methyltransferase in human prostate cancer. Biochem Biophys Res Commun, 2001, 287:705-713, doi:10.1006/bbrc.2001.5639
19. Patra, S. K., Deb, M., Patra, A. Molecular marks for epigenetic identification of developmental and cancer stem cells. Clin Epi- genetics, 2011, 2:27-53, doi:10.1007/s13148-010-0016-0
20. Halkidou, K., Gaughan, L., Cook, S., Leung, H. Y., Neal, D. E., Robson, C. N. Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate, 2004, 59:177-189, doi:10.1002/pros.20022
21. Weichert, W., Roske, A., Gekeler, V., Beckers, T., Stephan, C., Jung, K., et al. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br J Cancer, 2008, 98:604-610, doi:10.1038/sj.bjc.6604199
22. Velcich, A., Yang, W., Heyer, J., Fragale, A., Nicholas, C., Viani, S., et al. Colorectal cancer in mice genetically deficient in the
mucin Muc2. Science, 2002, 295:1726-1729,
doi:10.1126/science.1069094
23. Yamada, N., Hamada, T., Goto, M., Tsutsumida, H., Higashi, M., Nomoto, M., et al. MUC2 expression is regulated by histone H3 modification and DNA methylation in pancreatic cancer. Int J Can- cer, 2006, 119:1850-1857, doi:10.1002/ijc.22047
24. Hatayama, H., Iwashita, J., Kuwajima, A., Abe, T. The short chain fatty acid, butyrate, stimulates MUC2 mucin production in the human colon cancer cell line, LS174T. Biochem Biophys Res Com- mun, 2007, 356:599-603, doi:10.1016/j.bbrc.2007.03.025
25. Burger-van Paassen, N., Vincent, A., Puiman, P. J., van der Sluis, M., Bouma, J., Boehm, G., et al. The regulation of intestinal mu- cin MUC2 expression by short-chain fatty acids: implications for epithelial protection. Biochem J, 2009, 420:211-219, doi:10.1042/ BJ20082222
26. Schuler, S., Fritsche, P., Diersch, S., Arlt, A., Schmid, R. M., Saur, D., et al. HDAC2 attenuates TRAIL-induced apoptosis of pancreatic cancer cells. Mol Cancer, 2010, 9:80, doi:10.1186/1476- 4598-9-80
27. Fritsche, P., Seidler, B., Schuler, S., Schnieke, A., Gottlicher, M., Schmid, R. M., et al. HDAC2 mediates therapeutic resistance of pancreatic cancer cells via the BH3-only protein NOXA. Gut, 2009, 58:1399-1409, doi:10.1136/gut.2009.180711
28. Senese, S., Zaragoza, K., Minardi, S., Muradore, I., Ronzoni, S., Passafaro, A., et al. Role for histone deacetylase 1 in human tumor cell proliferation. Mol Cell Biol, 2007, 27:4784-4795, doi:10.1128/
MCB.00494-07
29. Kaminskyy, V. O., Surova, O. V., Vaculova, A., Zhivotovsky, B. Combined inhibition of DNA methyltransferase and histone deacet- ylase restores caspase-8 expression and sensitizes SCLC cells to TRAIL. Carcinogenesis, 2011, 32:1450-1458, doi:10.1093/carcin/ bgr135
30. Aguilera, D. G., Das, C. M., Sinnappah-Kang, N. D., Joyce, C.,
Taylor, P. H., Wen, S., et al. Reactivation of death receptor 4 (DR4) expression sensitizes medulloblastoma cell lines to TRAIL. J Neu- rooncol, 2009, 93:303-318, doi:10.1007/s11060-008-9788-x 31. Jung, K. H., Noh, J. H., Kim, J. K., Eun, J. W., Bae, H. J., Xie, H. J., et al. HDAC2 overexpression confers oncogenic potential to human lung cancer cells by deregulating expression of apopto- sis and cell cycle proteins. J Cell Biochem, 2012, 113:2167-2177, doi:10.1002/jcb.24090
32. Butler, L. M., Liapis, V., Bouralexis, S., Welldon, K., Hay, S., Thai le, M., et al. The histone deacetylase inhibitor, suberoylanilide hydroxamic acid, overcomes resistance of human breast cancer cells to Apo2L/TRAIL. Int J Cancer, 2006, 119:944-954, doi:10.1002/
ijc.21939
33. Shankar, S., Davis, R., Singh, K. P., Kurzrock, R., Ross, D. D., Srivastava, R. K. Suberoylanilide hydroxamic acid (Zolinza/vorino- stat) sensitizes TRAIL-resistant breast cancer cells orthotopically implanted in BALB/c nude mice. Mol Cancer Ther, 2009, 8:1596- 1605, doi:10.1158/1535-7163.MCT-08-1004
34. Peixoto, P., Castronovo, V., Matheus, N., Polese, C., Peulen, O., Gonzalez, A., et al. HDAC5 is required for maintenance of pericen- tric heterochromatin, and controls cell-cycle progression and sur- vival of human cancer cells. Cell Death Differ, 2012, 19:1239-1252, doi:10.1038/cdd.2012.3
35. Yasui, W., Oue, N., Ono, S., Mitani, Y., Ito, R., Nakayama, H.
Histone acetylation and gastrointestinal carcinogenesis. Ann N Y Acad Sci, 2003, 983:220-231,
36. Geng, H., Harvey, C. T., Pittsenbarger, J., Liu, Q., Beer, T. M., Xue, C., et al. HDAC4 protein regulates HIF1alpha protein lysine acetylation and cancer cell response to hypoxia. J Biol Chem, 2011, 286:38095-38102, doi:10.1074/jbc.M111.257055
37. Obach, M., Navarro-Sabate, A., Caro, J., Kong, X., Duran, J., Gomez, M., et al. 6-Phosphofructo-2-kinase (pfkfb3) gene pro- moter contains hypoxia-inducible factor-1 binding sites necessary for transactivation in response to hypoxia. J Biol Chem, 2004, 279:53562-53570, doi:10.1074/jbc.M406096200
38. Kress, S., Stein, A., Maurer, P., Weber, B., Reichert, J., Buch- mann, A., et al. Expression of hypoxia-inducible genes in tumor cells. J Cancer Res Clin Oncol, 1998, 124:315-320,
39. Liu, Y., Li, Y. M., Tian, R. F., Liu, W. P., Fei, Z., Long, Q.
F., et al. The expression and significance of HIF-1alpha and GLUT-
3 in glioma. Brain Res, 2009, 1304:149-154, doi:10.1016/j.
brainres.2009.09.083
40. Eot-Houllier, G., Fulcrand, G., Magnaghi-Jaulin, L., Jaulin, C.
Histone deacetylase inhibitors and genomic instability. Cancer Lett, 2009, 274:169-176, doi:10.1016/j.canlet.2008.06.005
41. Kim, I. A., Kim, J. H., Shin, J. H., Kim, I. H., Kim, J. S., Wu, H. G., et al. A histone deacetylase inhibitor, trichostatin A, enhances ra- diosensitivity by abrogating G2/M arrest in human carcinoma cells. Cancer Res Treat, 2005, 37:122-128, doi:10.4143/crt.2005.37.2.122
42. Miller, T. A., Witter, D. J., Belvedere, S. Histone deacetylase in- hibitors. J Med Chem, 2003, 46:5097-5116, doi:10.1021/jm0303094
43. Schemies, J., Sippl, W., Jung, M. Histone deacetylase inhibitors that target tubulin. Cancer Lett, 2009, 280:222-232, doi:10.1016/j.
canlet.2009.01.040
44. North, B. J., Verdin, E. Sirtuins: Sir2-related NAD-dependent protein deacetylases. Genome Biol, 2004, 5:224, doi:10.1186/gb- 2004-5-5-224
45. Westphal, C. H., Dipp, M. A., Guarente, L. A therapeutic role for sirtuins in diseases of aging? Trends Biochem Sci, 2007, 32:555- 560, doi:10.1016/j.tibs.2007.09.008
46. Fatkins, D. G., Zheng, W. Substituting N(epsilon)-thioacetyl- lysine for N(epsilon)-acetyl-lysine in peptide substrates as a general approach to inhibiting human NAD(+)-dependent protein deacety- lases. Int J Mol Sci, 2008, 9:1-11,
47. Biel, M., Kretsovali, A., Karatzali, E., Papamatheakis, J., Gi- annis, A. Design, synthesis, and biological evaluation of a small- mol- ecule inhibitor of the histone acetyltransferase Gcn5. Angew
Chem Int Ed Engl, 2004, 43:3974-3976,
doi:10.1002/anie.200453879
48. Solomon, J. M., Pasupuleti, R., Xu, L., McDonagh, T., Curtis, R., DiStefano, P. S., et al. Inhibition of SIRT1 catalytic activity increas- es p53 acetylation but does not alter cell survival following DNA damage. Mol Cell Biol, 2006, 26:28-38, doi:10.1128/MCB.26.1.28-
38.2006
49. Gui, C. Y., Ngo, L., Xu, W. S., Richon, V. M., Marks, P. A.
Histone deacetylase (HDAC) inhibitor activation of p21WAF1 in- volves changes in promoter-associated proteins, including HDAC1.
Proc Natl Acad Sci U S A, 2004, 101:1241-1246, doi:10.1073/
pnas.0307708100
50. Medda, F., Russell, R. J., Higgins, M., McCarthy, A. R., Camp- bell, J., Slawin, A. M., et al. Novel cambinol analogs as sirtuin inhib- itors: synthesis, biological evaluation, and rationalization of activity. J Med Chem, 2009, 52:2673-2682, doi:10.1021/jm8014298 51. Sandor, V., Senderowicz, A., Mertins, S., Sackett, D., Sausville, E., Blagosklonny, M. V., et al. P21-dependent g(1)arrest with down- regulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. Br J Cancer, 2000, 83:817-825, doi:10.1054/bjoc.2000.1327
52. Wharton, W., Savell, J., Cress, W. D., Seto, E., Pledger, W. J.
Inhibition of mitogenesis in Balb/c-3T3 cells by Trichostatin A.
Multiple alterations in the induction and activation of cyclin-cyclin- dependent kinase complexes. J Biol Chem, 2000, 275:33981-33987, doi:10.1074/jbc.M005600200
53. Bolden, J. E., Peart, M. J., Johnstone, R. W. Anticancer activi- ties of histone deacetylase inhibitors. Nat Rev Drug Discov, 2006, 5:769-784, doi:10.1038/nrd2133
54. Espino, P. S., Drobic, B., Dunn, K. L., Davie, J. R. Histone mod-
ifications as a platform for cancer therapy. J Cell Biochem, 2005, 94:1088-1102, doi:10.1002/jcb.20387
55. Tu, C. Y., Chen, C. H., Hsia, T. C., Hsu, M. H., Wei, Y. L., Yu, M. C., et al. Trichostatin A suppresses EGFR expression through induction of microRNA-7 in an HDAC-independent manner in lapatinib-treated cells. Biomed Res Int, 2014, 2014:168949, doi:10.1155/2014/168949
56. Kelly, W. K., Richon, V. M., O’Connor, O., Curley, T., Mac- Gregor-Curtelli, B., Tong, W., et al. Phase I clinical trial of histone deacetylase inhibitor: suberoylanilide hydroxamic acid administered intravenously. Clin Cancer Res, 2003, 9:3578-3588,
57. Bhalla, K. N. Epigenetic and chromatin modifiers as targeted therapy of hematologic malignancies. J Clin Oncol, 2005, 23:3971- 3993, doi:10.1200/JCO.2005.16.600
58. Dokmanovic, M., Marks, P. A. Prospects: histone deacetylase in- hibitors. J Cell Biochem, 2005, 96:293-304, doi:10.1002/jcb.20532
59. Marchion, D. C., Bicaku, E., Daud, A. I., Richon, V., Sullivan, D. M., Munster, P. N. Sequence-specific potentiation of topoisomer- ase II inhibitors by the histone deacetylase inhibitor suberoylanilide hydroxamic acid. J Cell Biochem, 2004, 92:223-237, doi:10.1002/
jcb.20045
60. Gore, S. D., Baylin, S., Sugar, E., Carraway, H., Miller, C. B., Carducci, M., et al. Combined DNA methyltransferase and histone deacetylase inhibition in the treatment of myeloid neoplasms. Can- cer Res, 2006, 66:6361-6369, doi:10.1158/0008-5472.CAN-06- 0080
61. Maslak, P., Chanel, S., Camacho, L. H., Soignet, S., Pandolfi, P. P., Guernah, I., et al. Pilot study of combination transcriptional modulation therapy with sodium phenylbutyrate and 5-azacytidine
in patients with acute myeloid leukemia or myelodysplastic syn- drome. Leukemia, 2006, 20:212-217, doi:10.1038/sj.leu.2404050 62. Garcia-Manero, G., Kantarjian, H. M., Sanchez-Gonzalez, B.,
Yang, H., Rosner, G., Verstovsek, S., et al. Phase 1/2 study of the combination of 5-aza-2’-deoxycytidine with valproic acid in pa- tients with leukemia. Blood, 2006, 108:3271-3279, doi:10.1182/
blood-2006-03-009142
63. Nemunaitis, J. J., Orr, D., Eager, R., Cunningham, C. C., Wil- liams, A., Mennel, R., et al. Phase I study of oral CI-994 in combina- tion with gemcitabine in treatment of patients with advanced cancer. Cancer J, 2003, 9:58-66,
64. Undevia, S. D., Kindler, H. L., Janisch, L., Olson, S. C., Schil- sky, R. L., Vogelzang, N. J., et al. A phase I study of the oral com- bination of CI-994, a putative histone deacetylase inhibitor, and capecitabine. Ann Oncol, 2004, 15:1705-1711, doi:10.1093/annonc/
mdh438
65. Yu, C., Rahmani, M., Conrad, D., Subler, M., Dent, P., Grant, S. The proteasome inhibitor bortezomib interacts synergistically with histone deacetylase inhibitors to induce apoptosis in Bcr/Abl+
cells sensitive and resistant to STI571. Blood, 2003, 102:3765-3774, doi:10.1182/blood-2003-03-0737
66. Fiskus, W., Pranpat, M., Bali, P., Balasis, M., Kumaraswamy, S., Boyapalle, S., et al. Combined effects of novel tyrosine kinase in- hibitor AMN107 and histone deacetylase inhibitor LBH589 against Bcr-Abl-expressing human leukemia cells. Blood, 2006, 108:645-
652, doi:10.1182/blood-2005-11-4639
67. Chen, Z. X., Mann, J. R., Hsieh, C. L., Riggs, A. D., Chedin, F.
Physical and functional interactions between the human DNMT3L protein and members of the de novo methyltransferase family. J Cell Biochem, 2005, 95:902-917, doi:10.1002/jcb.20447
68. Allfrey, V. G., Faulkner, R., Mirsky, A. E. Acetylation and Meth- ylation of Histones and Their Possible Role in the Regulation of Rna Synthesis. Proc Natl Acad Sci U S A, 1964, 51:786-794, 69. Kleff, S., Andrulis, E. D., Anderson, C. W., Sternglanz, R. Iden- tification of a gene encoding a yeast histone H4 acetyltransferase. J Biol Chem, 1995, 270:24674-24677,
70. Allis, C. D., Chicoine, L. G., Richman, R., Schulman, I. G.
Deposition-related histone acetylation in micronuclei of conjugating Tetrahymena. Proc Natl Acad Sci U S A, 1985, 82:8048-8052, 71. Ruiz-Carrillo, A., Wangh, L. J., Allfrey, V. G. Processing of new- ly synthesized histone molecules. Science, 1975, 190:117-128, 72. Parthun, M. R., Widom, J., Gottschling, D. E. The major cyto- plasmic histone acetyltransferase in yeast: links to chromatin repli- cation and histone metabolism. Cell, 1996, 87:85-94,
73. Yuan, H., Marmorstein, R. Histone acetyltransferases: Rising ancient counterparts to protein kinases. Biopolymers, 2013, 99:98- 111, doi:10.1002/bip.22128
74. Cullis, P. M., Wolfenden, R., Cousens, L. S., Alberts, B. M.
Inhi- bition of histone acetylation by N-[2-(S-coenzyme A)acetyl]
spermi- dine amide, a multisubstrate analog. J Biol Chem, 1982, 257:12165-
12169,
75. Balasubramanyam, K., Altaf, M., Varier, R. A., Swaminathan, V., Ravindran, A., Sadhale, P. P., et al. Polyisoprenylated benzophe- none, garcinol, a natural histone acetyltransferase inhibitor, repress- es chromatin transcription and alters global gene expression. J Biol Chem, 2004, 279:33716-33726, doi:10.1074/jbc.M402839200
76. Balasubramanyam, K., Varier, R. A., Altaf, M., Swaminathan, V., Siddappa, N. B., Ranga, U., et al. Curcumin, a novel p300/
CREB-binding protein-specific inhibitor of acetyltransferase, re- presses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. J Biol Chem, 2004, 279:51163-51171, doi:10.1074/jbc.M409024200
77. Stimson, L., Rowlands, M. G., Newbatt, Y. M., Smith, N. F., Raynaud, F. I., Rogers, P., et al. Isothiazolones as inhibitors of
PCAF and p300 histone acetyltransferase activity. Mol Cancer Ther, 2005,
4:1521-1532, doi:10.1158/1535-7163.MCT-05-0135
78. Nagy, Z., Tora, L. Distinct GCN5/PCAF-containing complex-