國 立 交 通 大 學
生物資訊及系統生物研究所
碩士論文
靈長類特有之微小核醣核酸叢集的比較基因體學分析
Comparative analysis of C19MC in primate genomes
研 究 生:潘承宗
指導教授:林勇欣
中華民國九十九年六月
靈長類特有之微小核醣核酸叢集的比較基因體學分析
Comparative analysis of C19MC in primate genomes
研 究 生:潘承宗
Student: Cheng‐Tsung Pan
指導教授:林勇欣
Advisor: Yeong‐Shin Lin
國
立
交
通
大
學
生物資訊及系統生物研究所
碩 士 論 文
A Thesis
Submitted to Institute of Bioinformatics and Systems Biology
College of Biological Science and Technology
National Chiao Tung University
in Fulfillment of the Requirements
for the Degree of Master
in
Bioinformatics and Systems Biology
June 2010 Hisinchu, Taiwan, Republic of China 中華民國九十九年六月
Chinese Abstract
Alu 單元和微小核醣核酸﹝microRNA﹞是兩個截然不同的基因體序列。在近幾年, 兩者皆被廣泛研究。Alu 單元是專一出現在靈長類動物的短散布重複因子﹝SINE﹞中佔 多數的因子之一,且它們不轉譯出有功能的蛋白質。在最近的研究中指出Alu 單元可能 和癌症的產生有所關聯。其他的研究也認為Alu 單元可能影響基因的功能。基於這些發 現,可以合理的認為,Alu 單元在靈長類的演化中可能扮演某種很重要的角色。在人類 第19 號染色體上的微小核醣核酸叢集﹝C19MC﹞中的 Alu 單元和微小核醣核酸,被發 現有一種很特殊的位置上的靠近。這個不尋常的片段可能是起因於一連串的複製 ﹝duplication﹞事件。在這篇研究中,我們分析了這些可能是複製單元的序列,無論是 在同物種的同源基因﹝paralogs﹞之間,或是異物種同源基因對﹝ortholog pairs﹞之間, 希望能辨識出序列中每個位置所遭受的選擇壓力﹝selection pressure﹞。我們發現在恆 河猴 C19MC 中的同物種同源基因序列,相對於人類有較高的變異度。同時我們的全面 性和區域性的分析也指出,無論在人類或恆河猴的 C19MC 中,不同的序列區域遭受到 不同的選擇力量。在表現子﹝exon﹞區域有最高的變異性,然而在中間區域靠近微小核 醣核酸的週邊,則有較高的保留度。此外,先前的研究指出Alu 可能會被微小核醣核酸 所抑制,因為Alu 單元的擴張可能會使基因體受損。在此篇研究的第二部份,我們假設 降低Alu 單元的表現程度,可能會使人類基因體有選擇優勢。因此我們認為在靈長類的 演化中,C19MC 可能是主要的防禦機制,用來對抗 Alu 單元的擴張。我們初步的結果指 出,無論在人類或恆河猴的基因體中,C19MC 中的微小核醣核酸傾向針對 AluS 而非 AluJ 和AluY。這個發現支持了我們的假說,C19MC 的出現可能是在 Alu 擴張之後的一種防 禦反應。English Abstract
Alu elements and microRNAs are two different types of genomic sequences. Each of them has been extensively studied in recent years. Alu is one of the most abundant SINEs (short interspersed nuclear element) discovered specifically in primates and it does not encode a functional protein. In recent studies, Alu was reported to be related to cancer. Some other studies further suggested that Alu may influence gene functions. Based on these findings, it is reasonable to speculate that Alu might play some important roles during primate evolution. An unusual cluster of positional proximity of Alu and microRNA was found in human chromosome 19 microRNA cluster (C19MC). This unusual fragment seems to be derived from a serious of duplication events. In this study, we analyzed the sequences of the possible duplication unit either in paralogs within a species or ortholog pairs between species to identify the selection pressures on each nucleotide site. We found that the sequences of rhesus C19MC are more diverged than their paralogs in human. Meanwhile, our global and local analyses revealed that in both human and rhesus C19MC, different regions are under different selection forces. The exons are more diverged, while the internal flanking regions, which are adjacent to microRNAs, are more conserved. In addition, previous studies suggested that Alu elements in human might be repressed by microRNAs, because Alu expansion might damage human genome. In the second part of our study, we hypothesized that reducing the expression level of Alu might have selectively advantage for human genome. We suggested that this C19MC cluster might be a major defender against Alu expansion during primate evolution. Our preliminary results indicate that microRNAs in C19MC tend to target AluS rather than AluJ and AluY in human and macaque genomes. This finding supports the hypothesis that the appearance of C19MC might follow Alu expansion as a response of defense.Acknowledgement
經過兩年非常愉快的碩士班,終於要畢業邁入下一個階段了,在新竹的兩年過得非 常的愉快,除了有很棒的實驗室夥伴和老師之外,還有在新竹認識很多親切的朋友們, 都讓我感覺研究生的生活甚至比大學更快樂。還沒有玩遍新竹的各地名勝,還沒有吃遍 新竹的各色餐廳,不知不覺地就要離開新竹了,除了畢業的喜悅外,更多的是依依不捨, 我想未來有機會我應該會經常回來交大這個令人懷念的地方。 首先當然要先感謝我的父母,這兩年來讓我全心全意在新竹生活,雖然比較少回家, 但還是能體諒我,無論經濟上或生活上,都讓我沒有後顧之憂。 也要感謝應逵,你是個很好的夥伴,兩年前暑假我們剛到新竹,還沒找到房子、實 驗室裡什麼設備什麼生活用品都沒有,兩個人一起睡在實驗室,一起動手施工一起討論 要怎麼建立起這個實驗室。整個過程中你都一直很努力,也不斷忍受我的嘮叨,為了讓 實驗室運作得更好,從買耗材買儀器,到自己做燈箱自己釘櫃子,雖然消耗了一年多, 但卻很開心也學到很多,這些成就感不是言語可以形容的。每天一起吃飯一起生活,也 讓我們有很多時間討論程式的寫法,學術的問題,甚至生活中的大小事,讓我學習到很 多不同的觀點和想法。還要感謝我們的另一位室友重延,因為我們常不在家,家裡的繳 費打掃等幾乎都由重延一手包辦,感謝他讓我們的家一直維持恰到好處的乾淨。 當然,能夠大膽嘗試這一切,無憂無慮的建立起實驗室,也要感謝林勇欣老師的大 力支持,無論是經費上或是精神上,有他對我們的信任和支援,我們才能在短短一年內 做這麼多事。雖然現在林先生有女朋友比較忙碌,不過這兩年還是跟我們到處玩,去香 山看夕陽,去中南部玩,還有一直想拍沒拍到的太空站,還有很多想吃的餐廳還沒吃, 兩年實在過得很快,每天在實驗室待到清晨的日子一下就過去了。除了學術上的指導之 外,更難得的是跟林老師的友誼,除了傳統的師生關係,更多的是像朋友間的幫忙和鼓 勵。 感謝志欽,雖然你常常被使喚,但為了實驗室的傳承,也只好繼續使喚你了。實驗 室的建立並不容易,希望你能從中學到很多,並把實驗室繼續的維護下去。也謝謝你在口試當天的幫忙跑腿,讓我可以專心在現場做準備。 感謝勤政,沒有你實驗室就沒有了娛樂,除了你本身就是個笑點之外,也要謝謝你 推薦電影,推薦新產品,還有在實驗室的維護上分憂解勞,協助我很多。感謝羿喬學長, 在人生規畫上和研究上的建議,還有感謝你的座位在我大混亂時暫時堆放東西。感謝家 豪學長在研發替代役申請時的協助,雖然你在實驗室待得時間太短暫,想學釣魚也還沒 學到,但我想之後還有很多機會找你教我釣魚。 感謝黃兆祺老師以及黃老師實驗室的成員們,你們是我兩年來最熟的一間實驗室, 也是在二館以外可以去泡茶聊天的唯一去處。黃老師給了我很多鼓勵和幫忙,和林勇欣 老師的嘴砲式鼓勵不同,您給我了滿多實質上的建議。 感謝熊昭老師,雖然只上過您兩門課,但還是熱心的來幫我口試,也讓我第一次有 機會去參觀一下國衛院,口試上的許多建議和叮嚀對我未來在學術上的思維很有幫助。 還要感謝交大攝影社的朋友們,雖然我只來了兩年,只參加了幾次出遊,但還是讓 我一起跟團拍照,讓我有機會辦成果展;感謝國倫一起討論和場佈,完成巨大的光牆作 品。還要感謝德芬當我的外拍模特兒,在兩年新竹生活中第一次拍出滿意的作品,雖然 妳的新竹生活不太順遂,但還是要加油,一定可以撐過去。 要特別感謝介伶,兩年來一有時間就到新竹陪我,也在不知不覺中似乎要變成實驗 室的成員之一,雖然實質幫助並不多,當作精神食糧也沒有很好吃,但還是盡責地做為 頂級食材,補充海怪能量。兩年來你也過得很辛苦,第一年要重考,第二年的研究生活 不愉快,但終於妳順利地考上新學校,也要展開新生活,希望妳之後兩年的研究生活也 能過得很平安如意。 最後謝謝交大土地公,雖然每次去拜拜我都忘記帶夠零錢,也都只請祢一罐仙草蜜, 但我相信祢不會太介意,除了謝謝祢保佑我兩年平安之外,也謝謝祢保佑全交大師生。 承宗 2010/06/24 於實驗室
Table of Contents
Chinese Abstract ……… i
English Abstract ……… ii
Acknowledgment ……… iii
Table of Contents ……… v
List of Figures ……… vii
List of Tables ……… viii
List of Appendix ……… ix
Abbreviations ……… x Chapter 1 General Introduction 1.1 Transposable Elements is not Just Junks Transposable Elements is a Kind of Genome Parasites ……… 1 Evolution of Alu Subfamilies ……… 3 Impact of Alu on Gene Regulation ……… 3 Alu Elements is a Source of Microsatellites ……… 4 1.2 Primate Specific microRNA Clusters Biogenesis and Function of microRNAs ……… 4 Primate Specific microRNA Cluster ……… 5 Consideration of Transposable Elements in Epigenetics ……… 6 Chapter 2 Substitution Analysis of C19MC 2.1 Backgrounds and Hypothesis ……… 7 2.2 Materials and Methods
2.2.1 Sequence Collection and Annotations ……… 8 2.2.2 Define C19MC Homologs between Human and Rhesus ……… 10 2.2.3 Nucleotides Substitution Analysis of C19MC ……… 11 2.3 Results and Discussion 2.3.1 MicroRNAs within C19MC Apeear a Characteristic Duplication Unit …… 13 2.3.2 Homology Definition and Genetic Composition of C19MC ……… 13 2.3.3 Substitution Analysis ……… 15 Chapter 3 C19MC probably as a defender against Alu Elements 3.1 Backgrounds and Hypothesis ……… 18 3.2 Materials and Methods 3.2.1 Determine Mature microRNA Expression Profile from Next‐Generation Sequencing Data ……… 19 3.2.2 Whole Genome Scanning (From Seed Candidates’ Point of View) ……… 21 3.2.3 Whole Genome Scanning (From Alu elements’ Point of View) ……… 22 3.3 Results and Discussion 3.3.1 C19MC has Extremely Different Expression Levels ……… 23 3.3.2 The Biased Distribution of C19MC Target Ability against Alu Elements ……… 23
3.3.3 Analysis of Alu Elements Targeted by C19MC ……… 25
Figures ……… 27
Tables ……… 55
References ……… 58
List of Figures
Figure 1 The Distribution and Structure of Transposons ……… 27
Figure 2 Distribution of Alu Subfamilies in Primate Genomes ……… 28
Figure 3 Evolution of Alu Subfamilies ……… 29
Figure 4 Biogenesis and Structure of microRNA ……… 30
Figure 5 The microRNA Clusters of Human ……… 31
Figure 6 Remarkable Duplication Unit of C19MC ……… 32
Figure 7 Two Different Models of C19MC Transcription ……… 33
Figure 8 Procedure of Rainbow Plot ……… 34
Figure 9 Determine C19MC Homologs in Rhesus ……… 35
Figure 10 Process of Substitution Calculation ……… 36
Figure 11 Pairwise Similarity Distances of C19MC ……… 37
Figure 12 Duplication Hotspots ……… 38
Figure 13 The Special L1MB7 Sequences on C19MC ……… 39
Figure 14 Overall Scatter Plot of PSubstitution in Two Species ……… 40
Figure 15 Proportion of Substitution in Macroscopic (Global) View ……… 41
Figure 16 Substitution Analysis and Multiple Sequence Alignment ……… 42
Figure 17 Hypothetical Model of Co‐Evolution ……… 47
Figure 18 Alignment of Sequencing Reads with C19MC Annotated Mature Region 48 Figure 19 C19MC Seed Selection from Human Sequencing Data ……… 49 Figure 20 Rhesus Seed Candidates Selection ……… 50 Figure 21 Distribution of Alu Target Ability of each Seed ……… 51 Figure 22 Targeting Preference of Seed Candidates: Different Alu Subfamilies …… 52 Figure 23 Interchange Seeds to Target Genomes of Each Other ……… 53 Figure 24 Percentages of Alu Subfamilies Not Targeted by C19MC ……… 54
List of Tables
Table 1 Othrlog Pairs Defined by Rainbow Dotplot in This Study ……… 55 Table 2 Summary of Human C19MC Features ……… 57
List of Appendix
Appendix 1 Annotation of C19MC ……… 63
Abbreviation
C19MC Chromosome 19 microRNA cluster C14MC Chromosome 14 microRNA cluster TE(s) Transposable element(s) LTR Long terminal repeat LINE Long interspersed element SINE Short interspersed element SVA An element made by SINE, variable number of tandem repeats or Alu‐like region L1 LINE1, Long interspersed element 1 RNA Pol II RNA polymerase II RNA Pol III RNA polymerase III ORF Open reading frame FAM Free Alu monomer FRAM Free Alu right monomer FLAM Free Alu left monomer Myr Million years RISC RNA‐induced silencing complex hsa‐ prefix which means human mml‐ prefix which means rhesus pd‐ prefix which means prediction mir‐ prefix which means microRNAChapter 1
General Introduction
1.1
Transposable Elements is not Just Junks
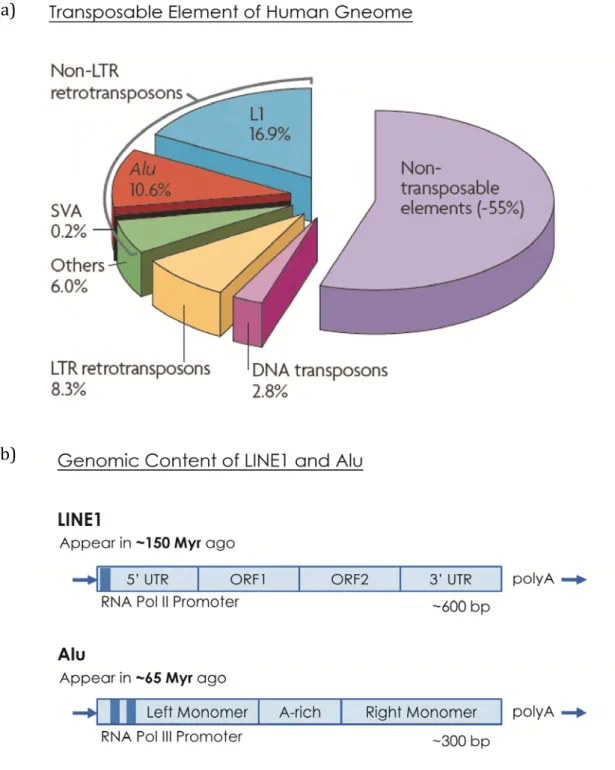
Transposable Elements is a Kind of Genome Parasites The genome composition was well understood after the genome project had completed. Our genome seems be occupied not only by the protein coding genes but also by other genomic elements. One element which is found ubiquitously and cosmically in most eukaryotic genomes is the transposable elements (TEs) due to their extent of genome contribution approximately to 45% or even more. (Lander, Linton et al. 2001) These TEs are often separated into two main categories, the DNA transposons and the retrotransposons, by their different biogenesis mechanisms. (Figure 1a) TEs from both categories could duplicate and move, so called “jumping”, within the genome by either genomic recombination or insertion of new copies into new positions. This special event could cause either positive results via triggering useful gene expansion or negative results via disrupting normal gene function and inducing diseases. Besides the DNA transposons occupied 3% of human genome but stopped activating currently, the retrotransposons function as well. One type of retrotransposons, the LTR retrotransposon, is slightly inaction within human genomes. By contrast, another type of retrotransposons, the non‐LTR retrotransposon, including LINE (long interspersed element), SINE (short interspersed element) and SVA (An element made by SINE, variable number of tandem repeats or Alu‐like region) are expressed detectably and found uniformly among primates so that they might be considered as “alive” along with primate evolution. That might be a reason that non‐LTR retrotransposons are found approximately one‐third of human genome and responsible for many genetic disorders. (Mills, Bennett et al. 2007; Cordaux and Batzer 2009)
LINE1 (usually abbreviated to L1) is one of the abundant non‐LTR retrotransposons found in primateswhich contains a RNA polymerase II (RNA Pol II) promoter at its 5’ UTR to drive transcription and encodes RNA‐binding protein from its ORF1 (open reading frame 1) and other two proteins, the endonuclease and reverse‐transcriptase from ORF2.. (Swergold 1990) (Figure
1b) Although many L1 in human genome are truncated (Ostertag and Kazazian
2001), the proteins encoded by other active L1 elements continuously help the primary transposition and duplication mechanism for both LINEs themselves and even Alu elements. (Dewannieux, Esnault et al. 2003)
Alu element is another well‐investigated retrotransposon in addition to L1 elements. In contrast to L1 elements which are transcribed by RNA Pol II and encode proteins, Alu elements are transcribed by RNA polymerase III (RNA Pol III) and do not encod proteins. They compose by two similar but not identical monomers with an internal A‐rich linker and rely on L1 mechanism for transposition. Interestingly, Alu elements do not follow the RNA Pol III terminator for stopping transcription but extend their transcript into the downstream flanking sequence until a terminator is found or the activity of RNA Pol III is reduced. (Figure 1b) (Batzer and Deininger 2002) In the opinion of selfish theory which assumes genetic components or genes devoted to keep themselves alive without nature selection. According to this theory, Alu elements are probably the most successful TEs not only because of their large territory in primate genomes but also their economical transposition mechanism by hijack the L1 machinery. (Weiner 2002; Mills, Bennett et al. 2007; Comeaux, Roy‐Engel et al. 2009)
Evolution of Alu Subfamilies
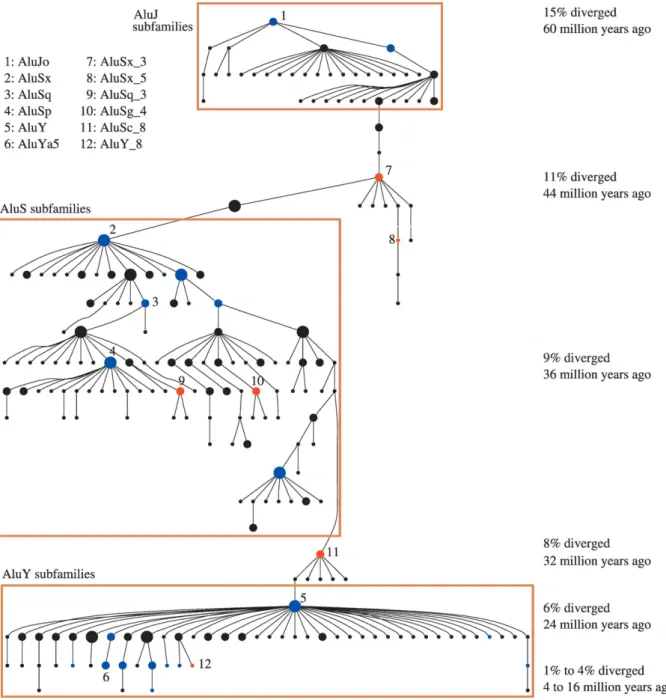
The oldest Alu‐like elements might be the 7SL RNA genederived monomer such as FAM, FRAM and FLAM, and then evolved into recent Alu elements which is a dimeric element made by two very similar but not identical monomers. The evolution of Alu elements expansion can be separated to 3 stages by evolutionary history. The first expansion of Alu elements started from AluJ which is the ancient Alu subfamily appeared about ~60 to 65 million years (Myrs) ago. They might accumulate many mutations and become truncation through the evolutionary time. After AluJ appearance, the AluS burst out its expansion after 15 Myrs later that caused itself rise to the major amount of Alu subfamily recently. (Figure 2) In our study, we assumed the AluS burst is the primary crisis of genome damage due to its huge amount of expansion and must be repressed by microRNAs. Finally, the youngest subfamily so far is AluY which continues in transposition and be polymorphic in population but inferior in amount. (Price, Eskin et al. 2004) (Figure 3)
Impact of Alu Elements on Gene Regulation
More and more studies found evidences that transposable elements have an important role and impact on gene regulatory networks. For example, L1 could facilitate the emersion of new regulatory proteins and transcription factors as well as move the regulated sequences (Belancio, Roy‐Engel et al. 2008); Alu elements might donate itself as the target sites of small RNA regulation (microRNAs, piwiRNAs or other kinds of RNAi). (Shankar, Grover et al. 2004) Those investigations suggest extensive functions of transposable elements which involved in gene regulatory network, epigenetics and even the popular topics, the small RNA regulation. (Polak and Domany 2006; Faulkner, Kimura et al. 2009) Most of the non‐coding RNAs, such as piwiRNA, are not well described, not to mention their interact function with transposable elements.
Studies indicated that not only well‐known microRNAs but also the novel piwiRNAs are probably as the defender against selfish transposons. (Brennecke, Malone et al. 2008; Halic and Moazed 2009) Here, we hypothesized that the phenomenon of Alu inserting into genes and changing the original regulatory mechanism might be dangerous without control. A defense mechanism of preventing Alu from over‐expansion and a monitoring mechanism must exist, and probably guarded by microRNAs inhibition. (Detail in chapter 3)
Alu Elements are Probably a Source of Microsatellites
Non‐LTR retrotransposons, especially Alu elements could generate microsatellites by their capacity of homopolymeric tract, which means a DNA sequence made of tandem repeats with same nucleotides. Each new copy of Alu could offer microsatellite source from its middle A‐rich linker region or 3’ A‐rich tail. (Arcot, Wang et al. 1995; Jurka and Pethiyagoda 1995) This concept was stronger while ~20% of all microsatellites shared by human and chimpanzee lie within Alu elements were found. (Kelkar, Tyekucheva et al. 2008) We expected to demonstrate an analysis of microsatellite generated by Alu elements within C19MC duplication units to be an approach to reconstruct C19MC microRNAs duplication in the future. (Not in this study)
1.2
Primate Specific microRNA Clusters
Biogenesis and Function of microRNAs
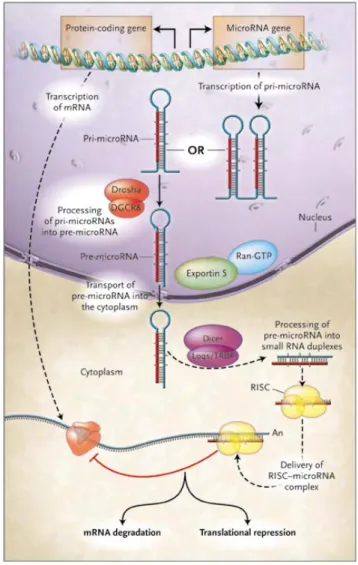
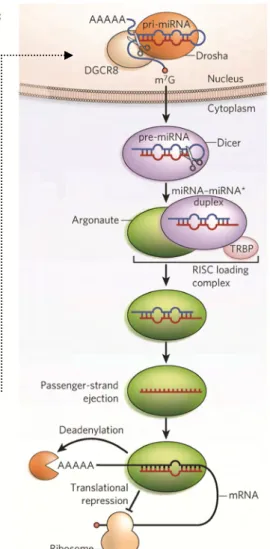
Typically, microRNA genes are derived from a precursor microRNAs transcribed by RNA polymerase II as same as normal protein coding genes. These precursors then are processed into 60‐70 nt long pre‐microRNA by an enzyme called Drosha. After the pre‐microRNA had transported into the cytoplasm, another enzyme called Dicer cleaves the pre‐microRNAs by cutting
its stem loop structure and generates a hybridized RNAs with two strands. These trimmed hybridized RNAs without stem loop are mature form of microRNAs. Either one strand or both strands could function as mature microRNAs to complementary hybridize with their mRNA target sites. This hybridization (perfect match of full‐length mature sequence is not necessary) recruits an enzyme complex called RISC (RNA‐induced silencing complex) to bind and induce the degradation of mRNA or inhibition of translation. (Neilson and Sharp 2008; Ghildiyal and Zamore 2009) (Figure 4) The mRNA degradation and translation inhibition process typically depend on the match of seed region (The nucleotides from second to eighth sites at 5’ end of mature microRNAs. (Kertesz, Iovino et al. 2007; Bartel 2009) This characteristic of microRNA is widely used in target site predication by bioinformatics approch. We also used this fundamental principle in our study to determine whether an Alu element is targeted by C19MC in chapter 3. C19MC is a Primate Specific microRNA Cluster Vast surveys of novel microRNA discovery indicated that many microRNA genes prefer to form clusters in a related shorter genomic distance to the normal genes rather than being random distributed throughout the genomes. (Lagos‐Quintana, Rauhut et al. 2003) A famous one microRNA cluster located in chromosome 19 is called C19MC which means chromosome 19 microRNAs cluster. This concentration of microRNA genes as a cluster was especially found in placental mammals (Glazov, McWilliam et al. 2008) that implies the importance and uniqueness of C19MC (the main research object in our study) in primates. Furthermore, the rapid evolution and functional diversification of two primate clusters in chromosome 19 and chromosome X were studied as a view of primate evolution. (Zhang, Peng et al. 2007; Zhang, Wang et al. 2008; Li, Liu et al. 2010) Another major character of the C19MC is that a huge amount of Alu
elements insertion into the flanking region of microRNAs. It’s totally different while compared with C14MC (Another rare investigated cluster in somatic chromosome) which has plentiful microRNAs but rare transposon insertions.
(Figure 5) Here, we focused on the C19MC and studied in‐depth by more local
concepts with comparative sequences analysis in nucleotides level.
Consideration of Transposable Elements in Epigenetics
The transposons activity and small RNA expression were studied and believed to be connected with epigenetics. (Costa 2008) As we mentioned that human C19MC are primary expressed in the placenta and embryonic stem cell and switched off in somatic cells, thus it makes sense to connect the C19MC into regulatory network in development. We believed that C19MC must play an importance role in primates’ development. A further study provided more evidence and confidence since they found C19MC are expressed in human cancer cell by an unusual control via epigenetic mechanism. (Tsai, Kao et al. 2009) According to our speculation, some important nucleotide sites with highly substitution we identified in this study could be the dominant regulatory sites. Although these sites were not reported in transcriptional level in previous C19MC studies, they might have some role in epigenetic level so that they encounter the selection force. In short, we believed these highly substituted sites may be involved in the epigenetic control rather than the mRNA level (such as RNA structure or transcription factor binding sites), so that the selection force on these sites was preserved.
Chapter 2
Substitution Analysis of C19MC
2.1
Background Introduction and Hypothesis
Controversial Transcription Mechanism of C19MC
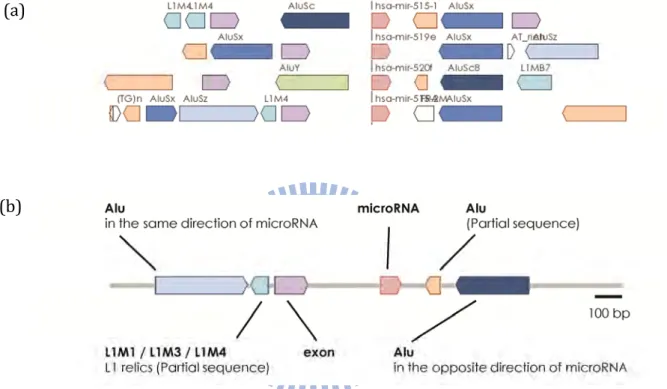
In recent years, more and more studies discovered the remarkable microRNA clusters arrangement in genome from bacteria to primates. (Lagos‐Quintana, Rauhut et al. 2003) Within the primate‐specific microRNA clusters called C19MC, microRNA genes are averagely located within the cluster and separated by the proximal Alu elements by approximately 100 bp spacing. (Bentwich, Avniel et al. 2005) This special rearrangement of microRNAs and Alu elements forms a distinctive duplication unit or a kind of tandem repeat.
(Figure 6) We thought they are probably a duplication unit, but the mechanism
of this unit both explained by molecular evolution or by cellular biology were still unknown even microRNA is a popular research topic. The transcriptional mechanism of mammalian microRNAs by RNA polymerase II is well known. (Cai, Hagedorn et al. 2004; Lee, Kim et al. 2004) However, Borchert reported these proximal Alu inside the duplication unit in C19MC could donate its own promoter for its neighbor microRNA for RNA polymerase III transcription. (Borchert, Lanier et al. 2006) (Figure 7B) This amazing finding not only suggests the new transcription model of microRNAs but also indicates the novel contribution of Alu elements: the upstream Alu elements could express downstream microRNAs. They finally showed and proved the miR‐515‐1, miR‐517a, miR‐517c and miR‐519a‐1 in C19MC are truly expressed using Pol III via Alu promoter in vitro.
Interestingly, an extremely opposite finding of C19MC transcription was reported after three years of Borchert’s discovery. (Bortolin‐Cavaille, Dance et al. 2009) They found a unique exon sequence which is located in the flanking of most C19MC microRNAs and is processed by RNA polymerase II via alternative splicing mechanism in vivo. They proved that C19MC microRNAs are the intron‐encoded genes of a novel RNA Pol II transcript and their expressions depend on the alternative splicing of the novel RNA Pol II transcript. (Figure
7A)
2.2
Materials and Methods
2.2.1 Sequence Collection and Annotations
C19MC
C19MC sequences (or so called genomic context) including microRNA genes and flanking regions were fetched from UCSC with version hg19 of human genome and version rheMac2 of rhesus (monkey) genome. (Rhead, Karolchik et al. 2010) Sequence lengths of human and rhesus C19MCs were 102301‐nt between position 54168188 and 54270488 in human chromosome 19, 105001‐nt between position 59771000 and 59876000 in rhesus chromosome 19 respectively. All selected range of C19MC sequences were confirmed by our Rainbow Dotplot program roughly to make sure the full‐length of C19MC had been selected.
Alu Elements
The annotation of Alu elements within C19MC were followed by the Repeat Masker. (Smit 1996‐2010) Only the SINE/Alu element category of records was kept in our study and the other categories such as simple repeat, FAM and etc. were removed. The possible ancestor of Alu elements, the FAM
families, was eliminated because they are too distant to affirm what their function and activation is in primate lineage. In the section of determining the ability of seed candidates targeting Alu elements, we included all Alu elements scanned and recorded by Repeat Masker. The primary role we analyzed in this section is the seed candidates so that we did not filter Alu element, since all of them could have possibility of targeted by C19MC either currently or in the evolutionary history. Although some of them are partial fragments as relics, they still might be targeted in long time ago.
However, in the section involving the Alu elements insertion within genes, we filtered those Alu elements with length shorter than 200‐nt. This criterion implies that only the Alu elements with at least one monomer were kept, because the normal full length of Alu elements is about 300‐nt. Although those shorter Alu elements were under the selection force as same as the others, they are incomplete and probably inactive currently. This incompleteness implies that they might be not involved in the gene regulation and not reserved by natural selection. Annotated standard sequences of Alu subfamilies and the standard consensus ancient sequences of Alu and other TEs were fetched from Repbase, GIRI. (Jurka, Kapitonov et al. 2005)
Exons
The consensus exon sequence of C19MC in primates is defined by previous study (Bortolin‐Cavaille, Dance et al. 2009), which indicated a ~123‐nt long, spliced exon with strong sequence similarity among primates. All exons defined in our study were searched and found using a local alignment against the consensus exon sequence by our own program and set similarity larger than 80%, i.e. a fragment with more than 99‐nt are identical to the 123‐nt consensus exon was defined as another exon.
2.2.2 Define C19MC Homologs between Human and Rhesus Rainbow Dotplot
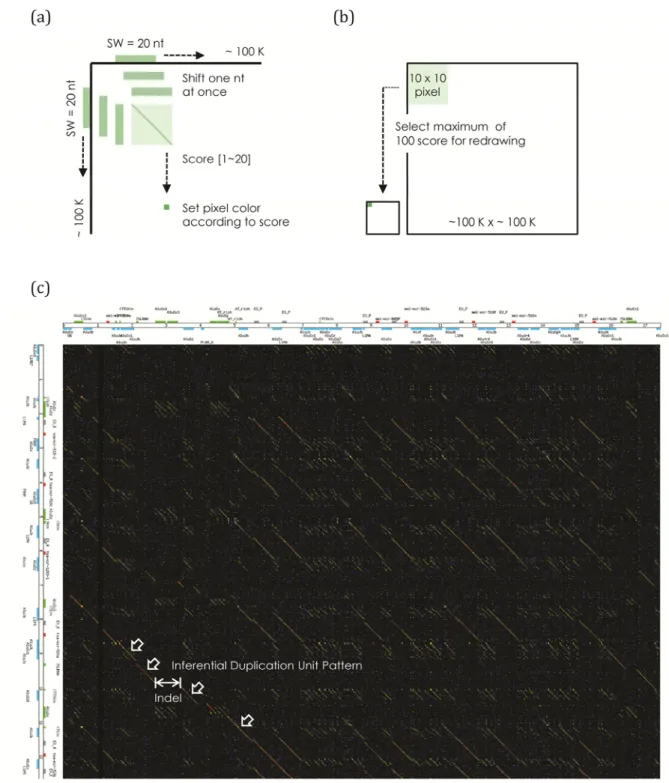
For determining the homology of C19MC among primates, we performed an analysis with our homemade visualization program called “Rainbow Plot”. The new idea of multiple‐color indicators was integrated to this program for intuitively observing the sequence similarity. This dotplot not only represents the relationship among paralogs or orthologs on a diagonal straight line but also the similarity on different color of each line.
As the normal dotplot program, sequences of C19MC from human and rhesus were put on two axes and scanned by 20‐nt sliding window. We also tested different sliding window sizes to find the optimal criterion. The resolution of dotplot will decrease if size is larger than 20‐nt. Meanwhile, because the multi‐color gradient depends on similarity scores, the number of colors will too few to present if sliding window size is smaller than 20‐nt. The sliding windows moved along with two axes by one nucleotide shift at each time, the score was counted as one while match pair and zero as mismatch pair. Thus, the possible scores were calculated from zero to twenty in each movement. After counting of full length square (~100K x ~100K), a reduction scoring process was done for reduce picture size. The maximum score of each 10 x 10 square area was selected as the final drawing score for representing the similarity of this square area. (Figure 8)
Homologs Definition
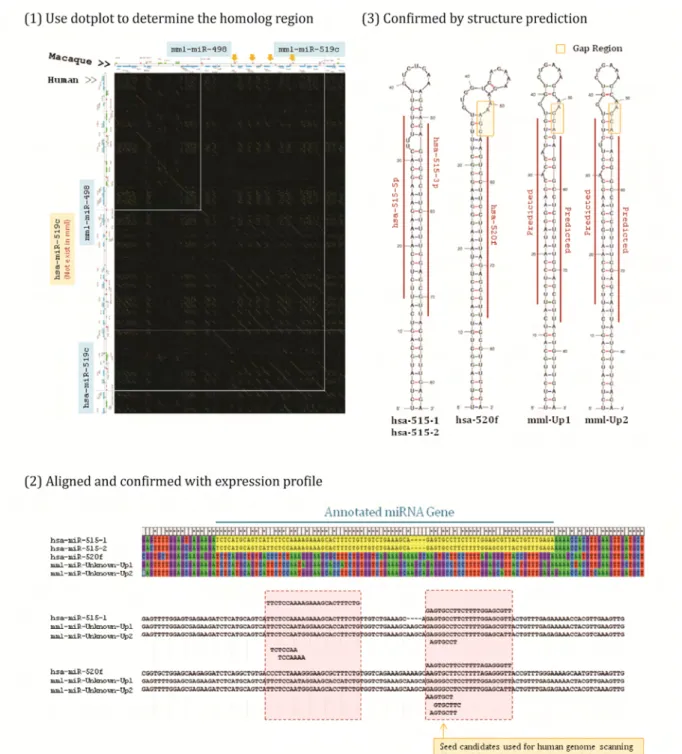
We used Rainbow Dotplot to rebuild the C19MC annotation in rhesus genome. The fragments of rhesus microRNA homolog genes were selected from dotplot by visualization and then processed pairwise sequence alignment with corresponding human microRNA for confirmation. We also included mature
microRNA sustained by Deep Sequencing data as a reference alignment sequence (see section 3.2.1 for detail) and combined MFold program. The MFold could predict RNA structure to double check the possible mature region and to confirm the nucleotides where the rhesus microRNA gene start and end. (Figure 9) 2.2.3 Nucleotides Substitution Analysis of C19MC Multiple Sequence Alignment and Aligned Sequences Trimming
Interested sequences region between each exon and microRNA were fetched from C19MC genomic context of human and rhesus. Each selected sequences were started from the first nucleotide of our defined exon and ended at the last nucleotide of microRNA gene annotated by miRBase in human and defined by our homologs definition process in rhesus (see section 2.2.2). Due to some sequences are lacks of internal transposable elements, all internal TEs were completely excised from the selected sequences before alignment. However, the internal flanking sequences except transposons were kept to gain information and improve analysis. Multiple sequence alignment was done by MEGA4 (Tamura, Dudley et al. 2007) and followed by manual check to improve gap reliability. One human sequence with downstream LTR13 flanking the exon rather than microRNA was excised, 4 rhesus sequences without downstream microRNA were also removed. Unreliable aligned regions of each sequence were also eliminated. Finally, a 456‐nt length (with gap) alignment of 49 interested paired ortholog sequences were chosen for further analysis. We kept sequences appear only in one species and set a blank sequence to its pair because these sequences still contain information within paralogs even though they do not have paired one.
Site Substitution Calculation
After multiple sequence alignment and trimming, each nucleotide site of ortholog pairs between two species was calculated by a proportion. The proportion of substitution was formulated by the number of pairs with substitution divided by the total number of pairs. If one sequence of a pair contains a gap in the site, it was deducted from the total number of pairs. For example, we have 49 ortholog pairs, synonymous with 98 sequences, for calculating. A site was found where are 30 pairs be identical, 7 pairs with substitution and 12 pairs have a gap or a blank sequence on one sequence, it was scored as 7 substitution divided by 37. Every sequence within a species, i.e. paralog, was calculated by determining whether the site differ to the nucleotide of majority between species. (Figure 10)
MicroRNA Similarity Distance within C19MC
In order to observe the similarity among microRNAs, we calculated the similarity distance of microRNA with its 6 neighbor micrRNAs, three from upstream and three from downstream. Used fragments of each microRNA were those described above in this section, exon and internal flanking region were included for distance calculation to improve reliability. Pairwise distance calculation was done by MEGA4 with Kimura 2‐parameter model and complete deletion, and then plotted the scores along with the length of C19MC. Six dots on a certain x‐axis position represent the distance between the local microRNAs and its six adjacent microRNAs; smaller the radius, more distant the neighbor. The least three similar microRNAs, mir‐512‐1, mir‐512‐2 and mir‐498 of two species were not included in this analysis, because they are too different to compare with the others. (Figure 11)
2.3
Results and Discussion
2.3.1 MicroRNAs within C19MC Appear a Characteristic Duplication Unit
The annotation of rhesus C19MC in miRBase is insufficient, so that we re‐annotated this region in our study. Only the transposon annotations were followed by Repeat Masker without any change. Both microRNA genes and exons in either human or rhesus genomes were re‐defined and manual confirmed in this study. By our visualized annotation (Appendix 1), we found a special unit with a regular patter including exon, Alu elements and microRNAs. Although we did not find evidence and mechanism of gene duplication, we believe this unusual pattern is must as a duplication unit which relates to the expansion of microRNA within C19MC. Moreover, because the transposition of Alu needs proteins offered by L1 element, we speculated the L1MB7 within C19MC also might have a key role in the start of duplication.
2.3.2 Homology Definition and Genetic Composition of C19MC
Dotplot is used for determining homologs extensively; we also performed this approach to find the homologs between human and rhesus and further to detect the evolutionary events such as insertion, deletion or recombination. Unlike normal dotplot program defined only one color for presentation, our new program shows straightforward information intuitively, friendly and visually by adding rainbow colors. Each color represents the degree of conservation within a sliding window size: red is the most conserved and followed by orange, yellow, green, and dark blue or purple is the least conserved, black represents no similarity found. (Figure 8c)
L1MB7 are Located nearby Two Rearrangement Hot Spots
We found two regions with multiple duplication events, usually called a hotspot, where display as many similar symmetrical diagonal lines within a narrow square region on the dotplot picture. (Figure 12) It is noteworthy that L1MB7, a kind of LINE 1 truncated form, are located at the proximal region of the hotspot. We speculated that the locations of L1MB7 correspond to the facilitation of C19MC duplication since L1 offers the primary mechanism of gene duplication, of cause including Alu elements, in most mammalian genomes. (Esnault, Maestre et al. 2000; Dewannieux, Esnault et al. 2003; Han and Boeke 2005) The special L1MB7 pattern was found at 3 regions on rhesus C19MC at 7K, 75K and 103K (related positions start within fetched C19MC sequence); meanwhile, we identified their 4 homologs in human C19MC. Two of them (at 12K and 17.5K) are probably caused by duplication because the Rainbow Dotplot shows a beautiful red line as high similarity. (Figure 13)
MicroRNA Homologs Definition and Annotation
Because the annotation of C19MC on rhesus genome is insufficient, we need to reconstruct the annotation of rhesus C19MC for further analysis. Forty‐seven human C19MC microRNAs were confirmed including one which is not annotated by miRBase (we denoted it by a prefix “hsa‐pd‐“). On the rhesus genome, 46 orgholog pairs were defined in our study, most of which are not annotated by miRBase, even though they did, many of their annotations are probably wrong. (Table 1) For example, some rhesus microRNA position are annotated at the homolog site to human, but their name is inconsistent to human, such as hsa‐mir‐520b is connected to mml‐mir‐519a. We kept all the names as is annotated in miRBase but re‐annotated them to the right genomic position. The rhesus microRNAs not exist in miRBase but predicted and used in our study is named with prefix “mml‐pd‐“ (means prediction). An undoubted
duplication event of human miR‐512 from rhesus one was observed. A copy lost of miR‐526a on human related to rhesus which has two copies was also indicated.
2.3.3 Substitution Analysis
We paid more attention to the consistent unit sequence which positions from exon to microRNA in C19MC for studying which nucleotide is important and significant to a certain extent of evolution. In the meantime, we expected to observe the fragment detergence of the sequence among different species. Sequence comparison using nucleotide substitution analysis was done by comparing within paralogs within one organism and comparing orgholog pairs between two species. Both line chart for macroscopic tendency of substitution (global view in full length of C19MC) and scatter plot for microscopic variation (local view in a certain region within C19MC) were used for visual presentation in our analysis.
The overall substitution of rhesus is more frequent than human among three different sequence regions. According to the scatter plots, the pattern of three regions in paralogs substitution related to ortholog pairs is the same inside species between human and rhesus: exon is the most divergent, followed by internal flanking region and the microRNA is more conserved. However, when this pattern was compared between two species instead inside species, we found rhesus has less conservation than human. These facts exclude the possible sampling bias caused by pseudogene; meanwhile, the result suggests that stronger selection force occurred in rhesus and caused more divergent in its paralogs. (Figure 14)
The Consistent Fluctuation of Global Tendency
The fluctuated peak along the line chart shows the difference among three proportions, human almost has lower substitution proportion related to the ortholog pairs’ one than rhesus in each site. The overall tendency indicates that human reached more adaptation and reduce its variety. The exon region expresses the unstable fluctuated related to the internal flanking and microRNA region. The internal flanking region, especially the part near to microRNA, shows the least fluctuant difference within paralogs in both human and rhesus. However, few nucleotids of microRNA have high proportion of substitution related to average low proportion of other nucleotides; this phenomenon is in line with the known microRNA conservation rules correspond to its structures.
In addition, the substitution pattern, including the difference and trend, of internal flanking region closed to exon is similar to exon region. It seems as a transitional progress of substitution gradient from varied exon region to the conserved flanking of microRNA but rose again in microRNA sequence. (Figure
15)
Substitution Variety in a Global View
These three proportions of each site were next drawn on an X‐Y scatter plot per 20‐nt length to investigate the local trend of substitution. The substitution proportion of ortholog pairs was plotted on X‐axis as a reference value corresponds to the two paralog proportion on Y‐axis. Therefore, this plot could reflect the different degree of substitution in a site within paralogs in one species to which between ortholog pairs in two species.
Most spots centralized at bottom‐left area and presented the similar intercept of Y display the site consistency between human and rhesus. However, few sites are biased and located at top‐right area suggest the higher substitution
occurred both within paralogs and between orthologs. The sites we had interested are those located at top‐right and existed higher intercept distance between two paralogs, since these sites have more variation between species.
(Figure 16)
The results were separated according to different components again. It is consistent with macroscopic analysis that most interest sites belong to exon region and only few are located in internal flanking. Even though many top‐right spots observed in microRNA region, but they are much closed between two species instead of the phenomena found in exon. It implied even the sites within microRNA have high divergence between species, it is conserved within paralogs. In other words, the microRNAs in C19MC may not only evolve specifically and separately after speciation but also be conserved to maintain some important gene regulation within a species.
Chapter 3
C19MC probably as a defender against
Alu Elements
3.1
Backgrounds and Hypothesis
The duplication of microRNA is related to repeat elements
Mammalian microRNAs derived from two inverted repeat elements via transposition were reported. (Smalheiser and Torvik 2005)
The Impact of Gene Expression by Alu Elements
It is found in recent research that Alu elements contain transcription factor binding sites might modulate gene expression (Shankar, Grover et al. 2004; Polak and Domany 2006) even function as promoters in C19MC. (Borchert, Lanier et al. 2006) Moreover, the major rules of Alu or other transposable element involved in the evolution of eukaryotic gene regulatory network were reviewed. (Feschotte 2008) However, this convenient and efficient mechanism of regulatory network evolution could be lethal due to unexpected, uncontrolled and rapid expansion of Alu. We believed a counteracting mechanism must be present to control the expansion of transposable element.
MicroRNA as a Guard of Genome against to Transposons
In the two cases we described in 2.1, some novel microRNAs governed by upstream Alu elements and transcribed by RNA polymerase III were indentified. (Gu, Yi et al. 2009) It suggests Alu elements involved in small RNA regulation. However, more and more studies suggested that transcription suppression by small RNAs is probably the means of killing transposons. (Malone and Hannon 2009)
The CoEvolution Model of C19MC Expansion
We also followed this hypothesis that one role of C19MC might be the guard of genome and maintain its completeness. The selfish transposons are devoted in their expansion; at the same time, microRNAs strive to repress their expansion. The microRNA duplication could be caused while transposons expand themselves; TEs facilitate the growth of their enemy while extending unintentionally. More microRNAs were duplicated as the defenders, more transposons were occurred as the occupiers, meanwhile induced more microRNA duplicated again. We expected to discover some evidences supporting this infinitive loop. (Figure 17) If this circle existed, maybe some Alu elements escaped from microRNA repression could be existed and discovered. The youngest AluY was found as this situation that the percentage of targeting AluY is reduced than AluS. It implies that the AluY probably find out the salvation.
3.2
Materials and Methods
3.2.1 Determine Mature microRNA Expression Profile from NextGeneration SequencingIt’s difficult to determine the expression of C19MC since many studies found the C19MC is only expressed in few cell lines including placenta cell, germ line cell and embryonic stem cell. Fortunately, we found a study might imply the data we want because they used the next‐generation sequencing (also called deep sequence) to discover novel microRNAs in human embryonic stem cell. The microRNA expression profiles of human embryonic stem cell were fetched from the Solexa sequencing data published by Morin (Morin, O'Connor et al. 2008), who collected the appropriate length of mRNA by electrophoresis and then processed for sequencing. We collected all the sequences with longer than
10‐nt length and counting their number of reads as the expression level. Because the next‐generation sequencing technique is powerful enough to detect mRNA even it has only one sequence, we could use this data to overcome the erratic mature sequence of microRNAs, which may occur nucleotide shift and not be identical in each time of expression. The residues of microRNA generation such as the loop cut by Dicer could be found in our alignment indicate the high‐resolution of this sequencing technique. (Figure 18)
Human Seed Candidates Selection
After redundant reads were removed, we aligned each selected reads, the possible mature microRNA, with annotated C19MC genes. We next confirmed the reads aligned with high possibility rather than random match. The sequencing reads which match to the mature region of microRNA (the arm structure of a microRNA) were kept, but the sequencing reads match to the loop region or other flanking region of microRNA rather than mature form were eliminated. In addition, the low number of sequenced reads still remained in our selection because we are not sure whether it functions or not. (Figure 19) Generally, it is believed that microRNA seeds are located on 2‐8 nucleotides counted from the 5’ end of mature sequence. Although it is not the golden rule, the seed are still the most important sites for determining target site interaction. (Bartel and Chen 2004; Kertesz, Iovino et al. 2007; Bartel 2009) Moreover, many studies involved in microRNA target sites prediction suggested the match of seed region is the major parameter while scanning. (Brennecke, Stark et al. 2005; Krek, Grun et al. 2005; Nakamoto, Jin et al. 2005; Xie, Lu et al. 2005) We also followed this consensus pattern by selecting seed from 2‐8 nucleotides of 5’ end mature sequence as our query candidates against genomes. Finally, 64 seed candidates were selected and subjected to further analysis.
Rhesus Seed Candidates Selection
Unfortunately, there was no C19MC expression data of rhesus reported so far so that we cannot use the same approach to determine real mature sequence of rhesus C19MC. To solve this problem, we combined the defined homologs described in 2.3.2 and the human mature sequences from real expression data to select rhesus seed candidates. Because the mature regions of each microRNA are not conserved among human C19MC and the mature sequence could be varied even within a microRNA, we relaxed our standards while choosing rhesus seeds. The standard mature sequences of rhesus were followed as the position aligned to human and considered 3‐nt shift of mature region. (Figure 20) Due to the relation of standards, 231 seed candidates were selected more than the human.
3.2.2 Whole Genome Scanning (From Seed Candidates’ Point of View)
The genome position perfectly matched with seed candidates were recorded after genomic scanning. Next, these positions were cross‐matched with the collated annotation of Alu elements described in the section 2.2.1.
(Figure 21)
Ability of Hitting Alu Elements of each Seed
Because the number of Alu targeted by microRNAs could be varied depend their specific seeds, the different amount of hit number may also reflect the repression ability of the seeds. To determine whether the numbers of Alu targeted by each seed of C19MC have a bias against normal distribution of random event, we generated a random dataset of seven‐nucleotide‐length seed candidates to simulate the 7‐nt length seed candidates we selected from the real datasets of human C19MC and rhesus C19MC. This generated random dataset contains 5000 seeds which are not included those seeds selected from real
C19MC in two species. All Alu elements annotated by Repeat Masker were used as target pool against by three seed datasets, seeds of human C19MC, seeds of rhesus C19MC and 5000 random generated seeds. Whole genome scan was done by our own program.
Interchange of Seed Candidates
To determine whether the seeds have specialization between two species and as a reference dataset, we next interchanged the seed candidates of one species to against the genome of another species. In other words, we used human seeds as queries to scan rhesus genome and vice versa. 3.2.3 Whole Genome Scanning (From Alu elements’ Point of View) Targeted Proportion among Different Alu Subfamilies Alu elements are raised in different evolutionary stages from the ancient AluJ to the most abundant AluS then the youngest AluY. Presumably, the AluJ is inactive now due to its long evolution age, but recent studies suggest the AluS could still maintain its activity. (Bennett, Keller et al. 2008) According to our original hypothesis, the C19MC we observe now probably have ability to repress AluS and AluY expansion which might damage the genomes, but lost function to inhibit the inactive AluJ which has no harm to the genome. Meanwhile, because AluS appear early and more amount than AluY, C19MC could evolve to against AluS primarily. Even if we cannot observe the expectation, the proportion of Alu targeted by C19MC might be different among 3 Alu subfamilies. Here, all Alu elements were considered in the analysis because all of them probably had been targeted in the evolutionary history no matter which positions or strands they are located recently.
Classification of Alu Elements by Inserted Position
Alu elements annotated by Repeat Masker were scaned and separated into 4 categories including insertion of 5’UTR, ORF, 3’UTR and non‐coding genes, by the reference gene annotation fetched from UCSC. We assumed this gene regulation involving by Alu element might depend on microRNA mechanism. Therefore, only the Alu elements inserted into genes in the same direction of mRNA transcription were selected. Only the Alu elements transcribed with genes into mRNA have opportunity to be targeted by microRNAs.
3.3
Results and Discussion
3.3.1 C19MC has Extremely Different Expression Levels The expression or microRNAs in C19MC ranges from extremely low level with less than 10 reads to high level with hundreds reads. We expected to find out the relationship between expression level and Alu elements, but achieved nothing. It may because the regulatory mechanism of C19MC could via epigenetics instead of mRNA level. The expression profiles of each mature microRNAs were summarized on the Table (Table 2).
3.3.2 The Biased Distribution of C19MC Target Ability against Alu Elements
Basically, although the nucleotides variation of a local genomic region could be different, the stochastically generated seeds with short length in the extreme should hit genomes in random and show a normal distribution. We used the 5000 random seeds to against genomes as a control to determine whether seeds of C19MC have different targeting ability to Alu elements.
(Figure 22, 23) The number of Alu targeted by each seed were plotted on a
logarithmic scale to reduce their extreme variation and shown on X‐axis; the Y‐axis represents how many seeds against Alu elements in the same amount. As
we expected, the trend of random seed dataset displayed a smooth normal distribution which indicates the seeds of microRNA, if they generated randomly and not encountered selection force, might target genomes fortuitously. Both human and rhesus seeds have biased distribution located on the high Alu element hits region (right side of x‐axis). We found that seeds belong to this distribution are complementary to the potential microRNA targeting sequence of Alu elements reported by previous study. This coincident complement between our biased seeds with the core target site of Alu reported by them indicates that the defense mechanism probably exists and selection force might occur. Biased Seed Candidates have Conserved Sequences These biased seeds of human and rhesus are the same mostly. It suggests that the defense mechanism against Alu elements maybe conserved in primate lineage. Based on our hypothesis, the C19MC are primary to defense the AluS expansion, thus they also duplicated themselves to achieve the enough ability of repression. If this hypothesis is real, we could find the conservation of seeds between human and rhesus due to their functional preservation.
Specialization of Human C19MC Seeds
While we interchanged C19 seed candidates of each species to against with the genome of another species, we found that some seed candidates present a biased pattern. (Figure 24) Almost seeds of two resources were lined up with a diagonal as equal Alu hits between two species. However, the human seeds seem be specialized against to human genome because they deviated from the diagonal line. This pattern indicates that the specialization occurred and selection force existed in human C19MC seed during primate evolution, thus the human C19MC provides specific seeds against most human Alu elements but
useless while against rhesus Ale elements. Also, it indicates the seeds might be functional in the past although they are not highly expressed now. Only they functioned during the primate evolution, the selection force could be existent. Interestingly, both human and rhesus seeds targeting AluY are deviated from the patterns of targeting AluJ or AluS and tend toward targeting more rhesus Alu elements than human ones. Rhesus also has two specific biased seeds not found in human and they deviated only in targeting AluY. 3.3.3 Analysis of Alu Elements Targeted by C19MC Preference of Alu Targeted by C19MC: Different Alu Subfamilies The slight different microRNA abilities of targeting Alu against three Alu subfamilies could be observed if the variation occurred in conserved target regions of AluY. In addition, because AluY was born after AluS, if C19MC originally recognizes the conserved sequence of AluS elements, the biased targeting number of AluY is still detectable. We demonstrate extended analysis that separated the number of targeted Alu elements into three different subfamilies in order to detect the biased target preference of C19MC among Alu subfamilies. (Figure 25) As our expectation, the AluJ shows the least hit by human C19MC. This result supported our two reasons: one is that many AluJ are truncated in sequences and accumulated mutations during evolution so that cannot be targeted by C19MC recently; another is that C19MC might appear while AluS expansion and originally evolve to against AluS. Although the results seems not be significant in rhesus, the trend among three subfamilies is similar. This is probably because of the overestimation in rhesus seeds selection which mixed many pseudo‐seeds and caused false positive.
Interestingly, AluY shows slightly reduction of targeting as we expected even we did not know the mechanism of its escapes. The few amount of AluY escaped from C19MC targeting could be induced by the target site mutation as same as AluJ. These mutated AluY might have more opportunities to survive and to duplicate under the C19MC defense force so that we observed this slightly decrease of targeting. The related low difference between AluS and AluY may because the evolution time is not enough for AluY to accumulate mutation and to encounter selection.
Preference of Alu Targeted by C19MC: Different Alu Inserted Position
Many genes are inserted by transposons during evolution process, especially Alu elements. Depends on the inserted position, we classified Alu elements into 4 categories, harbored on 5’UTR, ORF (open reading frame) and 3’UTR of genes and those inserted to the non‐coding genes. In our opinion, if the inserted Alu elements become the component of gene regulation by microRNA, the Alu harbored on 3’UTR of genes are more important and beneficial. While the inserted Alu provides the biological benefits and involved in regulatory network, the repression and inhibition of them are not necessary anymore. We examined this concept by analyzing the targeting proportion of Alu inserted into different position of genes in human and rhesus.
Figure 1 The Distribution and Structure of Transposons (a) Percentage of transposable elements in human genome. Alu elements approximately occupy the one‐third of genome content. Adapted from (Cordaux and Batzer 2009). (b) Genomic structure of L1 and Alu element. The main difference between them is whether protein encoded or not. Promoters are marked as dark blue. (a) (b)
Figure 2 Distributions of Alu Subfamilies in Primate Genomes
The current distributions of Alu subfamilies in primate genomes are shown. Genome versions are fetched from UCSC by hg19 for human, ponAbe2 for orangutan, rheMac2 for rhesus and panTro2 for chimpanzee. The other category represents the non‐Alu SINEs. 26.13% 57.50% 11.98% 4.39% Human Homo sapiens 23.84% 56.28% 11.49% 8.40% Orangutan Pongo abelii 22.26% 50.81% 19.12% 7.82% Rhesus Macaca mulatta 24.00% 56.04% 11.91% 8.05% Chimpanzee Pan troglodytes
Figure 3 Evolution of Alu Subfamilies
Evolutionary tree of 213 Alu subfamilies throughout the primate evolution were identified in previous study. Adapted from (Price, Eskin et al. 2004).