國 立 交 通 大 學
生 物 科 技 學 院
分子醫學與生物工程研究所
碩士論文
第二型血管收縮素轉換酶和基質金屬蛋白酶的
活性與肺部疾病之相關性
Activities of Angiotensin Conversion Enzyme II and
Matrix Metalloproteinases Associated with
Pulmonary Diseases
研 究 生: 廖燕秋
指導教授: 林志生
博士
第二型血管收縮素轉換酶和基質金屬蛋白酶的活性與肺部疾病之相關性
Activities of angiotensin conversion enzyme II and matrix metalloproteinases associated with pulmonary diseases
研 究 生 : 廖燕秋 Student : Yan-Chiou Liao
指導教授 : 林志生 博士 Advisor : Chih-Sheng Lin Ph.D.
國立交通大學 生物科技學院
分子醫學與生物工程研究所 碩士論文
A Thesis
Submitted to Institute of Medicine and Bioengineering College of Biological Science and Technology
National Chiao Tung University In partial fulfilment of the requirements
For the degree of master In
Biological Science and Technology July 2013
Hsinchu, Taiwan, Republic of China
Acknowledgement
時間過得很快,相對來說是很充實的兩年碩士研究生活,其中的酸甜苦辣回憶,也 是值得細細品味的。一轉眼也輪到我寫致謝文,很開心自己加入了一個充滿歡樂又不失 專業的實驗室,有著一群很棒的實驗室夥伴們以及一位非常照顧學生的指導老師。 首先最要感謝的是心臟組已經畢業的大學長-阿關關,謝謝你的細心指導與嚴格訓 練,使得我可以獨當一面的做實驗,真的很感激!學長你是在實驗室裡的超級重要角色, 不管在實驗上還是生活上,總是照顧我們這群小毛頭的需求,就連畢業了,也要隨時接 到我們的求救電話,所以應該趕快找個賢內助可以好好照顧你啦!千雅學姊,實驗室的 犀利女王,總是可以一針見血的說出要點,讓實驗室充滿笑聲,謝謝妳在很多事上面的 幫忙,可以少掉不必要的繁雜程序。也要預祝學姊順利畢業前途似錦。耶豆學長,學識 一流、電腦高手,而且講話很有趣,總能讓大家開懷大笑。戴樂,謝謝你多次陪我早上 去爬山,也教我許多投資理財的觀念,讓我在做實驗之虞,還可以學習到不同的東西! 偉修哥,謝謝你幫忙打掃動物房,讓大家可以順利做實驗。 芳沅,是一個很善良又聰明漂亮的女孩,在妳身上學到了很多,如處事觀點、品味 生活等,以及可以發現深夜的實驗室有許多好料的。謝謝妳與我互相扶持一起度過這兩 年的生活,以及妳的女兒 Betty 豐富我的生活,畢業後我們各奔東西,但我們的友誼是 不受時間空間影響的。祝妳能完成自己的夢想亦或是找到一份令人稱羨的工作與理想的 另一伴喔! 意涵,有時候是老婆婆狀態的思考動作,有時候是什麼都不怎麼明瞭的傻妞,但妳 是很棒的小助手,謝謝妳實驗的幫忙,也謝謝妳在平常的幫忙,請妳幫忙的事總是能順 利完成。葛麗,獨立又聰明,笑點有很獨特,交代給妳的事總是不用擔心,讓人很信任。 碧珊,有時候是少一根筋的模式,做實驗需要多點思考與謹慎,升上碩二會更加辛苦, 加油囉!采郁,是嚴重潔癖與很會烹飪的陳媽媽,處理事情很快速,藻類組就靠妳囉! 郁彬,是聲音低沉且步調慢慢的大叔,因為你,我們晚上才不會挨餓,謝拉! 也要感謝大學部學弟妹們的幫忙,佩衡、一華、明慧,你們能在大學活采多姿的生 活之餘,抽空來實驗室學習研究,此用心已勝過大多數人,前程似錦、無可限量,祝福 你們。相處不久的碩一學弟妹們張蓉、日升以及子仁,我還來不及與大家熟悉就要離開, 但我很榮幸能教導各位和妳們解說我的實驗,碩班和大學有很大不同,好好適應好好加 油! 當然,我最感謝的是我的指教教授-林志生教授。老師對於研究上的指導,常常在 觀念上給予我重要的啟發。在實驗的細微環節上,也總不吝惜地提出以往自己的實驗經 驗給我學習參考。更多的是在做人做事的態度、方法和責任感上有深深地影響。也感謝 老師在忙綠之餘還抽空對我的論文進行修改及建議。由衷的感謝老師的包容與指導,對 老師的感謝已不是一句感恩就能道盡。到目前的訓練使我羽翼漸豐,即將翱翔,但對於 未來未知且更多險惡的環境顯然是不足的,將來我也將謹記老師的教誨,更上一層樓。 我要非常感謝吳介信老師、吳希天老師能來擔任我的口試委員,因為老師們蒞臨指 導及給予相當有價值的建議,讓我能順利的取得學位。另外,還有在此研究中犧牲奉獻 的動物們,沒有你們就沒有這篇論文,更遑論人類醫療的進步。 最後,感謝我親愛的家人,謝謝你們在這兩年對我的支持與鼓勵,以及讓我在這個 物價頗高的新竹生活衣食無缺,使我能無後顧之憂地順利完成學業,謝謝。 廖燕秋 謹誌 交通大學分子醫學與生物工程研究所碩士班 中華民國一○二年七月
第二型血管收縮素轉換酶和基質金屬蛋白酶的活性
與肺部疾病之相關性
研究生 : 廖燕秋
指導教授 : 林志生 博士
國立交通大學
生物科技學院
分子醫學與生物工程研究所
碩 士 論 文
中文摘要
在腎素-血管收縮素系統中(Renin-angiotensin system; RAS),已知血管收縮素轉化 酶(angiotensin-converting enzyme; ACE)/血管收縮素II(angiotensin II; Ang II)途徑 (ACE/Ang II axis)與許多肺部疾病有關,但在RAS系統中的另一個ACE類似酵素,第 二型血管收縮素轉化酶(angiotensin-converting enzyme II; ACE2)的生理功能卻較少被 探討,ACE2可將Ang II水解成血管收縮素1-7(angiotensin 1-7; Ang 1-7),而Ang 1-7被認 為有拮抗Ang II的功能。在我們過去研究結果中顯示,ACE2/Ang 1-7 axis的異常與心臟 組織纖維化病程有關,而此與基質金屬蛋白酶(matrix metalloproteinases; MMPs)和基 質金屬蛋白酶組織抑制因子(tissue inhibitors of MMPs; TIMPs)的調控失衡有關。
肺部疾病中常見的肋膜積液(pleural effusion)是指肋膜腔中積存過量的液體,為 一種臨床上常見的病徵,其形成機制主要為心臟衰竭、炎症、惡性腫瘤、肺結核等病症 所造成不同程度的病程。過去的研究中顯示肋膜積液病程中所誘發免疫反應的發炎機制, 會進而導致肋膜腔纖維化。另外肺纖維化,臨床上稱之為原發性肺纖維化(idiopathic pulmonary fibrosis),是一種起因不明,長期漸進發展且不可逆轉的致命性肺部疾病。 在本研究中,我們欲探討肺部ACE2和MMPs/TIMPs平衡調控與肺部疾病和肺部組 織纖維化的關係性。本研究的第一部分為探討ACE/ACE2和MMPs的變化,是否與特定 之肋膜積液病變有關,進而發展可用於臨床診斷的生物性指標;研究的第二部分為利用 博萊酶素(bleomycin)的胸腔注射誘發小鼠肺纖維化,用以探討肺臟組織纖維化病程中,
ACE/ACE2與MMPs/TIMPs活性表現的差異性。另外本研究也利用ACE2基因剔除小鼠 (ACE2 knockout (KO) mice)探討ACE2在肺纖維化病變病程中所扮演的角色。
在第一部分的臨床肋膜積液研究中,相較於濾出型積液(transudates effusion),在 滲出型積液(exudates effusion)中,可測得顯著較高ACE/ACE2之活性比值與MMPs活 性。再則,在滲出型積液中,肺結核患者肋膜積液中的ACE/ACE2比值、腺核苷去氨酶 (adenosine deaminase; ADA)活性及MMP-9活性均顯著高於肺炎與腺癌病患肋膜積液中 所測得之值。本項研究之重要結論為RAS和MMP活性與肋膜積液病變有關,而肋膜積液 中ACE/ACE2比值、ADA活性及MMP-9活性可做為肺結核患者之診斷生物標誌。 在第二部分的肺纖維化研究中,我們成功建立胸腔注射1 U/kg bleomycin方式誘發 實驗小鼠產生肺部纖維化病症之模式,而在小鼠誘發肺部疾病病程中(bleomycin處理後 第3、7及28天),小鼠肺臟組織中所測得的ACE/ACE2與MMP-9/TIMP-1活性有顯著改變, 此即實驗小鼠在注射bleomycin後第7天時,其肺部組織中的ACE、MMP-9和TIMP-1皆有 顯著上升,且組織切片觀察有顯著白血球的浸潤和膠原蛋白累積的病變;再則於 heterozygous、homozygous及hemizygous ACE2 KO小鼠實驗中,結果顯示在缺乏ACE2 的小鼠中,於注射bleomycin的早期(第3天)就會使肺組織中的MMP-9活性有顯著表現, 以及TIMP-1含量顯著下降。本項研究之重要結論為ACE2在bleomycin誘發肺臟纖維化病 程中,扮演的功能與MMP-9/ TIMP-1的表現調節有關。
【關鍵詞】 肺部疾病、肋膜積液、肺纖維化、第二型基質金屬蛋白酶、第九型基質金 屬蛋白酶、第二型血管收縮素轉換酶
Activities of Angiotensin Conversion Enzyme II and Matrix
Metalloproteinases Associated with Pulmonary Diseases
Graduate student: Yan-Chiou Liao
Advisor: Chih-Sheng Lin Ph.D.
Institute of Molecular Medicine and Bioengineering
College of Biological Science and Technology
National Chiao Tung University
Abstract
Angiotensin converting enzyme (ACE)/angiotensin II (Ang II) axis in renin-angiotensin system (RAS) is associated with the development of several pulmonary diseases; however, much less is known about the functions of angiotensin converting enzyme II (ACE2), an ACE homologue that hydrolyses Ang II to angiotensin 1-7 (Ang 1-7), a peptide that exerts the actions opposite to those of Ang II. In our previous studies, we showed that ACE2
dysregulation and unbalanced matrix metalloproteinases (MMPs)/tissue inhibitors of MMPs (TIMPs) are highly associated with the fibrotic damage in cardiovascular diseases. In this project, we aimed to study the molecular mechanism of ACE2 regulation on pulmonary fibrosis by experimental mouse models. We also attempted to elucidate the roles of
ACE2/Ang 1-7 axis in pulmonary function and the hypothesis whether part of the antifibrotic effects of ACE2/Ang 1-7 axis is via the balancing regulation of MMPs/TIMPs?
Pleural effusion is a common medical problem in the chest and involves accumulation of an abnormal amount of pleural fluid in the pleural space. Several diseases, such as congestive heart failure, liver cirrhosis, tuberculosis, adenocarcinoma and pneumonia, are common diseases that cause pleural effusions, but there is less rapid and accurate diagnostic method for pleural effusion. Recent studies showed that the inflammatory response induced by these chest diseases would cause pleural fibrosis. The major enzymes, ACE and ACE2 in RAS, may be involved in the mechanism of pleural fibrosis and pleural effusion. Another lung disease such as idiopathic pulmonary fibrosis has been known as a chronic, progressive, irreversible, and usually lethal lung disease of unknown cause.
MMPs/TIMPs balance in pulmonary disease and pulmonary fibrosis. The first part of the present studies was to detect ACE/ACE2 and MMPs activities in pleural effusions. It can identify diagnosis based on clinical variables to differentiate from pleural effusions. For the second part of the present studies, we took mice to induce pulmonary fibrosis with bleomycin in lung cavity to investigate the pathogenesis of pulmonary fibrosis, and the balancing activity ACE/ACE2 as well as MMPs/TIMPs. In addition, we used ACE2 knockout (KO) mice to investigate the role of ACE2 in the pathogenesis of pulmonary fibrosis.
In the first part, the study of clinical pleural effusions, the ACE/ACE2 ratios and MMPs activity in exudate effusions were significantly higher than in transudate effusions.
Furthermore, in exudative effusion, ACE/ACE2 ratio, adenosine deaminase (ADA) and MMP-9 activities in tuberculosis effusions were significantly higher than those of pneumonia effusions and adenocarcinoma effusions. The major results of the part I are that the RAS and MMPs activity are important in abnormal pleural fluid. ACE/ACE2 ratio, ADA activity and MMP-9 activity can be used as biomarkers in the diagnosis of tuberculosis patients.
In the second part, we have successfully established the mouse model of pulmonary inflammation and fibrosis induced by 1 U/kg bleomycin injection. The profiling of lung ACE/ACE2 and MMP-9/TIMP-1 activity in the mice challenged with bleomycin at 3-day, 7-day and 28-day after the treatments have been performed. The data showed that ACE, MMP-9 activities and TIMP-1 concentration in the lung tissue were significantly increased in wild-type (WT) mice injected bleomycin at 7-day, and the histological sections showed significant leukocyte infiltration and collagen accumulation. Furthermore, in heterozygous, homozygous and hemizygous ACE2 KO mice, the results showed that the lung tissue MMP-9 activity is significantly higher than Control in early stage (3-day), and TIMP-1 concentration is significantly decreased. The major results of the part II indicate that the important role of ACE2 on bleomycin-induced pulmonary fibrosis may be via the regulation of
MMP-9/TIMP-1 expression.
Keywords: pulmonary disease, pleural effusion, pleural fibrosis, matrix metalloproteinase 2,
Abbreviation
ACE Angiotensin-converting enzyme ACE2 Angiotensin-converting enzyme II
ACEIs Angiotensin-converting enzyme inhibitors Ad Adenocarcinoma
ADA Adenosine deaminase
Ang II Angiotensin II(Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-His-Leu) Ang-(1-7) Angiotensin-(1-7) (Asp-Arg-Val-Tyr-Ile-His-Pro)
Ang-(1-9) Angiotensin-(1-9) (Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-His) AT1R Angiotensin II type I receptor
AT2R Angiotensin II type II receptor Ble Bleomycin
CRP C-reactive protein ECM Extracellular matrix
IPF Idiopathic pulmonary fibrosis MAPK Mitogen-activated protein kinase MEK Mitogen-activated/ERK kinase MMP-2 Matrix metalloproteinase 2 MMP-9 Matrix metalloproteinase 9 MMPs Matrix metalloproteinases
NADPH Nicotinamide adenine dinucleotide phosphate NF-B Nuclear factor-kappa B
PE Pleural effusion Pn Pneumonia
RAS Renin-angiotensin system ROS Reactive oxygen species TB Tuberculosis
Tet Tetracycline
TGF-1 Transforming growth factor-beta 1 TIMPs Tissue inhibitors of metalloproteinases TNF- Tumor necrosis factor-alpha
Content
Acknowledgement... i
Chinese Abstract... iii
English Abstract... v
Abbreviation... vii
Content... viii
List of Tables... xi
List of Figures... xii
I. Literature Review... 1
1-1. Pleural effusions... 1
1-1-1. Physiology of the pleural space... 1
1-1-2. Pathogenesis of pleural effusions... 1
1-1-3. Clinical manifestations and diagnose tests... 2
1-2. Pleural inflammation and fibrosis... 3
1-2-1. The mesothelium and regulation of pleural inflammation... 3
1-2-2. Pleural fibrosis... 4
1-3. Pulmonary fibrosis... 6
1-4. Renin angiotensin system... 7
1-5. Angiotensin converting enzyme II... 8
1-6. Matrix metalloproteinase... 10
1-6-1. Gelatinase A (MMP-2, Type II collagenase)... 12
1-6-2. Gelatinase B (MMP-9, Type V collagenase)... 13
1-7. ACE2 and gelatinase (MMP-2 and MMP-9)... 13
II. Research Purpose and Strategy... 23
III. Materials and Methods... 26
3-2. Pleural effusion samples collection and analyses... 26
3-3. ADA activity... 27
3-4. ACE and ACE2 activity assay... 27
3-5. Gelatin zymography assay... 27
3-6. Enzyme-linked immunosorbent assay (ELISA)... 28
3-7. ACE2 knockout mice... 28
3-8. RNA isolation and quantification... 30
3-9. Reverse transcription-polymerase chain reaction (RT-PCR) and Real time polymerase chain reaction... 30
3-10. Animal model, induction of lung fibrosis and treatment of mice with Lenti-ACE2... 31
3-11. Sample preparation... 32
3-11-1. Protein extraction... 32
3-11-2. Histological determination of fibrosis... 32
3-12. Western blotting... 33
3-13. Statistical analysis... 33
IV. Results... 34
Part I: MMPs/TIMPs and ACE/ACE2 in pleural effusions... 34
4-1-1. General characteristics of pleural effusions... 34
4-1-2. ACE and ACE2 activities in transudates and exudates... 34
4-1-3. MMP-2 and MMP-9 activities in transudates and exudates... 34
4-1-4. ACE and ACE2 levels in exudates from patients with different diseases …….. 35
4-1-5. MMP-2, MMP-9, and ADA levels in exudates from patients with different diseases... 35
4-1-6. ELISA for TGF-β1 concentration in pleural transudative and exudative effusions... 36
4-1-7. ELISA for TNF-α concentration in pleural transudative and exudative effusions... 36 Part II MMPs/TIMPs and ACE/ACE2 in pulmonary fibrosis... 46
4-2-1. ACE2 knockout mice... 46
4-2-2. ACE and ACE2 activity in WT mouse lung... 46
4-2-3. MMP-9 and TIMP-1 activities in WT mouse lung... 47
4-2-4. ACE activity in ACE2 KO mouse lung…... 47
4-2-5. MMP-9 activities in ACE2 KO mouse lung... 48
4-2-6. TIMP-1 concentration in ACE2 KO mouse lung... 48
4-2-7. Histological section staining of mouse lung tissue... 48
4-2-8. ACE2 activity in ACE2 KO mice after lenti-ACE2 injection... 49
V. Discussion... 66
VI. Conclusions... 72
List of Tables
Table 1-1. General causes of pleural effusions... 15
Table 1-2. Diagnose of transudates and exsudates... 16
Table 1-3. The cellular responses of cytokines and growth factors implicated in the
progression of pleural fibrosis... 17
Table 1-4. Types of different matrix metalloproteinases and their substrate specificity.. 18
Table 1-5. Functional properties of the tissue inhibitors of metalloproteinases………... 19
Table 4-1-1. Pleural fluid characteristics of the study population... 37
Table 4-1-2. Pleural fluid characteristics of tuberculosis, pneumonia and
List of Figures
Fig. 1-1. Family of enzymes and proteins belonging to the ACE family of proteins... 20
Fig. 1-2. Schematic diagram of the role of the RAS in acute lung failure and the proposed action of the SARS-coronavirus... 21
Fig. 1-3. Family of enzymes and proteins belonging to the MMPs family of proteins... 22
Fig. 2-1. The flowchart of research strategy of Part I... 24
Fig. 2-2. The flowchart of research strategy of Part II... 25
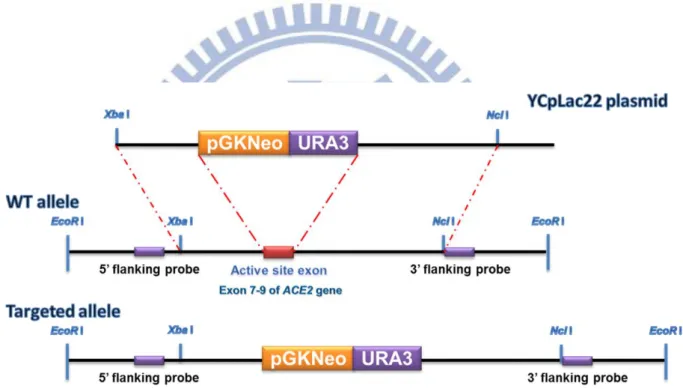
Fig. 3-1. Strategy for producing targeted disruption of the ace2 gene... 29
Fig. 4-1-2. Enzymatic activity of ACE and ACE2 in pleural transudative and exudates effusions... 39
Fig. 4-1-3. Correlations between ACE and ACE2 activity in pleural effusion... 40
Fig. 4-1-4. MMP-2 and MMP-9 activities in pleural transudative and exudative effusions... 41
Fig. 4-1-5. ACE and ACE2 activities in exudative effusions from patients with tuberculosis, pneumonia, and adenocarcinoma... 42
Fig. 4-1-6. MMP-2, MMP-9, and ADA levels in exudative effusions from patients with tuberculosis, pneumonia, and adenocarcinoma... 43
Fig. 4-1-7. ELISA for TGF-β1 concentration in pleural transudative and exudative effusions... 44
Fig. 4-1-8. ELISA for TNF-α concentration in pleural transudative and exudative effusions... 45
Fig. 4-2-1. PCR genotyping protocol for ACE2 knockout mouse... 50
Fig. 4-2-2. ACE2 expression and activity in the ACE2 knockout mice... 51
Fig. 4-2-3. ACE and ACE2 activity in the lung tissue of mice challenged with bleomycin... 52
Fig. 4-2-4. ACE and ACE2 activity in the lung tissue of mice challenged with tetracyclin... 53
Fig. 4-2-5. MMP-9 and TIMP-1 in the lung tissue of mice challenged with bleomycin.... 54
Fig. 4-2-7. ACE activity in the lung tissue of ACE2 KO mice challenged with
bleomycin……….. 56
Fig. 4-2-8. ACE activity in the lung tissue of ACE2 KO mice challenged with
tetracycline... 57
Fig. 4-2-9. MMP-9 activity in the lung of ACE2 KO mice treated with bleomycin... 58
Fig. 4-2-10. MMP-9 activity in the lung of ACE2 KO mice treated with tetracycline... 59
Fig. 4-2-11. TIMP-1 concentration in the lung of ACE2 KO mice treated with
bleomycin... 60
Fig. 4-2-12. TIMP-1 concentration in the lung of ACE2 KO mice treated with
tetracycline... 61
Fig. 4-2-13. Histological examinations of the lung tissue of wild-type mice treated with
bleomycin... 62
Fig. 4-2-14. Histological examinations of the lung tissue of wild-type mice treated with
tetracycline... 63
I. Literature Review
1-1. Pleural effusions
1-1-1. Physiology of the pleural space
The pleural space is the coupling system between the lung and the chest wall;
accordingly, it is a crucial feature of the breathing apparatus. The pressure within the pleural space (the pleural pressure) is important in cardiopulmonary physiology, because it is the pressure at the outer surface of the lung and the heart and the inner surface of the thoracic cavity. The pleural mesothelium is a monolayer of cells that vary from a flattened ovoid shape to columnar or cuboidal cells that lie loosely over the underlying substructure. The connective tissue of the pleural basement membrane is a complex structure that underlies the surface layer of mesothelial cells and is involved in inflammation of the pleural space (Jantz et al., 2008). Fluid that enters the pleural space can originate in the pleural capillaries, the interstitial spaces of the lung, the intrathoracic lymphatics, the intrathoracic blood vessels, or the
peritoneal cavity. It is believed that normal fluid enters the pleural space originates in the capillaries in the parietal pleura (Nahid et al., 2003). The normal pleural fluid production is approximately 15 mL in 50 kg individual human beings.
1-1-2.Pathogenesis of pleural effusions
Pleural fluid accumulates when the rate of pleural fluid formation exceeds the rate of pleural fluid absorption. The main factors that lead to increased pleural fluid formation or decreased pleural fluid absorption are tabulated in Table 1-1. Normally, a small amount (0.01 mL/kg/hour) of fluid constantly enters the pleural space from the capillaries in the parietal pleura. Almost all of this fluid is removed by the lymphatics in the parietal pleura, which have a capacity to remove at least 0.20 mL/kg/hour. Note that the capacity of the lymphatics to remove fluid exceeds the normal rate of fluid formation by a factor of 20.
1-1-3.Clinical manifestations and diagnose tests
The presence of moderate to large amounts of pleural fluid produces symptoms and characteristic changes on physical examination. The symptoms of a patient with a pleural effusion are mainly dictated by the underlying process causing the effusion. Many patients have no symptoms referable to the effusion. When symptoms are related to the effusion, they arise either from inflammation of the pleura, from compromise of pulmonary mechanics, from interference with gas exchange, or on rare occasions, from decreased cardiac output.
The accumulation of clinically detectable quantities of pleural fluid is distinctly
abnormal. Pleural effusions have classically been divided into transudates and exudates (Light et al., 1972). A transudative pleural effusion develops when the systemic factors influencing the formation or absorption of pleural fluid are altered so that pleural fluid accumulates. The pleural fluid is a transudate. The fluid may originate in the lung, the pleura, or the peritoneal cavity (Broaddus et al., 1992). The permeability of the capillaries to proteins is normal in the area where the fluid is formed. In contrast, an exudative pleural effusion develops when the pleural surfaces or the capillaries in the location where the fluid originates are altered such that fluid accumulates. The pleural fluid is an exudate. The most common causes of exudative pleural effusions are pleural malignancy, parapneumonic effusions, and pulmonary embolism.
An accurate diagnosis of the cause of the effusion, transudate versus exudate, relies on a comparison of the chemistries in the pleural fluid to those in the blood, using Light's criteria. According to Light's criteria (Light et al., 1972), a pleural effusion is likely exudative if at least one of the following exists. Also see Table 1-2.
1. The ratio of pleural fluid protein to serum protein is greater than 0.5 2. The ratio of pleural fluid LDH and serum LDH is greater than 0.6
3. Pleural fluid LDH is greater than 0.6 or 2/3 times the normal upper limit for serum. Different laboratories have different values for the upper limit of serum LDH, but examples include 200 and 300 IU/L.
The sensitivity and specificity of Light's criteria for detection of exudates have been measured in many studies and are usually reported to be around 98% and 80%, respectively (Romero et al., 2000; Joseph et al., 2002).
1-2. Pleural inflammation and fibrosis
1-2-1. Mesothelium and regulation of pleural inflammation
Pleura is lined by a monolayer of mesothelial cells which secrete glycosaminoglycans and other surfactant-like molecules to lubricate the pleural surface (Roth, 1973; Beavis et al., 1994). The mesothelium has a number of other important functions including movement of fluid, particulates and cells across the pleural cavity; release of pro- and anti-inflammatory and other immunomodulatory mediators; antigen presentation; secretion of factors that promote both deposition and clearance of fibrin; and synthesis of growth factors and ECM proteins to aid in serosal repair (Mutsaers 2002; Mutsaers et al., 2004).
The onset of pleural infections and injury is characterized by a massive influx of leukocytes from the vascular compartment into the pleural space (Brauner et al., 1993). This is likely to be initiated on the surface of the mesothelial cell. For example, bacteria, talc and asbestos fibres are phagocytosed by mesothelial cells which induce cell activation and release of chemokines such as interleukin (IL)-8 (Tanaka et al., 2000). Furthermore, mediators released from activated macrophages also stimulate mesothelial cells to release potent inducers of neutrophil and monocyte chemokines including IL-8, growth-related oncogene (GRO)-α, interferoninducible protein (IP)-10, monocyte chemoattractant protein (MCP)-1
(Betjes et al., 1993; Nasreen et al., 2001). Secretion of these chemokines is polarised toward
the cell apical surface, creating a chemotactic gradient from the basolateral to the apical side of the mesothelial cell (Li et al., 1998). By secreting chemokines in a polarized manner, mesothelial cells promote directed transmesothelial migration of both neutrophils and monocytes.
Movement of leukocytes from the circulation to the site of inflammation is facilitated by the expression of integrins and adhesion molecules. Mesothelial cells express several cell adhesion molecules including intercellular adhesion molecule (ICAM)-1, vascular cellular adhesion molecule (VCAM)-1, E-cadherin, Ncadherin, CD49a, CD49b and CD29 and can be induced by IL-1b (Rossen et al., 2001), tumor necrosis factor- (TNF-) and interferon- (IFN-) in vitro (Hausmann et al., 2000). Leukocytes express the β2 integrin family members, lymphocyte function associated antigen (LFA)-1 (CD11a/CD18), and Mac-1 (CD11b/CD18) on their surface, which are counter receptors for ICAM-1. Interaction between LFA-1/ Mac-1
and ICAM-1 leads to cell-cell adherence and results in transmigration of leukocytes across mesothelial cell monolayers (Zeillemaker et al., 1996).
Resolution of inflammation and repair of the pleura without fibrosis requires a
down-regulation of the inflammatory response, including leukocyte clearance and inhibition of fibroblast proliferation and collagen production. Mesothelial cells are likely to contribute to controlling inflammation both in normal and inflamed tissue as they have cyclooxygenase (Cox) activity, expressing both Cox-1 and Cox-2 mRNA and metabolise arachidonic acid to release prostaglandins and prostacyclin (Hott et al., 1994). Mesothelial cells also participate in regulating efflux of inflammatory cells from serosal cavities via stomata and the draining lymphatics. Recently we demonstrated that macrophage emigration from inflamed peritoneum is controlled through specific integrin mediated regulation of macrophage-mesothelial cell interactions involving very late antigen (VLA)-4 andVLA5 (Bellingan et al., 2002).
1-2-2. Pleural fibrosis
During the process of wound healing, formation of a transitional fibrin neomatrix
contributes to tissue organization and fibrotic repair (Dvorak et al., 1986). In a similar context, disordered pathways of coagulation and fibrinolysis that favor intrapleural fibrin deposition are characteristic of pleural inflammation and pleurodesis (Idell et al., 1995). The observation of intrapleural fibrin in association with pleural inflammation lends further support to the notion that the fibrinous neomatrix is involved in the evolution of pleural repair and location (Bignon et al., 2001). The rapid appearance of intrapleural fibrin resembles that of
intrapulmonary fibrin deposition in the progression of accelerated pulmonary fibrosis, as may occur in severe cases of acute respiratory distress syndrome (ARDS) (Idell et al., 1995). Disordered local fibrin turnover, therefore, occurs in both settings and represents an early tissue response that sets the stage for progressive remodeling and may lead to fibrotic repair in the face of protracted injury. Extravascular tissue fibrin deposition, as occurs in the setting of pleural injury, follows a progression recapitulating that which occurs in the setting of wound healing and the desmoplastic response associated with solid forms of neoplasia. As a function of increased microvascular permeability, plasma is extravasated and enters the parenchyma of inflamed tissues, where it encounters tissue factor (TF). TF forms a complex with activated factor VII to initiate coagulation at these extravascular sites, thereby initiating the formation of transitional fibrin, which may undergo progressive remodeling subject to the
influence of locally elaborated mediators of inflammation and the products of inflammatory and parenchymal cells. A wide array of myeloid and parenchymal cells, including mesothelial cells, can produce components of the fibrinolytic response as well as inhibitors of the
fibrinolytic system, including urokinase (uPA), its receptor (uPAR), and plasminogen activator inhibitor (PAI)-1. The responses of these cells may thereby influence whether transitional fibrin is cleared or maintained, based on the balance of the expression of the components of the fibrinolytic system and their net influence on local fibrinolytic activity. As reviewed elsewhere, there is considerable interplay between the coagulation and
fibrinolytic systems and other inflammatory pathways that may potentiate local inflammation and reparative responses (Idell et al., 2001; Ingelfinger et al., 2003).
Resident pleural cells express uPA, uPAR and PAI-1, as well as tPA, and all of these components can be detected in pleural fluid. Plasminogen, a substrate for both tPA and uPA, is likewise present in pleural fluids, and as such plasmin can be generated locally (Idell et al., 1991). Pleural fluids harvested from patients with congestive heart failure demonstrate detectable fibrinolytic activity, but the fibrinolytic activity of patients with exudative pleural effusions is subject to inhibition by PAI, in particular PAI-1, and antiplasmins. In these situations, much of the uPA expressed in pleural fluids is bound to PAI-1 (Idell et al., 1991). The decrement in fibrinolytic capacity associated with these disorders underlies the deposition of fibrin between the visceral and parietal pleural surfaces in these conditions. While
inhibition of fibrin clearance is implicated in the development of accelerated pleural
organization and fibrosis, the alterations are also observed in exudative effusions that do not locate. It is, therefore, likely that other alterations act in synergy to promote intrapleural organization in conditions such as evolving pleurodesis and complicated parapneumonic effusions or organizing haemothoraces. For example, cytokines implicated in the pathogenesis of pleural injury, including TNF-α, can upregulate uPAR expression at the surface of cell types involved in pleural injury and thereby influence local remodeling of transitional fibrin. Exposure of mesothelial cells to asbestos can also influence uPAR expression (Perkins et al., 1999). The control of the fibrinolytic system may occur at multiple regulatory levels, such as at gene transcription and post-transcriptionally, with respect to mRNA stability and
translational control.
Over years, a large number of locally produced peptides have been identified and shown to play a role in the maintenance and regulation of cell and tissue function. These peptides,
including cytokines and polypeptide growth factors, are mediators produced by one cell that have the capacity to govern either their own cellular function (autocrine) or the function of other cells (paracrine). Cytokines and growth factors have been implicated in the regulation of intracellular and intercellular signaling events responsible for the maintenance of normal healthy pleural tissue and in the induction and progression of pathogenic states of pleural disease. The involvement of these proteins in the processes of pleural injury, repair and fibrosis is the subject of ongoing investigations. Mesothelial cells initiate the healing process by attracting inflammatory cells, fibroblasts, and other cells to the site of injury. These cells, in turn, release factors which regulate cell proliferation, migration, differentiation, and ECM production (Table 1-3).
1-3. Pulmonary fibrosis
Pulmonary fibrosis is a lung disease that is refractory to treatment and carries a high mortality rate. It includes a heterogeneous group of lung disorders characterized by the progressive and irreversible destruction of lung architecture caused by scar formation that ultimately leads to organ malfunction, disruption of gas exchange, and death from respiratory failure (Wynn, 2011).
Idiopathic pulmonary fibrosis (IPF) may induce by chronic inflammation of the alveolar lung injury or alveolar epithelial cell damaged continuously to become the structural
remodeling and lung tissue fibrosis. Pathology and lung fibrosis related diseases about the type of sarcoma, radiation pneumonitis, chronic obstructive pulmonary disease (COPD) and ARDS. Generally, pulmonary fibrosis is the final phase of many severe lung injuries , characterized by lung cells damaged, the invasion of immune cells, releasing the promote fibrosis factor, fibroblast proliferation, differentiation and activation, and increased extracellular matrix production (Kuwano et al., 2001).
Repair of damaged tissues is a fundamental biological mechanism that allows the ordered replacement of dead or damaged cells after injury, a process critically important for survival (Wynn, 2007). However, if this process becomes dysregulated, it can lead to the development of a permanent fibrotic “scar,” which is characterized by the excess
accumulation of ECM components (e.g., hyaluronic acid, fibronectin, proteoglycans, and interstitial collagens) at the site of tissue injury.
Although the relative importance of inflammation in the progression of pulmonary fibrosis has been debated, it was induced to become a strong inflammatory response by many forms of the disease (Crystal et al., 2002). Even if some types of pulmonary fibrosis maintain a significant inflammatory component throughout the course of the disease, other forms like IPF are often characterized as exhibiting highly progressive fibrotic disease in the absence of detectable inflammation (Thannickal et al., 2004).
The fibrosis inducing side effect associated with bleomycin therapy observer in the human population has been exploited by researchers attempting to develop murine models of human interstitial pneumonias (Chua et al., 2005). Nevertheless, many important advances have been generated from rodent models, which have been dominated by transgenic and knockout mice that display either enhanced or decreased susceptibility to pulmonary fibrosis.
A variety of experimental models have been generated to study the mechanisms of pulmonary fibrosis (Moore et al., 2008). However, the mouse bleomycin model has garnered the most attention, perhaps because it is a well- characterized and clinically relevant model of pulmonary fibrosis. Nevertheless, although it successfully models the early proinflammatory stages of the disease, because of the transient nature of the bleomycin response and the
reversibility of the fibrosis, it is unclear whether this model can truly replicate the chronic and progressive forms of the disease seen in humans.
1-4. Renin angiotensin system
The renin-angiotensin system (RAS) is a classically hormonal system consists of endocrine, paracrine and intracrine system (Fyhrquist et al., 2008). The manly function of RAS involved the balance of salt and water, blood pressure and natriuresis, it also plays an important local role to regulate regional blood flow and nutrition in several target organs such as heart (Guimarães et al., 2012), blood vessels (Khakoo et al., 2008), and lungs (Imai et al., 2008; Shrikrishna et al., 2012; Wong et al., 2012).
Recently, a local tissue-based RAS has also been described and appears to play a key role in injury/repair responses (Marshall, 2003; Wösten-van et al., 2008). The expression of RAS components and the elevation of angiotensin converting enzyme (ACE) in a number of lung diseases suggest the existence of a pulmonary RAS and that Ang II could mediate, at least in part, the response to lung injury. The central role of ACE is to generate Ang II from
Ang I. Ang II is a key effector peptide of RAS that causes vasoconstriction and exerts
multiple biological functions. Ang II can also cause the development of interstitial fibrosis via physiological regulation of MMPs expression and/or activation (Chen et al., 2008).
The angiotensin pathway is involved in the pathogenesis of many fibrotic diseases and in idiopathic pulmonary fibrosis an innate overexpression of Ang II, a potent TGF-β1 inductor has been demonstrated. Ang II therapeutic blockade could be therefore a promising
antifibrotic approach (Jeffery et al., 2001; Mandegara et al., 2004; Antoniu, 2008). The molecular mechanism may be one of the parameters of pulmonary fibrosis. There are a lot of unknown about the pathway of Ang II/AT1R axis duration of pulmonary fibrosis (Budinger, 2011). Ang II has another signaling pathway to active Smads in cell (Rodríguez-Vita et al., 2005; Yang et al., 2009). Moreover, ROS (Reactive Oxygen Species) may be induced via Ang II to promote tissue fibrosis (Okada et al., 2009).
1-5. Angiotensin converting enzyme II
There is abundant expression of RAS components in lung, including ACE and ACE2. ACE2 was cloned as a first homolog of human ACE and mapped to the X chromosome by two independent research groups in 2000 (Donoghue et al., 2000; Tipnis et al., 2000). The ACE2 is an 805 amino acid zinc-metallopeptidase and type I integral membrane glycoprotein encoded from 18 exons with a molecular weight of approximately 120 kDa (Turner et al., 2002), it is predominantly observed in the heart, kidneys and testes (Tipnis et al., 2000) such as cardiomyocytes (Gallagher et al., 2008), luminal surface of tubular epithelial cells
(Donoghue et al., 2000; Tipnis et al., 2000) and adult Leydig cells (Douglas et al., 2004). In addition, ACE2 also had been confined at a lower level in a wide variety of tissues including the brain (Xia et al., 2008; Xu et al., 2011), liver (Lambert et al., 2008; Pereira et al., 2009) and lung (Kuba et al., 2006; Imai et al., 2008).
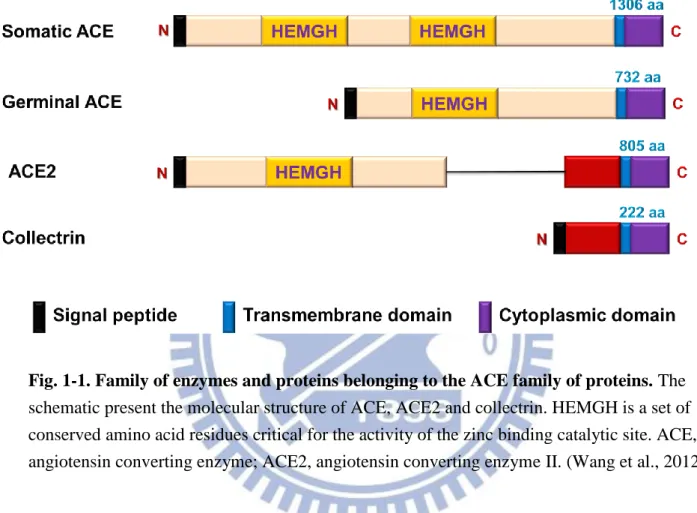
In molecular structure, the human ace2 gene comprise 18 exons, the first 12 exons of
ace2 is similar to the first 11 exons of the ace gene. Moreover, the zinc-binding motif
(HEMGH) of ACE2 is located within exon 9, compared to exon 8 of the ace gene (Donoghue et al., 2000; Tipnis et al., 2000). As like ACE, ACE2 has two domains of the amino-terminal catalytic domain and the carboxy terminal domain, shares 42% sequence identity and 61% sequence similarity with the catalytic-domain of ACE (Donoghue et al., 2000; Tipnis et al.,
2000; Douglas et al., 2004). Unlike somatic ACE, ACE2 only contains a single catalytic site with the prototypical zinc-binding HEMGH motif, and functions as a carboxymonopeptidase removing a single C-terminal residue from peptide substrates whereas ACE acts as a
carboxydipeptidase (peptidyldipeptidase), removing a C-terminal dipeptide (Clarke et al., 2012). In addition, the carboxy-terminal domain of ACE2 shows 48% sequence identity with collectrin, which was a non-catalytic protein that has a critical role in amino acid absorption in the kidney (Danilczyk et al., 2006; Malakauskas et al., 2007), pancreatic beta-cell
proliferation (Akpinar et al., 2005) and insulin exocytosis (Fukui et al., 2005) (Fig. 1-1). ACE2, a close homologue of ACE, functions as a negative regulator of the angiotensin system and was identified as a key receptor for severe acute respiratory syndrome coronavirus infections (Kuba et al., 2005; Imai et al., 2010). ACE2 reduces the generation of Ang II by catalyzing the conversion of Ang I to Ang 1-9 and facilitating hydrolysis of Ang II to Ang 1-7. Ang 1-7 has been recognized as a potential endogenous inhibitor of the classical RAS cascade (Mercure et al., 2008) (Fig. 1-2). Hence, the ACE2/Ang 1-7 axis may be an important
negative modulator of Ang II bioactivity, counteracting the effects of ACE in determining net tissue Ang II levels. Abnormally elevated ACE combined with decreased ACE2 expression may be involved in fibrotic processes in vitro and in vivo, and the mechanism may involve expression and activation of specific MMPs (Pan et al., 2008).
In recent years, ACE2/Ang 1-7 / Mas axis have been explored in pulmonary hypertension disease (Ferreira et al., 2009; Yamazato et al., 2009). Moreover, the first research was found that ACE2 associated with pulmonary fibrosis directly in 2008 at the AJP-Lung Cell Mol Physiol (Li et al., 2008). Li et al have foundACE2 mRNA, protein and enzymatic activity were severely decreased in human IPF and in both rat and mouse models of experimental lung fibrosis induced by bleomycin. In addition, the mice with lung fibrosis were given recombinant ACE2 protein and reduced lung fibrosis. (Shenoy et al., 2010). ACE2 KO mice do not have lung abnormalities when compared with their wild-type littermates (Turner et al., 2002). However, it was recently shown that loss of ACE2 expression precipitates severe acute lung failure. Moreover, injection of recombinant human ACE2 attenuates acute lung failure in Ace2 KO as well as in wild-type mice (Fourrier et al., 1985).
With respect to the possible role of ACE2 in the lung in relation to acute lung injury in particular, the relationship to the ACE2 expression at the alveolar capillary interface is of interest. It is tempting to speculate that increased ACE2 may play a role in reducing the initial
leakage over the alveolar capillary interface. This would then slow down the vicious circle that often occurs after a damaging effect to this interface and leads to the clinical pathological picture of diffuse alveolar damage with intra-alveolar oedema and fibrin deposits. These results support a critical role for the intrapulmonary RAS in the pathogenesis of acute lung injury and show that ACE2 is a key molecule involved in the development and progression of acute lung failure (Inge et al., 2007).
1-6. Matrix metalloproteinase
The tissue fibrosis is caused by excessive accumulation of ECM components, especially types I and III collagen, in various pathological manifestation diseases (LeRoy et al., 1974; Uitto et al., 1979). The balance of ECM components is maintained by MMPs and TIMPs (Clutterbuck et al., 2009). MMPs are essential components for various normal biological processes such as embryonic development, morphogenesis, reproduction tissue resorption and remodeling (Szarvas et al., 2011), they also implicated in a number of key pathologic
processes including inflammation, fibrosis, arthritis, pulmonary diseases and cancer
(Amălinei et al., 2010), because of the abnormally ECM deposition that imbalance MMPs and the TIMPs caused.
Compared to the cardiovascular disease, it reported less about the balance of MMPs and TIMPs activity in respiratory diseases less. However, the research results showed MMPs and TIMPs in some respiratory disease in the past (Gaggar et al., 2011). Researchers can measure higher activity of MMP-3 (Yamashita et al., 2011) and MMP-7 (Fujishima et al., 2010) in patients with pulmonary fibrosis. The results of experimental animals pulmonary fibrosis induced by bleomycin, TGF-ß1 or radiation irradiation, showed abnormal activity of MMP-3 (Yamashita et al., 2011), MMP-12 (Kang et al., 2007.), MMP-13 (Flechsig et al., 2010) related to the course of pulmonary fibrosis. Furthermore, it was also detected the abnormal performance of TIMPs or the imbalance activity of MMPs/TIMPs in experimental animal pulmonary fibrosis (Manoury et al., 2007). Interestingly, a lot of research about MMPs gelatinase (MMP-2 and MMP-9) has been reported in cardiovascular research but less in the lung tissue and pulmonary fibrosis.
MMPs had been discovered by Gross and Lapiere in 1962, are a group of Zn2+ and calcium dependent endopeptidases of common significant peptide chain sections, however
glycosylated in different amount and different locations (Sternlicht et al., 2001). MMPs comprise a large family of protease and share several similarities in terms of their structure, regulation and function (Nagase et al., 1999; Bode et al., 2001). Up to now, 28 types of MMPs have been identified, and they are further divided into six major subfamilies based on structure and substrate specificity, including collagenases, gelatinases, stromelysins,
matrilysins, membrane-type MMPs and other MMPs (Table 1-4; Vargová et al., 2012). The major structure of all MMPs consists of three domains: N-terminal hydrophobic signal sequence, a propeptide domain region and a catalytic domain (Nagase, 1997; Visse and Nagase, 2003). The N-terminal hydrophobic signal sequence decides the MMPs which been released out or maintained in the cell membrane.Furinrecognition motifs are seen in multiple MMP isoforms’ prodomain regions, and type II fibronectin repeats are seen in gelatinase (MMP-2 and -9) catalytic domains. These regions seem to have important implications for the interaction of the MMPs with its substrate. The molecular structure of different type of MMPs was present in Fig. 1-3.
MMPs have the combined ability to degrade essentially all connective tissue components. MMPs are involved in many normal homeostatic mechanisms, the expression and activity is commonly elevated in conditions where inflammation and tissue remodeling/repair are operative (McCaw et al., 2007).
In addition to regulation of activation, there are numerous inhibitors of MMPs.
Although TIMPs are often thought of as the predominant group of inhibitors for MMPs, they are really the most specific endogenous MMP inhibitors. The majority of MMP-related inhibition in vivo occurs through relatively nonspecific MMP inhibitors such as α2 macroglobulin (McCaw et al., 2007). TIMPs are a group of four small (20–24 kDa)
MMP-specific inhibitors that bind to MMPs in a 1:1 stoichiometric relationship (Zucker et al., 1998). All four mammalian TIMPs have many basic similarities, but they exhibit distinctively structural features, biochemical properties and expression patterns (Table 1-5). Animal studies, in addition to human studies in adults, support a role for MMPs and an imbalance between MMPs and TIMPs in the pathogenesis of several well-recognised pulmonary disorders, such as COPD (Finlay et al., 1997) and asthma (Tanaka et al., 2000).
The ECM decreased by MMPs is mainly impressed by TIMPs, MMPs and TIMPs play a critical role in maintaining the balance between ECM deposition and degradation in
and -4, have been identified (Cruz-Munoz et al., 2008), these TIMPs are secreted by a variety of cell lines such as smooth muscle cells and macrophages. TIMPs also involved in the process of inflammation and fibrosis, their activity is increased by PDGF and TGF-β and either increased or decreased by different ILs (Jones et al., 2003). In addition, evidences suggest that fibrotic livers have high expression of the TIMP-1 and TIMP-2, and thus the combination of low expression of MMPs and high TIMPs may prevent the degradation of the fibrillar collagens.
Recently, abnormal pleural MMP levels, MMP-2 and MMP-9, have been reported, and the association between MMPs and development of pleurisy has been investigated (Iglesias et al., 2005; Oikonomidi S et al., 2010).
1-6-1. Gelatinase A (MMP-2, Type II collagenase)
In 1978, Sellers et al. were first to separate a gelatinase activity from collagenase and stromelysin in the culture medium from rabbit bone (Sellers et al., 1978). A similar enzyme, acting on basement membrane type IV collagen was reported by Liotta et al. (1979) in the following year. Gelatinase was purified from human skin, mouse tumor cells, rabbit bone, and human gingival. The completed sequence of the human MMP-2 except for the signal peptide was reported by Collier et al. (2001). Gelainase A has a triple repeat of fibronectin type I domains inserted in the catalytic domain; this domain participates in binding to the gelatin substrates of the enzyme. MMP-2 is ubiquitously expressed in the cells which comprise the heart and is found in normal cardiomyocytes, as well as in endothelium, vascular smooth muscle cells and fibroblasts (Coker et al., 1999).
MMP-2 (EC 3.4.24.24) is a protease with gelatinolytic activity (hence its alternate name, gelatinase A), which is found to be expressed constitutively in various cell types found in the lungs. This enzyme has a broad spectrum of substrates and is involved in modulating diverse cellular functions, including angiogenesis (Brown et al., 2003), tissue remodelling and
potentiation of inflammatory response (Kumagai et al., 1999). MMP-2 is activated in a unique membrane-type MMP-dependent manner, demonstrating a classic example of MMP-to-MMP activation. MMP-2 is thought to contribute to the pathogenesis of a variety of pulmonary disorders, including COPD, asthma, lung cancer and interstitial pulmonary fibrosis (Chakrabarti et al., 2005).
1-6-2. Gelatinase B (MMP-9, Type V collagenase)
In 1972, Harrwas and Krane detected a gelatinase activity in rheumatoid synovial fluid. Sopata et al. described a gelatinase from human polymorphonuclear leukocytes. Rabbit macrophages produce a very similar enzyme which is able to digest type V collagen (Horwitz et al., 1977). The neutrophil collagenase and gelatinase were resolved in 1980 (Murphy et al., 1980). Purification of MMP-9 protein was achieved in 1983 and sequencing of the cDNA was completed in 1989. An interesting phenomenon, still not fully understood, is the binding of TIMP-1 to proMMP-9 to form a complex (Stetler-Stevenson et al., 1989). Human neutrophil MMP-9 commonly occurs as a complex with lipocalin. A series of papers concerned a 95 kDa protein in plasma that binds to gelatin culminated in the identification of this protein as
MMP-9 (Makowski et al., 1998).
MMP-9 (EC 3.4.24.35), or gelatinase B, is broadly expressed in a variety of cells in the lung, including inflammatory (PMNs, macrophages), epithelial and endothelial cells. This observed redundancy belies important location-specific functions of MMP-9, some of which seem in opposition to other MMP-9 functions. For example, MMP-9 has observed important pro-inflammatory effects by generating PGP (Gaggar et al., 2008) and increasing the
chemokine potency of IL-8 (Van den Steen et al., 2000), but MMP-9 also plays an important role in the regulation of granuloma formation in tuberculosis (Volkman et al., 2010).
Similarly, MMP-9 had been thought to lead to matrix breakdown, but recently it has been suggested that MMP-9 may have a role in matrix repair (Bove et al., 2007; Gagger et al., 2011).
1-7. ACE2 and gelatinase (MMP-2 and MMP-9)
ACE2 is a newly identified component of RAS and plays a negative regulator of Ang II in the RAS. Most of published papers reveal that Ang II could break the balance of MMPs expression in heart and induce heart remodeling (Brassard et al., 2005; Yaghooti et al., 2011), but the relative between ACE2 and MMPs are still unknown. In 2009, Kassiri’s group utilized the left anterior descending artery ligation and ACE2 KO mice to investigate the role of ACE2 in MI (Kassiri et al., 2009). In wild-type mice, ACE2 was persistent increased in the infarct zone of heart, ACE2-deficient was increased interferon-γ, interleukin-6, phosphorylation of
ERK1/2 and JNK1/2 signaling pathways and MMP-2 and MMP-9 levels in response to MI. Loss of ACE2 also associated with the increased expression and phosphorylation of p47phox, Ang II levels, NADPH oxidase activity, and superoxide generation, which could lead to enhanced MMP-mediated degradation of the extracellular matrix in ACE2-deficient
myocardium and eccentric remodeling, increased pathological hypertrophy, and worsening of systolic performance (Bodiga et al., 2011; Patel et al., 2012).
Interesting, ACE2 overexpression inhibited cell growth, MMP-2 and MMP-9 expression, VEGFa production, and ACE and AT1R expression in human lung cancer xenografts and A549 cells in vitro (Feng et al., 2011). These evidences reveal that Ang II mediated AT1R to induce NADPH oxidase and MMP activation, AT1R blocker and Ang 1-7 supplementation inhibited NADPH oxidase and MMP activation (Kassiri et al., 2009; Bodiga et al., 2011). Furthermore, these results suggest that ACE2 serves as a protective mechanism and associated with MMPs expression, especially MMP-2 and MMP-9, but the detail signal pathway still not clear.
Table 1-1. General causes of pleural effusions
Processes occurring Caused References
Increased pleural fluid formation
Increased interstitial fluid in the lung Left ventricular failure, pneumonia, and pulmonary
embolus Wiener et al., 1993 Increased intravascular pressure in pleura Right or left ventricular failure, superior vena caval
syndrome Light et al., 1980 Increased permeability of the capillaries in the
pleura
Pleural inflammation
Increased levels of vascular endothelial growth factor
Cheng et al., 1999; Thickett et al., 1999 Increased pleural fluid protein level
Decreased pleural pressure Lung atelectasis or increased elastic recoil of the lung Eid et al., 1999 Increased fluid in peritoneal cavity Ascites or peritoneal dialysis Kirschner et al., 1998 Disruption of the thoracic duct
Disruption of blood vessels in the thorax
Decreased pleural fluid formation
Obstruction of the lymphatics draining the
parietal pleura Stephen, 2004 Elevation of systemic vascular pressures Superior vena caval syndrome or right ventricular
Table 1-2. Diagnose of transudates and exsudates
Transudate Exsudate Reference
Main causes Increased hydrostatic pressure, ecreased colloid osmotic pressure
Inflammation Marel et al., 1993 Appearance Clear Cloudy
Specific gravity < 1.012 > 1.020 Light et al., 1979 Protein content < 25 g/L > 35 g/L Heffner et al., 1997 fluid protein / serum protein < 0.5 > 0.5 Light et al., 1972 Difference of albumin content
with blood albumin
> 1.2 g/dL < 1.2 g/dL Roth et al., 1990
fluid LDH / upper limit for serum < 0.6 or < 2/3 of normal > 0.6 or > 2/3 of normal Light et al., 1972 Cholesterol content < 45 mg/dL > 45 mg/dL Heffner et al., 1997
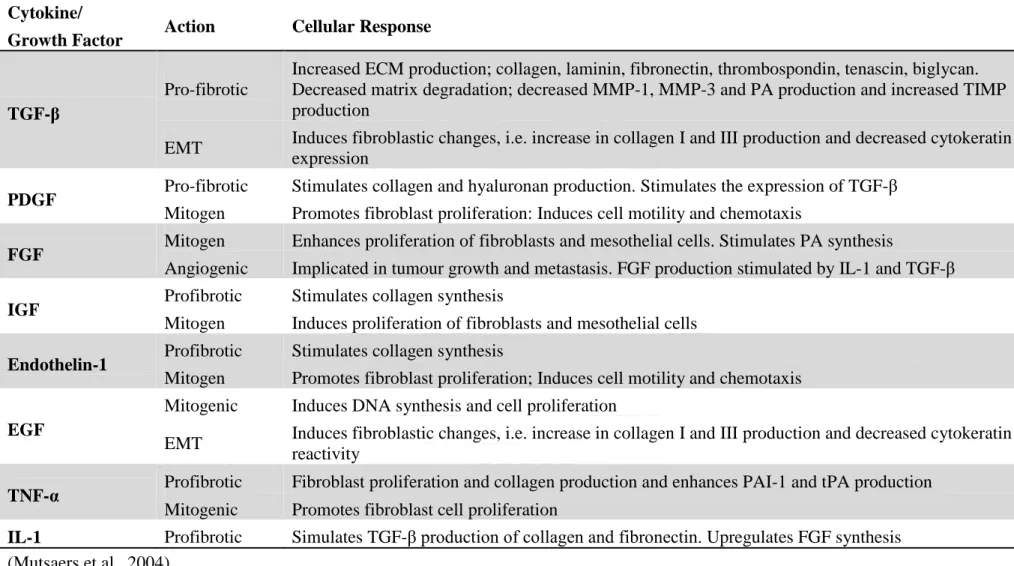
Table 1-3. The cellular responses of cytokines and growth factors implicated in the progression of pleural fibrosis Cytokine/
Growth Factor Action Cellular Response
TGF-β
Pro-fibrotic
Increased ECM production; collagen, laminin, fibronectin, thrombospondin, tenascin, biglycan. Decreased matrix degradation; decreased MMP-1, MMP-3 and PA production and increased TIMP production
EMT Induces fibroblastic changes, i.e. increase in collagen I and III production and decreased cytokeratin expression
PDGF Pro-fibrotic Stimulates collagen and hyaluronan production. Stimulates the expression of TGF-β
Mitogen Promotes fibroblast proliferation: Induces cell motility and chemotaxis
FGF Mitogen Enhances proliferation of fibroblasts and mesothelial cells. Stimulates PA synthesis
Angiogenic Implicated in tumour growth and metastasis. FGF production stimulated by IL-1 and TGF-β
IGF Profibrotic Stimulates collagen synthesis
Mitogen Induces proliferation of fibroblasts and mesothelial cells
Endothelin-1 Profibrotic Stimulates collagen synthesis
Mitogen Promotes fibroblast proliferation; Induces cell motility and chemotaxis
EGF
Mitogenic Induces DNA synthesis and cell proliferation
EMT Induces fibroblastic changes, i.e. increase in collagen I and III production and decreased cytokeratin reactivity
TNF-α Profibrotic Fibroblast proliferation and collagen production and enhances PAI-1 and tPA production
Mitogenic Promotes fibroblast cell proliferation
IL-1 Profibrotic Simulates TGF-β production of collagen and fibronectin. Upregulates FGF synthesis (Mutsaers et al., 2004)
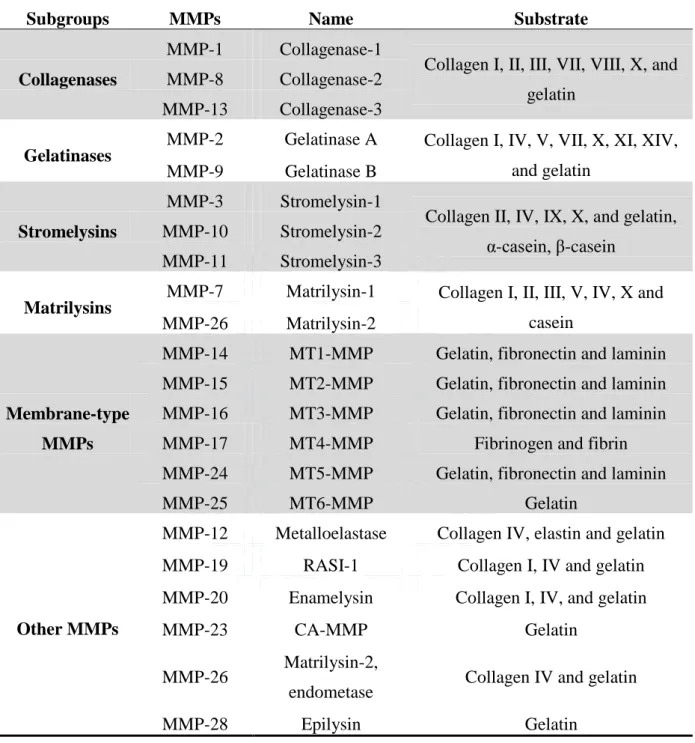
Table 1-4. Types of different matrix metalloproteinases and their substrate specificity Subgroups MMPs Name Substrate Collagenases
MMP-1 Collagenase-1
Collagen I, II, III, VII, VIII, X, and gelatin
MMP-8 Collagenase-2 MMP-13 Collagenase-3
Gelatinases
MMP-2 Gelatinase A Collagen I, IV, V, VII, X, XI, XIV, and gelatin
MMP-9 Gelatinase B
Stromelysins
MMP-3 Stromelysin-1
Collagen II, IV, IX, X, and gelatin, α-casein, β-casein
MMP-10 Stromelysin-2 MMP-11 Stromelysin-3
Matrilysins MMP-7 Matrilysin-1 Collagen I, II, III, V, IV, X and
casein MMP-26 Matrilysin-2
Membrane-type MMPs
MMP-14 MT1-MMP Gelatin, fibronectin and laminin MMP-15 MT2-MMP Gelatin, fibronectin and laminin MMP-16 MT3-MMP Gelatin, fibronectin and laminin MMP-17 MT4-MMP Fibrinogen and fibrin MMP-24 MT5-MMP Gelatin, fibronectin and laminin MMP-25 MT6-MMP Gelatin
Other MMPs
MMP-12 Metalloelastase Collagen IV, elastin and gelatin MMP-19 RASI-1 Collagen I, IV and gelatin MMP-20 Enamelysin Collagen I, IV, and gelatin MMP-23 CA-MMP Gelatin
MMP-26 Matrilysin-2,
endometase Collagen IV and gelatin MMP-28 Epilysin Gelatin
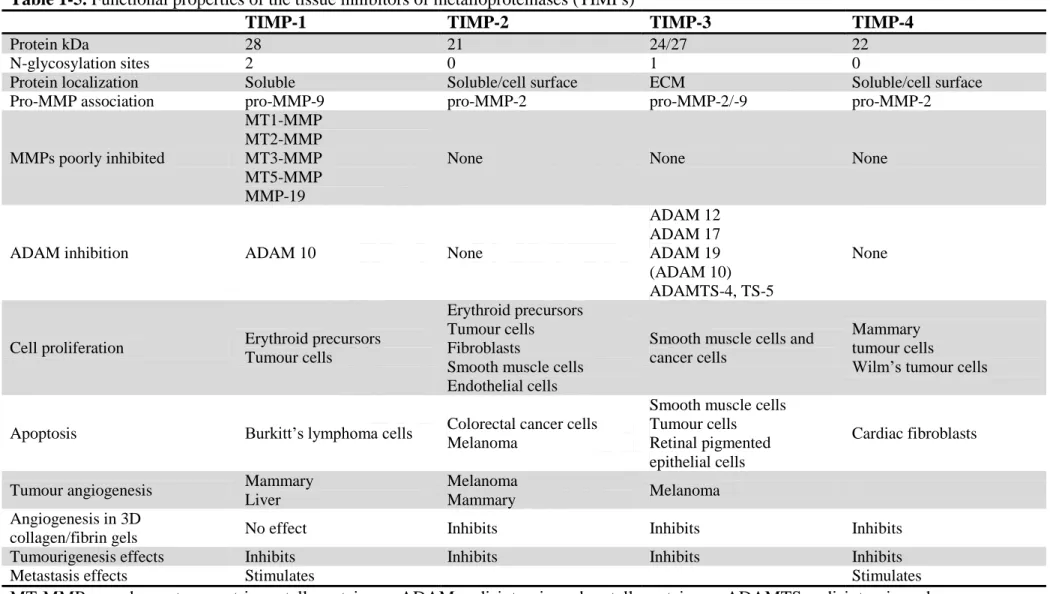
Table 1-5. Functional properties of the tissue inhibitors of metalloproteinases (TIMPs)
TIMP-1 TIMP-2 TIMP-3 TIMP-4
Protein kDa 28 21 24/27 22 N-glycosylation sites 2 0 1 0
Protein localization Soluble Soluble/cell surface ECM Soluble/cell surface Pro-MMP association pro-MMP-9 pro-MMP-2 pro-MMP-2/-9 pro-MMP-2
MMPs poorly inhibited MT1-MMP MT2-MMP MT3-MMP MT5-MMP MMP-19
None None None
ADAM inhibition ADAM 10 None
ADAM 12 ADAM 17 ADAM 19 (ADAM 10) ADAMTS-4, TS-5 None
Cell proliferation Erythroid precursors Tumour cells
Erythroid precursors Tumour cells Fibroblasts
Smooth muscle cells Endothelial cells
Smooth muscle cells and cancer cells
Mammary tumour cells
Wilm’s tumour cells Apoptosis Burkitt’s lymphoma cells Colorectal cancer cells
Melanoma
Smooth muscle cells Tumour cells Retinal pigmented epithelial cells
Cardiac fibroblasts Tumour angiogenesis Mammary
Liver
Melanoma
Mammary Melanoma Angiogenesis in 3D
collagen/fibrin gels No effect Inhibits Inhibits Inhibits Tumourigenesis effects Inhibits Inhibits Inhibits Inhibits Metastasis effects Stimulates Stimulates
MT-MMP, membrane-type matrix metalloproteinase; ADAM, a disintegrin and metalloproteinase; ADAMTS, a disintegrin and metalloproteinase thrombospondin type.( Bokarewa et al., 2005)
Fig. 1-1. Family of enzymes and proteins belonging to the ACE family of proteins. The
schematic present the molecular structure of ACE, ACE2 and collectrin. HEMGH is a set of conserved amino acid residues critical for the activity of the zinc binding catalytic site. ACE, angiotensin converting enzyme; ACE2, angiotensin converting enzyme II. (Wang et al., 2012)
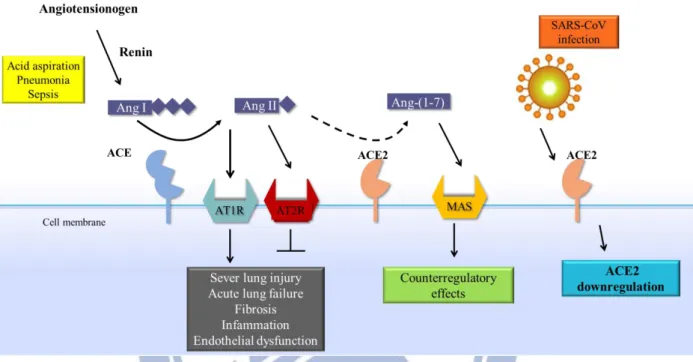
Fig. 1-2. Schematic diagram of the role of the RAS in acute lung failure and the
proposed action of the SARS-coronavirus (SARS-CoV). In acute lung injury, such as acid
aspiration, pneumonia or sepsis, the generation of angiotensin II (Ang II) from angiotensin I (Ang I) is enhanced by angiotensin-converting enzyme (ACE), and Ang II induces ALI through stimulation of the AngII type 1 receptor (AT1R), whereas ACE2 and AngII type 2 receptor (AT2R) negatively regulate this pathway and are protective. On the other hand, SARS-CoV infection is mediated through binding of the SARS–spike protein to ACE2 ,which downregulates the protective molecule ACE2, and thus leads to severe lung injury and acute lung failure. (Imai et al., 2010)
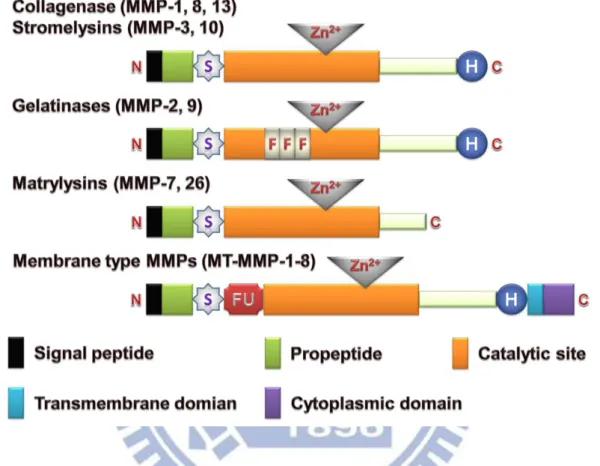
Fig. 1-3. Family of enzymes and proteins belonging to the MMPs family of proteins. The
schematic present the mainly molecular structure of collagenases, stromelysins, gelatinases, matrylysins and membrane type MMPs. S, cysteine switch; FU, intracellular furin-like serine proteinases; F, collagen-binding type II repeats of fibronectin; Zn2+, zinc-binding site; H, hemopexin domain. (Vargová et al., 2012)
II. Research Purpose and Strategy
Remodeling of the lung architecture is a hallmark of many lung diseases, for example, loss of alveolar walls in emphysema, subepithelial fibrosis in asthmatic airways, IPF, cavity formation in tuberculosis, and bronchiectasis in cystic fibrosis. ACE2 is a novel element in RAS and plays a significant role against the harmful effect of Ang II induced. The Ang II has been degraded by ACE2 and form Ang 1-7 to suppress the tissue remodeling that Ang II induced. All of these pathologic changes involve extensive alterations of lung ECM. MMPs or TIMPs have been proposed to be key in causing these changes because of their capacity to cleave structural proteins such as collagens and elastin.
The aim of this study is investigating the regulatory mechanism of ACE/ACE2 and MMPs/TIMPs in pulmonary diseases, and the association between ACE2 and gelatinase (MMP-2 and MMP-9) in the process of lung fibrosis. This study includes two parts. The first part aimed to identify diagnosis based on clinical variables to differentiate TB, Pn and Ad from pleural effusions. The Pleural effusions from 125 patients were processed in our laboratory from Mackay Memorial Hospital among August 2010 to December 2011.We investigated the ratio of ACE/ACE2 and MMPs in the pathogenesis of pleural effusions. The second part aimed to establish the animal model of pulmonary fibrosis, wild-type mice and ACE2 knockout (ACE2+/-, ACE2-/y and ACE2-/-) were received 1 U/kg bleomycin (Rey-Parra et al., 2012) and 20 mg/kg tetracycline (Dryzer et al., 1993) in chest cavity to induce
inflammatory and fibrosis response and sacrificed after 3-, 7- and 28-day, respectively. Through the detection of MMP activity and the level of fibrosis by Histological section, we can understand the ACE2 regulation associated with the balanced expression and activity of MMPs/TIMPs.
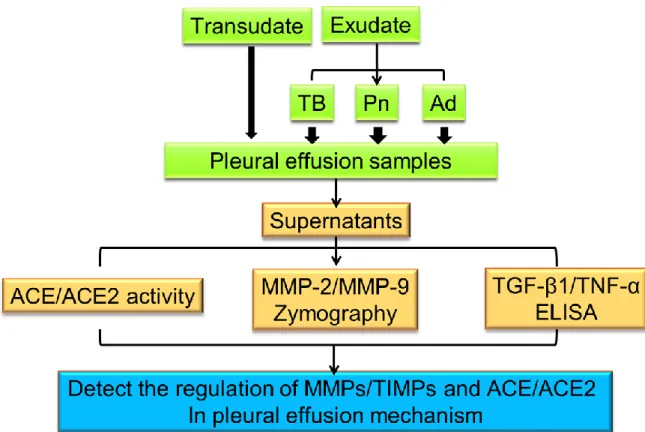
Fig. 2-1. The flowchart of research strategy of Part I. The purpose of this study is to
identify the association of two key enzymes in RAS, ACE and ACE2, with MMP-2 and MMP-9 in the pleural fluid of patients. The Pleural effusions from 125 patients were processed in our laboratory from Mackay Memorial Hospital among August 2010 to December 2011.We also detail discussed whether abnormal RAS, accompanied with high levels of ACE/ACE2, might be a cause of elevated MMP-9 activity in tuberculous pleural effusion.
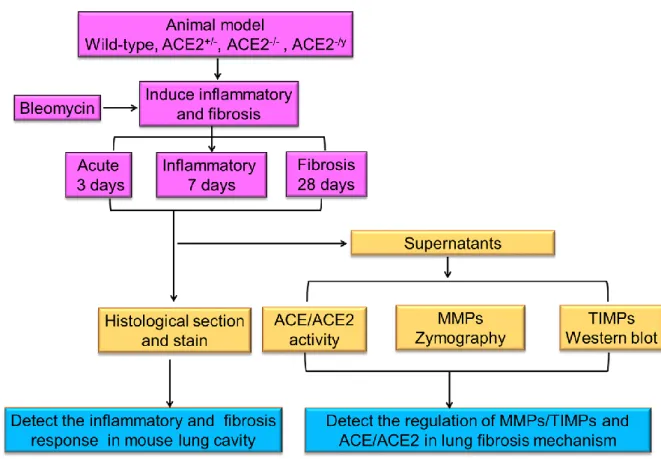
Fig. 2-2. The flowchart of research strategy of Part II. The purpose of this study is to identify the association of two key enzymes in RAS, ACE and ACE2, with MMP-2 and MMP-9 in pulmonary fibrosis. We establish a pulmonary fibrosis animal model and ACE2 knockout (ACE2+/-, ACE2-/y and ACE2-/-) mice by injecting 1 U/kg bleomycin and 20 mg/kg tetracycline, an antibiotic, that is generally used to induce inflammatory and fibrosis response to understand the association between ACE2 and MMPs.
III. Materials and Methods
3-1. Chemicals and reagents
The Monoclonal Anti-b-Actin IgG, glyceraldehyde-3-phosphate-dehydrogenase antibody (AC-15; #A5441), horseradish peroxidase (HRP) labeled secondary antibodies (donkey anti-goat IgG and goat anti-rabbit IgG; #sc2020 and #sc2004), and the rabbit polyclonal IgG, Ikaros antibody (H-100; #sc13039), were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Anti-ACE2 (#ab59351) antibodies were purchased from Abcam (Cambridge, MA, USA). Bleomycin and Tetracycline were purchased from Sigma-Aldrich (St.Louis, MO, USA). The ACE2 overexpression lentiviruses were purchased from Vectorite Biomedica Inc. (Vectorite Biomedica, Taipei, Taiwan). The ACE2 inhibitor, DX600, ACE and ACE2 fluorescence substrate, Mca-APK (Dnp), were purchased from Ana Spec (Fremont, CA, USA). All other reagents were obtained from Sigma-Aldrich.
3-2. Pleural effusion samples collection and analyses
Pleural effusions from 125 patients were processed in our laboratory from August 2010 to December 2011. All pleural effusions were designated as transudates or exudates according to Light’s criteria. Definitive diagnosis of tuberculous, pneumonia, or adenocarcinoma
effusions for the exudate was verified by examining effusion biochemistry, cytology, acid-fast staining, and clinical follow-up. The study was performed with the approval of the
Institutional Review Board of Mackay Memorial Hospital. Informed consent was obtained from all patients.
According to Light’s criteria, pleural effusions were divided into 45 transudative pleural effusions and 80 exudative pleural effusions. The exudative pleural effusions were further divided into tuberculous pleural effusions (20 patients), pneumonia pleural effusions (32 patients), and malignant pleural effusions (28 patients). Fresh pleural fluid was collected in sterile tubes without anticoagulant reagents to prevent the release of gelatinases during platelet activation, and the tubes were immediately centrifuged at 3,000 × g for 30 min to separate the supernatants and cell pellets. The supernatants were aliquoted and stored with the cell pellets at −80°C until further use.
For each pleural effusion sample, the routine pleural analyses included total protein, lactate dehydrogenase (LDH), glucose, and white blood cell (WBC) count. In addition, the activities of ACE, ACE2, ADA , gelatinases (MMP-2 and MMP-9), TGF-β1 and TNF-α in the pleural effusions were measured.
3-3. ADA activity
ADA activity was measured or determined in the pleural fluids with a commercial colorimetric assay kit (Bio Quant, San Diego, CA, USA) (Valdés et al., 1996). Adenosine deaminase (ADA) isoenzyme analysis in pleural effusions: Diagnostic role and relevance to the origin of increased ADA in tuberculous pleurisy. The absorbance of the quinone dye generated in the last step of the ADA reaction series was monitored by absorbance at 550 nm with an ELISA reader (Bio-Rad model 550; Hercules, CA, USA).
3-4. ACE and ACE2 activity assay
ACE and ACE2 activities were assayed with the fluorogenic substrates Mca-YVADAPK and Mca-APK-Dnp (AnaSpec, San Jose, CA, USA), according to Vickers et al. with slight modifications (Vickers et al., 2002). The assay was performed in a microquartz cuvette with 20 μL pleural fluid, 50 μM fluorogenic substrate and protease inhibitor cocktail (1: 200; Sigma-Aldrich) in a final volume of 100 μL in ACE or ACE2 assay buffer. The reaction was followed kinetically for 1 hour using a fluorescence reader at an excitation wavelength of 330 nm and an emission wavelength of 390 nm. All samples were fitted and plotted using Grafit v. 4.0 (Sigma-Aldrich), and enzyme activity was expressed as RFU/hour/mL. Parallel samples were incubated with the above mentioned reaction mixture in the presence of 1 μM captopril (Sigma-Aldrich), a specific ACE inhibitor for determining specific ACE activity, or 1 μM DX600 (AnaSpec), a specific ACE2 inhibitor for determining specific ACE2 activity.
3-5. Gelatin zymography assay
The MMP-2 and -9 activities were detected by gelatin zymography utilized