行政院國家科學委員會專題研究計畫 成果報告
鉛對神經膠原細胞的腫瘤壞死因子α過量表現之調控機制
探討
計畫類別: 個別型計畫
計畫編號: NSC91-2314-B-006-154-
執行期間: 91 年 08 月 01 日至 92 年 07 月 31 日
執行單位: 國立成功大學工業衛生科暨環境醫學研究所
計畫主持人: 劉明毅
報告類型: 精簡報告
處理方式: 本計畫涉及專利或其他智慧財產權,1 年後可公開查詢
中 華 民 國 92 年 9 月 29 日
行政院國家科學委員會補助專題研究計畫 成 果 報
告 □期
中進度
報 告
(計畫名稱)
鉛對神經膠原細胞的腫瘤壞死因子 á 過量表現之調控
計畫類別: 個別型計畫 □ 整合型計畫
計畫編號:NSC 91-2314B-006-154
執行期間:91 年 8 月 1 日至 92 年 7 月 31 日
計畫主持人:劉明毅
共同主持人:
計畫參與人員:
成果報告類型(依經費核定清單規定繳交): 精簡報告 □完整報
告
本成果報告包括以下應繳交之附件:
□赴國外出差或研習心得報告一份
□赴大陸地區出差或研習心得報告一份
□出席國際學術會議心得報告及發表之論文各一份
□國際合作研究計畫國外研究報告書一份
處理方式:除產學合作研究計畫、提升產業技術及人才培育研究
計畫、列管計畫及下列情形者外,得立即公開查詢
附件一
□涉及專利或其他智慧財產權,□一年□二年後可公開
查詢
執行單位:
國立成功大學工業衛生科暨環境醫學研究所中 華 民 國 92 年 9 月 28 日
可供推廣之研發成果資料表
□ 可申請專利 □ 可技術移轉 日期:92 年 9 月 28 日
國科會補助計畫
計畫名稱:鉛對神經膠原細胞的腫瘤壞死因子 á 過量表現之調控機制 探討
計畫主持人:劉明毅
計畫編號: 91-2314-B-006-154- 學門領域:環境醫學與公 共衛生
技術/創作名稱
發明人/創作人
技術說明
中文:鉛是一種具有神經毒性的重金屬,在低劑量且長期的暴露之 下,鉛除了會直接對腦神經細胞造成損害之外,鉛也會藉由傷害神經 膠原細胞導致腦血障蔽的破壞,間接的影響神經系統,造成腦部的發 育與功能上的障礙。本研究目的在探究鉛對神經膠原細胞 TNF-á 基因 的過度表現的調控機制。本研究之工作結果如下: 鉛的暴露會誘發神 經膠原細胞上的腫瘤壞死因子 á (TNF-á)過量產生,並呈現劑量與反 應的關係。B6 小鼠經腹腔注射含 12.5 mg/kg body weight 之醋酸鉛溶 液,於注射後 24 小時犧牲。其腦部組織經冷凍組織切片及免疫染色 法觀察 TNF-á 得知小鼠在暴露鉛的狀態下,腦部之神經膠原細胞及神 經細胞會表現過量之 TNF-á。並同樣之腦組織經 TUNEL 染色後得 知,鉛暴露並不會造成腦部細胞的凋亡。
附件二
英文:Lead (Pb) affects not only neurons but also glial cells, and causes neurological impairment in the brain. We have shown an enhancement of tumor necrosis factor-α (TNF-α) in U-373MG glial cells after exposure of Pb. To determine whether Pb-induced TNF-α triggers glial cell death, the apoptotic response was investigated in glial cells both in vitro and in vivo. In vitro, Pb ranging from 0.1 to 10 µM increased the expression of TNF-α in a dose-dependent manner in U-373MG cells. However, exposure to Pb did not affect apoptosis in U-373MG cells detected by flow cytometric analysis with merocyanine 540 staining and propidium idodine staining. In B6 mice, glial cells in the brain were immunostained positive for TNF-α after an intraperitoneal administration of Pb (12.5 mg/kg). Consistent with the findings in the cell culture system, few apoptotic cells were detected in glial and neurons by terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) in the brain of B6 mice. We conclude that induction of TNF-α alone by Pb does not cause cell apoptosis in cells of glial origin.
可利用之產業
及
可開發之產品
技術特點
推廣及運用的價值
1.每項研發成果請填寫一式二份,一份隨成果報告送繳本會,一份送
貴單位研發成果推廣單位(如技術移轉中心)。
2.本項研發成果若尚未申請專利,請勿揭露可申請專利之主要內容。
3.本表若不敷使用,請自行影印使用。
Enhancement of TNF-α expression does not trigger apoptosis upon
exposur e of glial cells to lead and lipopolysacchar ide
Abstr act
Lead (Pb) and lipopolysaccharide (LPS) affect not only neurons but also glial cells, and cause neurological impairment in the brain. We have previously shown an enhancement of tumor necrosis factor-α (TNF-α) in U-373MG glial cells after exposure of Pb and LPS. To determine whether Pb- or LPS-induced TNF-α triggers glial cell death, the apoptotic response was investigated in glial cells both in vitro and in vivo. In vitro, Pb ranging from 0.1 to 10 µM increased the expression of TNF-α in a dose-dependent manner in U-373MG cells. Similarly, LPS (500 ng/ml) stimulated TNF-α expression in the same cells. A combination of LPS and Pb did not cause higher mRNA TNF-α expression than LPS or Pb alone. However, exposure to Pb and LPS did not affect apoptosis in U-373MG cells detected by flow cytometric analysis with merocyanine 540 staining and propidium idodine staining. In B6 mice, glial cells in the brain were immunostained positive for TNF-α after an intraperitoneal administration of Pb (12.5 mg/kg) or LPS (10 mg/kg). Consistent with the findings in the cell culture system, few apoptotic cells were detected in glial and neurons by terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) in the brain of B6 mice. We conclude that induction of TNF-αalone by Pb and LPS does not cause cell apoptosis in cells of glial origin.
1. Introduction
Both lead (Pb, a toxic metal) and lipopolysaccharide (LPS, an organic biopolymer) affect a wide range of physiological functions in the central nervous system (CNS). Pb is able to trigger neurological injuries and mental impairments even at very low levels of exposure (Murata et al., 1993; Sinha et al., 1993). In the CNS, astroglia accumulate more Pb than neuronal cells do, and Pb might damage neuronal cells indirectly through the action of glial cells (Lindahl et al., 1999). Pb alters the intracellular metal content (Tiffany-Castiglioni et al., 1988), and impairs glial
differentiation and functions in both glial and neuronal cells (Stark et al., 1992; Kern et al., 1993; Zawia and Harry, 1996). In addition, Pb induces the expression of tumor necrosis factor alpha (TNF-α) in various cells of glia origin (Selmaj and Raine, 1988;
Tiffany-Castiglioni, et al., 1988; Liu et al., 2000).
On the other hand, LPS released during gram-negative bacterial infection also triggers severe pathological alterations in the CNS (Baue, 1975). LPS is a powerful inducer of TNF-α (Goutet et al., 2000), and it is involved in septic shock syndrome during the course of bacterial meningitis (Cohen and Abraham, 1999). LPS causes death in tyrosine hydroxylase-positive neurons in mixed neuron/glia cultures (Bronstein et al., 1995; Kim et al., 1995). Also, neuronal apoptosis occurring in neuronal degeneration during inflammation can be mediated by TNF-α secreted by microglia (Kim et al., 2000).
U-373MG, a human glioblastoma, is susceptible to TNF-α induction by Pb (Liu et al., 2000) and LPS (Yang and Yang, 1998), and served as the in vitro target in this study to determine potential TNF-α-associated cell apoptosis. We have explored the TNF-α induction and analyzed whether this overexpressed TNF-α triggers apoptosis by LPS and Pb in glial cells.
2. Mater ials and methods 2.1. Cell culture
Human glioma cell line U-373MG was purchased from the American Type Culture Collection (Rockville, MD). Cells were cultured in Dulbecco modified Eagles' medium (DMEM) supplemented with 20% of fetal bovine serum (FBS), penicillin, and streptomycin (100 U/ml) in a humidified atmosphere of 5% CO2/95%
air at 37°C. To explore the effect of Pb, Pb acetate was added to the culture medium to final concentrations as indicated in the 24 h subculture. For the LPS treatment, 500 ng/ml LPS-O157 (kindly provided by Dr. S. Chen-Szu, National Institute of Health, USA) was used.
2.2. Semi-quantitative reverse transcription-polymerase chain reaction (RT-PCR) RNA was prepared from the RNeasy Total RNA kit (Qiagen, Hilden,
Germany). Total RNA was converted to cDNA using oligo-dT as a primer and StrataScriptTM
H-reverse transcriptase (Stragene, San Diego, CA). The mixture was incubated at 37°C for 1 h and then heated to 90°C for 5 min to inactivate the reverse transcriptase. The generated cDNA was subjected to PCR amplification using specific primers (Table 1) for β-actin or TNF-α gene on a DNA Thermal Cycler (Hybaid Omnigene, Middlesex, UK) (Liu et al., 2000). PCR products were fractioned by agarose electrophoresis and stained with ethidium bromide, then visualized by UV light. β-actin was used as a quantitative control for RT-PCR, because Pb does not affect β-actin gene expression (Zawia and Harry, 1996). All experiments were conducted at least three times.
Table 1. PCR condition
Gene Primer sequence Temperature (°C)
Length (bp)
TNF-α 5'- GAG TGA CAA GCC TGT AGC - 3' 55 363
5'- CCC TTC TCC AGC TGG AAG - 3'
β-actin 5'- AGC GGG AAA TCG TGC GTG - 3' 58 309 5'- CAG GGT ACA TGG TGG TGG TGC C - 3'
2.3. Bioactivity assay for TNF-á
U-373MG cells (24-well plates, 3×105 cells/well, 1 ml) were treated with LPS or Pb acetate for 48 h. The supernatant was harvested and assayed based on its cytotoxicity to L-929 cells in the presence of actinomycin D (Neale et al., 1989).
Briefly, L-929 cells were seeded at a density of 2×104 cells/well into a 96-well plate.
After cultured for 24 h, the medium was replaced with fresh RPMI medium containing actinomycin D at a final concentration of 1 ìg/ml. Cells were incubated further for another 1 h, and then the TNF-á containing supernatants were added in.
The plates were incubated for 24 h. Viable cells were determined MTT (0.5 mg/ml) by measurement of the produced formazan at 570 nm according to the manufacture’s description. Recombinant human TNF-á (15-500 pg/ml) was used as the standard to
calculate the amount of TNF-á in the supernatants.
2.4. Detection of cell apoptosis by merocyanine 540 (MC 540) and propidium idodine (PI) staining assays
MC 540 staining and PI exclusion staining were used to assess the induction of apoptosis by Pb and LPS. After being washed twice with phosphate-buffered saline (PBS) containing 1% bovine serum albumin (BSA), cells were stained directly with MC 540 (Acros, Fairlawn, New Jersey) in the dark for 10 min. Apoptotic cells are susceptible to MC 540 binding due to alteration of the surface membrane (Reid et al., 1996). At late stage of apoptosis, the integrity of the plasma membrane is lost to allow infusion of PI. Pb- or LPS-treated cells were washed with PBS and stained with PI (400 ng/ml) for 30 s in the dark (Yang and Yang, 1998). The fluorescence of stained cells was then analyzed by the FACScan analyzer (Becton Dickinson, Baltimore, MD) with logarithmic amplification.
2.5. Animal preparation
Male B6 mice, weighing 20-25 g, were obtained from and housed in our institution's Laboratory Animal Center. Mice were randomly grouped and kept in a room with a 12 h dark/light cycle and with central air conditioning (25°C, 70%
humidity). Mice were fed with standard food ad libitum and cared for according to appropriate nationally approved guidelines. Mice were intraperitoneally injected once with 1 ml solution containing (a) lead acetate (12.5 mg/kg body weight) (Hsu et al., 1998), (b) LPS (10 mg/kg body weight) (Uno et al., 1996), or (c) PBS as a control.
Mice were sacrificed 24 h post-injection, and their brain tissue was removed and embedded in Tissue-Tek OCT compound (Sakura Finetek, Torrance, CA), frozen in liquid nitrogen, and stored at −80°C until sectioning.
2.6. Immunohistochemistry
Brain sections of 5µm were fixed in 3% paraformaldehyde for 1 min and washed twice in Tris buffered saline (TBS) for 5 min each time. Hydrogen peroxide (0.3%) in TBS was applied for 30 min at room temperature to quench endogenous peroxidase. The non-specific binding was blocked using 3% BSA (Sigma, St. Louis,
MO) in TBS for 30 min at 37°C. Sections were incubated overnight at 4°C in the same solution containing a 1:200 dilution of rabbit polyclonal anti-murine TNF-α antibody (Endogene, Woburn, Massachusetts). Antibody detection was carried out using peroxidase conjugated goat anti-rabbit secondary antibody and
diaminobenzidine tetrahydrochloride solution (DAB) was used as a peroxidase substrate.
2.7. Detection of cell apoptosis by TUNEL
Apoptosis in the brain was detected with the ApopTag Detection Kit (Intergen, Purchase, New York). Brain section specimens were washed with PBS and soaked to quench the endogenous peroxidase in a mixture containing 3% H2O2 solution and then rinsed with PBS once more. To add the residues of the digoxigenin-nucleotide to the 3'-OH end of the DNA catalytically, terminal deoxynucleotidyl transferase (TdT) with reaction buffer were used and the specimens mounted for 1 h. The reaction was terminated by applying a stop solution for 30 min at 37°C and the specimens were washed with PBS. Before rinsing with PBS, the anti-digoxigenin-peroxidase solution was applied for 15 min to the sections to join the chromogenic substrates to the complex between the DNA and the digoxigenin-nucleotide. To detect positive staining, specimens were soaked in the DAB and counterstained with hematoxylin.
2.8. Statistical analysis
Results were analyzed by one-way ANOVA test, and then post-hoc comparisons were done with Dunnett's test (for cell growth and viability). Differences with a p-value < 0.05 were judged significant. All experiments were repeated at least three times.
3. Results
3.1. Pb and LPS induced TNF-α mRNA expressions in U-373MG cells
After treatment with 0.1-10µM of lead acetate, the level of TNF-αtranscripts detected by RT-PCR was elevated in U-373MG cells (Fig. 1: lanes 3, 5, and 7). The
induction of TNF-α by Pb showed a dose-dependent tendency (the higher Pb the concentration, the higher the level of induction). LPS (500 ng/ml) alone also strongly enhanced the transcription of TNF-α (Fig. 1: lane 2) and that was comparable to the effect of LPS plus Pb. This implied that LPS and Pb did not synergistically induce TNF-α(Fig. 1: lanes 4, 6, and 8).
Fig. 1. RT-PCR analysis on TNF-α genes in U-373MG glioma cells treated with lead alone and lead plus LPS. Lane 1: control, lane 2: LPS (500 ng/ml) alone, lane 3: 0.1 µM Pb, lane 4: LPS + 0.1 µM Pb, lane 5: 1.0 µM Pb, lane 6:
LPS + 1.0 µM Pb, lane 7: 10 µM Pb, lane 8: LPS + 10 µM Pb.
3.2. Effects of Pb and LPS on cell death on U-373MG cells
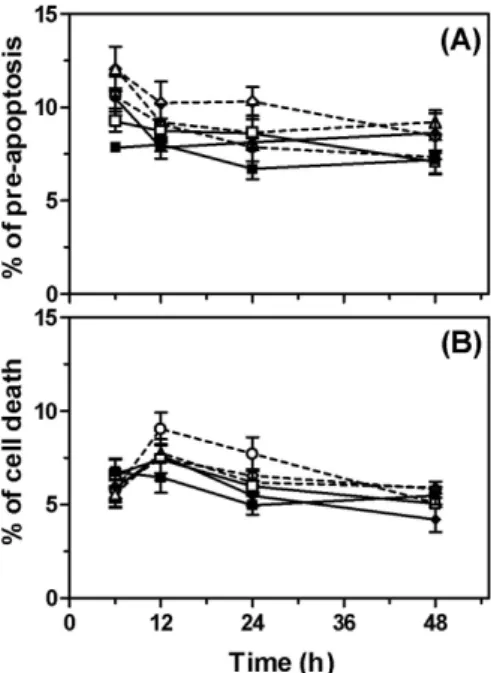
To analyze apoptosis, we measured the integrality of the cell membrane with MC 540- or PI-staining by flow cytometry. By MC 540-staining, the number of
pre-apoptotic cells in cultures treated with LPS for 6, 12, 24, or 48 h was similar to the number in cultures treated with Pb-treated cells (Fig. 2A). By PI-staining, Pb and LPS had no significant effect on apoptosis in U-373MG cells (Fig. 2B).
Fig. 2. Apoptosis detected by (A) MC 540-staining and (B) PI-staining in U-373MG cells. 0 µM Pb ( ■ ), LPS (500 ng/ml) alone ( □ ), 10 µM Pb ( ◆ ), 0.1 µM Pb + LPS ( △ ), 1.0 µM Pb + LPS ( ○ ), 10 µM Pb + LPS ( ◇ ).
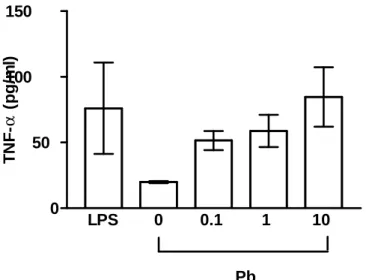
3.3 TNF-á bioactivity
U-373MG were stimulated with LPS (500 ng/ml) and Pb acetate (0.1-10 ìM) for 48 h to evoke TNF-α synthesis. TNF-α was determined by the cytotoxic activity on L-929 cells. After LPS stimulation, the TNF-á secretion was significantly elevated to 76 pg/ml compared to negative control (20 pg/ml). Similarly, Pb induced also the secretion of TNF-á in a dose-dependent manner. Among TNF-á secretions, 51 pg/ml, 58 pg/ml, and 84 pg/ml were detected in the culture supernatant when U-373MG was exposed to Pb of 0.1 ìM, 1 ìM, and 10 ìM, respectively.
LPS 0 0.1 1 10 0
50 100 150
Pb TNF-α(pg/ml)aa a
Fig. 3. L-929 bioassay of TNF-á bioactivity. LPS 500 ng/ml, 0: negative control, 0.1: Pb acetate 0.1 ìM, 1: Pb acetate 1 ìM, 10: Pb acetate 10 ìM
3.4. Effects of Pb and LPS on TNF-α expression in the B6 mouse brain
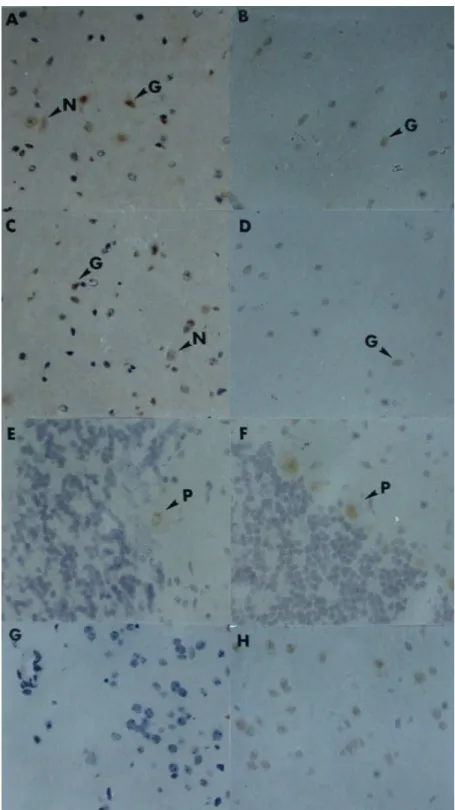
To investigate the induction of TNF-α in vivo, B6 mice were injected with Pb or LPS, and the brain section was analyzed by immunohistochemistry. Generally, TNF-α was not expressed in the brains of control mice (negative controls) (Fig. 4G) but was expressed in ischemic brains (positive controls) (Fig. 4H). In both Pb- and LPS-treated mice, TNF-α was detected on glial and neuronal cells in the cerebral cortex and subcortical white matter (Figs. 4A, 4B, 4C, and 4D), and on Purking cells in the cerebellum (Figs. 4E and 4F).
Fig. 4. Immunocytochemical analysis of the TNF-α expression in the B6
mouse brain.
LPS: A (cerebral cortex), C (subcortical white matter), E (cerebellum), Pb: B (cerebral cortex), D (subcortical white matter), F (cerebellum), Normal control: G, Ischemic brain for positive control: H. (G: glial cell, N: neuronal cell, P: Purking cell) (×400 in all fields)
3.5. Effects of Pb and LPS on cell apoptosis in the B6 mouse brain
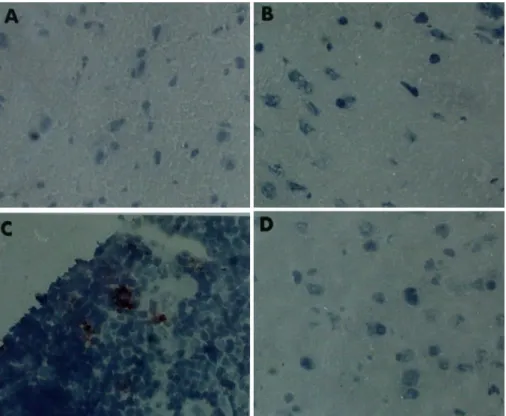
Apoptotic cells in brain tissue were stained by TUNEL assay. Although TUNEL-positive cells were observed in the thymuses of control mice (Fig. 5C), no TUNEL-positive cells were observed in the brains of the mice (Fig. 5D). In both Pb- and LPS-treated mice, no apoptotic cells were found in brain areas, including the cerebral cortex (Figs. 5A and 5B), subcortical white matter, and cerebellum (data not shown).
Fig. 5.TUNEL analysis of apoptosis in the B6 mouse brain. LPS: A (cerebral cortex), Pb: B (cerebral cortex), Mock control: C (thymus), Mock control: D (cerebral cortex)
4. Discussion
Glial cells are the major TNF-α-secreting cells in the neuronal system, and glia can exacerbate neuronal toxicity by the release of TNF-α (Viviani et al., 1998). When
neuronal injury such as ischemia or neurotoxicant damage occurs, TNF-αis released by glial cells (Dunn, 1992; Lee et al., 2000). Consistent with previous studies (Breder et al., 1994; Sawade et al., 1989; Liu et al., 2000), Pb stimulated the expression of TNF-α in cultured glial cells, increased both in mRNA detected by RT-PCR and in its bioactivity determined by toxicity on L-929 cells. TNF-α was also expressed in glial cells in the brains of B6 mice treated with Pb, and this in vivo finding was similar to results obtained from human glioma U-373MG in vitro. Notably, although TNF-α was expressed in neuronal cells in vivo after the injection of Pb or LPS, it was not found in cultured neuroblastomas (data not shown). This suggests that the production of TNF-α in neurons might require glial cells.
Traumatically induced damage in neurons and oligodendrocytes has been attributed to an excess production of TNF-α (Lee et al., 2000), and several pieces of evidence reveal that the excess amount of TNF-α is released by glial cells (Sawada et al., 1989; Kim et al., 2000). Since Pb and LPS stimulated the expression of TNF-α in U-373MG, an autocrine cytokine action was expected to modify cell growth and death. However, treatment with Pb, LPS, or both combined did not suppress the growth of U-373MG cells (data not shown). LPS administered directly to the brain has been shown to induce neuronal degeneration (Mouihate and Pittman, 1998;
Matsuoka et al., 1999), but TNF-α was able to prevent human peripheral blood monocytes from apoptosis (Mangan et al., 1991). In our system, intraperitoneally administered LPS consistently did not induce apoptosis in either neurons or glia.
Although peripheral administration of LPS induced the production of TNF-α in the brain, based on our data, it did not obviously damage cells. Besides neurotoxicity, the morphological change of endothelial cells in blood-brain barrier (BBB) or permeability increase of BBB are also related to excess production of TNF-á (Duchini et al., 1996; Tsao et al., 2001). Thus, the role of TNF-á in LPS and Pb induced neuronal damage may attribute to other pathological changes instead of a directly cell toxicity to neuronal or to glial cells. Because, in the present study, apoptosis was not induced by Pb and LPS through i.p. injection (Mouihate and Pittman, 1998), it seems reasonable to assume that there is at least one other factor required for its induction in
glial cells, both in vitro and in vivo. We hypothesize that cell type and growth condition are good candidates for this other factor. For example, the combination of interferon-γand LPS strongly induced neuronal apoptosis, and the apoptotic effect is mediated by nitric oxide released by microglia (Matsuoka et al.,1999). Further studies will have to be done to elucidate this point.
5. References
Baue, A.E., 1975. Arch. Surg. 110, 779-781.
Breder, C.D., Hazuka, C., Ghayur, T., Klug, C., Huginin, M., Yasuda, K., Teng, M.
and Saper, C.B., 1994. Proc. Natl. Acad. Sci. USA 91, 11393-11397.
Bronstein, D.M., Perez-Otano, I., Sun, V., Mullis Sawin, S.B., Chan, J., Wu, G.C., Hudson, P.M., Kong, L.Y., Hong, J.S. and McMillian, M.K., 1995. Brain Res. 704, 112-116.
Cohen, J. and Abraham, E., 1999. J. Infect. Dis. 180, 116-121.
Duchini, A., Govindarajan, S., Santucci, M., Zampi, G. and Hofman, F.M, 1996. J.
Invest. Med. 44. 474-482.
Dunn, A.J., 1992. Brain Res. Bull. 29, 807-812.
Goutet, M., Ban, M. and Binet, S., 2000. Toxicology 145, 15-26.
Hsu, P.C., Hsu, C.C., Liu, M.Y., Chen, L.Y. and Guo, Y.L., 1998. J. Toxicol. Environ.
Health 55, 45-64.
Kern, M., Audesirk, T. and Audesirk, G., 1993. Neurotoxicology 14, 319-327.
Kim, Y.S., Kennedy, S. and Tauber, M.G., 1995. J. Inf. Dis. 171, 1363-1368.
Kim, W.G., Mohney, R.P., Wilson, B., Jeohn, G.H., Liu, B. and Hong, J.S., 2000. J.
Neurosci. 20, 6309-6316.
Lee, Y.B., Yune, T.Y., Baik, S.Y., Shin, Y.H., Du, S., Rhim, H., Lee, E.B., Kim, Y.C., Shin, M.L., Markelonis, G.J. and Oh, T.H., 2000. Exp. Neurol. 166, 190-195.
Lindahl, L.S., Bird, L., Legare, M.E., Mikeska, G., Bratton, G.R. and Tiffany-Castiglioni, E., 1999. Toxicol. Sci. 50, 236-243.
Liu, M.Y., Hsieh, W.C. and Yang, B.C., 2000. Toxicology 147, 59-64.
Mangan, D.F., Welch, G.R. and Wahl, S.M., 1991. J. Immunol. 146, 1541-1546.
Matsuoka, Y., Kitamura, Y., Takahashi, H., Tooyama, I., Kimura, H., Gebicke-Haerter, P.J., Nomura, Y. and Taniguchi, T., 1999. Neurochem. Intern.
34, 91-99.
Mouihate, A. and Pittman, Q.J., 1998. Brain Res. 805, 95-103.
Murata, K., Araki, S., Yokoyama, K., Uchida, E. and Fujimura, Y., 1993. Environ.
Res. 61, 323-336.
Neale, M. L., Williams, B. D., and Matthews, N., 1989. Br. J. Rheumatol. 28:
104-148.
Reid, S., Cross, R. and Snow, E.C., 1996. J. Immunol. Methods 192, 43-54.
Sawada, M., Kondo, N., Suzumura, A. and Marunouchi, T., 1989. Brain Res. 491, 394-397.
Selmaj, K.W. and Raine, C.S., 1988. Ann. Neurol. 23, 339-346.
Sinha, S.P., Shelly, Sharma, V., Meenakshi, Srivastava, S. and Srivastava, M.M., 1993. Bull. Environ. Contam. Toxicol. 51, 490-493.
Stark, M., Wolff, J.E. and Korbmacher, A., 1992. Neurotoxicol. Teratol. 14, 247-252.
Tiffany-Castiglioni, E., Garcia, D.M., Wu, J.N., Zmudzki, J. and Bratton, G.R., 1988.
J. Toxicol. Environ. Health 23, 267-279.
Tsao, N., Hsu, H.P., Wu, C.M., Liu, C.C. and Lei. H.Y., 2001. J. Med. Microbio. 50, 812-821.
Uno, T., Tanaka, H. and Nagai, H., 1996. Prostaglandins 52, 447-461.
Viviani, B., Corsini, E., Galli, C.L. and Marinovich, M., 1998. Toxicol. Appl.
Pharmacol. 150, 271-276.
Yang, B.C. and Yang, T.L., 1998. J. Microbiol. Immunol. Infect. 31, 95-100.
Zawia, N.H. and Harry, G.J., 1996. Toxicol. Appl. Pharmacol. 138, 43-47.