Thermodynamics of uniaxial phase transition: Ab initio study of the diamond-to-

-tin transition in
Si and Ge
C. Cheng,*W. H. Huang,†and H. J. Li
Department of Physics, National Cheng Kung University, Tainan, Taiwan, Republic of China
共Received 23 June 2000; revised manuscript received 10 November 2000; published 27 March 2001兲 The diamond-to--tin phase transitions of Si and Ge under uniaxial compression are investigated with ab
initio calculations. A thermodynamic analysis is used to identify the transition pressures at uniaxial
compres-sion. The transition pressures were found to be pronouncedly lower than those at hydrostatic compression, i.e., 3.9 and 2.5 GPa compared to 11.4 and 9.5 GPa for Si and Ge, respectively. The different contributions to the phase transition are analyzed numerically and discussed.
DOI: 10.1103/PhysRevB.63.153202 PACS number共s兲: 64.70.Kb, 61.50.Ks, 05.70.Ce
Previous experimental studies have shown1,2that nonhy-drostatic compression effectively reduces the phase-transition pressure of diamond structured Si to the metallic
-tin phase. Theoretically, hydrostatic phase-transition pres-sures have been successfully calculated by using the en-thalpy function H⫽E⫹PV with ab initially evaluated inter-nal energy E(V) and pressure P (⫽⫺E/V) for several
systems.3 However, a similar ab initio approach has never been performed to determine the transition pressure under nonhydrostatic conditions as the enthalpy function is no longer the appropriate quantity for determining the relative stability between phases. The presently available theoretical studies of uniaxial compression were carried out by isobaric molecular dynamics based on an empirical interatomic potential.4
In this paper, we carry out a thermodynamic quantity analysis, similar to the use of the enthalpy difference in hy-drostatic cases, in order to distinguish the relative stability between phases under uniaxial compression. This is applied to the uniaxial phase transition between the diamond and
-Sn structures of Si and Ge using ab initio calculations. We find that uniaxial compression greatly lowers the phase tran-sition pressure for both Si and Ge, from 11.4 to 3.9 GPa for Si and 9.5 to 2.5 GPa for Ge. The anisotropic phase-transition pressures are qualitatively similar, in the low-pressure range, to the previous empirical molecular simulations.4
The hydrostatic phase-transition pressure of two solid phases can be determined by the enthalpy function H⫽E
⫹PV, i.e., the hydrostatic pressure P at which the system
transforms to the phase of lower H. To investigate the phase transition of the system under anisotropic stress we go back to the second law of thermodynamics. The principle of in-creasing entropy5tells us that the condition for a phase tran-sition 共from phase 1 to phase 2兲 to occur is
E2⫺E1⫹W⭐T共S2⫺S1兲, 共1兲
in which W is the work done by the system during the phase transitions and E1and E2are the internal energies of phases 1 and 2. The work W can be written as
W⫽Px
冕
1 2 lylzdlx⫹Py冕
1 2 lzlxdly⫹Pz冕
1 2 lxlydlz, 共2兲where the limits of the lx, ly, and lz in the integrals are the
lattice constants of the crystal phases 1 and 2 under a uni-form stress having diagonal components Px, Py, and Pz
only, which we shall for convenience refer to as ‘‘pres-sures.’’ Under the hydrostatic condition Px⫽Py⫽Pz, the three integrands of Eq.共2兲 can be combined to form an exact volume differential, i.e., dV. This is not true for systems
under nonhydrostatic condition. In the case of diamond and
-tin phases, we choose the eight-atom tetragonal unit cells for both phases. Under uniaxial compression, i.e., Px⫽Py, the inequality Eq. 共1兲 becomes
共E2⫹PxV2⫺TS2兲⫹共Pz⫺Px兲
冕
1 2
lxlydlz
⭐共E1⫹PxV1⫺TS1兲. 共3兲
At T⫽0 K, which corresponds to the situation of our ab
initio calculations, the inequality is further reduced to
关共E2⫺E1兲⫹Px共V2⫺V1兲兴⫹
冋
共Pz⫺Px兲冕
1 2
lxlydlz
册
⫽⌬H⫹⌬¯W⫽⌬¯H⭐0. 共4兲
The inequality is satisfied if phase 2 is relatively more stable than phase 1 and the phase-transition pressures are those when the equality condition is met. In addition to the ⌬H term, there is a term⌬¯W, which is not present under hydro-static condition. The magnitude of the ⌬¯W term depends on
the path of the integration in Eq. (2) and reduces to zero only under hydrostatic conditions. However, we will show that in
the diamond-to--tin phase transition of Si and Ge, the physically most sensible path is also the shortest path. After this work was completed, we learned that the existence of the path-dependent term in the free energy for lattices under nonhydrostatic applied load had been mentioned in a previ-ous paper by Wang et al.6Their studies were mainly on the elastic stability criteria of lattices under arbitrary but uniform external load. A recently published paper by Libotte and Gaspard7 studied the effects of nonhydrostaticity on the pressure-induced distortion of the -tin phase in silicon us-PHYSICAL REVIEW B, VOLUME 63, 153202
ing a different approach for the Gibbs free energy. It will be interesting to see whether using our approach will lead to similar results.
In our paper, the total energy of the diamond and -Sn structures of Si and Ge were calculated with the ab initio density-functional method,8and the generalized gradient ap-proximation 共GGA兲9 for the exchange and correlation func-tional was used. The calculations were performed using the
initio total-energy and molecular-dynamics Vienna ab-initio simulation program, developed at the Institut fu¨r
Ma-terial Physik of the Universita¨t Wien.10–12 Ultrasoft pseudopotentials13 were used for the electron-ion interac-tions. It has been demonstrated previously that ultrasoft pseudopotentials can be used for investigating phase transi-tions at high pressures.14 All present calculations were car-ried out in momentum space and the Kohn-Sham wave functions15were expanded in a plane-wave basis. The energy cutoffs for the plane-wave basis were 400 and 500 eV for Si and Ge, respectively. The integration over the Brillouin zone was approximated by k-point sampling using the Monkhorst-Pack method.16 The k-point sets of 64 and 1280 k points were used for the tetragonal 8-atom cell of the diamond structures and the metallic -Sn structure, respectively. The total energy, i.e. E(ax,az), was evaluated at various lattice
constants ax⫽ayand azof the tetragonal cells for every 0.05
Å . The range of lattice constants considered was between 5
共5.2兲 and 5.8 共6.0兲 Å for both axand az of diamond Si共Ge兲,
and between 6.3 共6.8兲 and 6.95 共7.45兲 for ax, and between 2.4共2.6兲 and 2.9 共3.4兲 for az of the-tin Si共Ge兲. The pres-sures were obtained from the partial derivatives of the
evalu-ated total-energy surface E(ax,az), i.e., Px⫽Py⫽
1 2axaz E共ax,az兲 ax
冏
a z and Pz⫽ 1 axax E共ax,az兲 az冏
a x . 共5兲The calculated lattice constants of the two phases at zero pressure were ax⫽ay⫽az⫽5.46 共5.76兲 Å for diamond Si
共Ge兲 and ax⫽ay⫽6.82 共7.37兲 Å and az⫽2.65 共2.87兲 Å , i.e.,
c/a⫽0.3886 共0.3894兲, for -tin Si 共Ge兲. They are in good
agreement with the experimental results17,18of 5.43共5.65兲 Å for diamond Si 共Ge兲 and c/a⫽0.3903 共0.3896兲 for-tin Si
共Ge兲. Calculations with a higher-energy cutoff of 500 eV in
the plane-wave basis expansion for diamond and -Sn Si were carried out for convergence tests. Similarly, a different k-point set of 576 k points for -Sn Si was considered. The change in transition pressure due to numerical errors is ex-pected to be less than 0.5 GPa.
The evaluated⌬¯H, using Eq. 共4兲, of Si from diamond to
-Sn phase under uniaxial compression, are shown in Fig. 1. We have taken the shortest path共Fig. 2兲 in the integral of the
⌬¯W term, which gives
⌬¯W⫽13共Pz⫺Px兲共az,⫺az,dia兲共ax,2⫹ax,ax,dia⫹ax,dia
2 兲,
共6兲
where the lattice constants共subscript ‘‘dia’’ and ‘‘’’ for the diamond and -tin phases, respectively兲 are those when the phases are under the uniaxial pressures Pxand Pz. The hy-drostatic phase-transition pressure is at 11.4 GPa, similar to
FIG. 1. The contour plot of the quantity ⌬¯H 关Eq. 共4兲兴 for the diamond phase共1兲 and-tin phase 共2兲 of Si at different pressures. The phase transition occurs at ⌬¯H⫽0, which is denoted with the bold line.
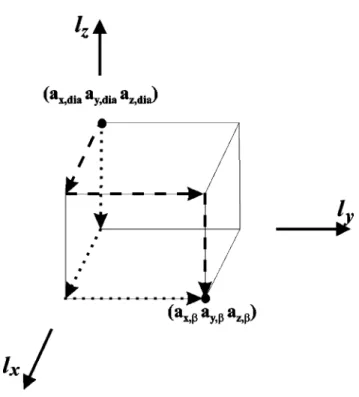
FIG. 2. Schematic plot showing the different paths for the evalu-ation of ⌬¯W 关Eq. 共4兲兴 in the present paper. The dotted path gives
⌬¯W⫽(Pz⫺Px)(az,⫺az,dia)(ax,dia)2while the dashed path gives
⌬¯W⫽(Pz⫺Px)(az,⫺az,dia)(ax,)2. The shortest path 关Eq. 共6兲兴
corresponds to the integral along the body diagonal connecting the two points (ax,dia,ay ,dia,az,dia兲 and (ax,,ay ,,az,兲.
BRIEF REPORTS PHYSICAL REVIEW B 63 153202
the result of previous GGA calculations19and in good accord with experimental results.17 The transition pressure is effec-tively reduced to 3.9 GPa ( Pz) when the lateral pressure Px⫽Py is zero. In fact, the transition pressures follow roughly the relation Pz⫽0.658Px⫹3.9 GPa. The aniso-tropic transition pressures have Pz⬎Px at pressures below the hydrostatic transition pressure 11.4 GPa, and Pz⬍Pxfor higher values. A similar trend to lower transition pressures was found in an isobaric molecular-dynamic simulation of this transition using an empirical interatomic potential.4The authors found the uniaxial transition pressures followed roughly the relation Pz⫽Px⫹9 GPa for pressures up to
Px⫽9 GPa, analogous to our relation above.
In order to see how different terms in Eq.共4兲 contribute to the phase stability at different pressures, the contour plots of
the⌬H and ⌬¯W terms for all the pressures considered in this
paper were investigated. It was found that for the diamond and-tin phases of Si, the ⌬¯W is essentially constant for a fixed value of pressure anisotropy, i.e., Pz⫺Px, while the
⌬H term depends little on the Pz pressure. These two facts
together lead to a nice behavior of⌬¯H whose value changes linearly as we move away from the transition-pressure line
共Fig. 1兲. Another interesting fact was found concerning the
lattice constants of the two phases at the phase-transition pressures. On the phase-transition line in Fig. 1, the az of
both phases was found to vary little compared to the varia-tion of the ax values. The variation of a3 is between 5.277
共2.550兲 and 5.283 共2.578兲 Å while that of a1is between 5.21
共6.584兲 and 5.51 共6.865兲 Å for diamond (-tin兲 Si between
the hydrostatic point and the point with Px⫽0 on the transi-tion line in Fig. 1.
In addition to the shortest path, two other possible paths were considered in order to see how far they can alter the transition pressure. We consider two paths along the edges of the tetragonal on which the two points (ax,dia,ax,dia,az,dia)
and (ax,,ax,,az,) are the body-diagonal corners共Fig. 2兲. One path reaches az, before ax, and the other ax, before az,. These two paths give, respectively, the minimum and
maximum⌬¯W under the condition that the values of lx and
lz along the path lie everywhere between the limits of inte-gration, i.e., min(ax,dia,ax,)⭐lx⭐max(ax,dia,ax,) and
min(az,dia,az,)⭐lz⭐max(az,dia,az,). We found the effect of these two paths is to tilt the original transition line, which was obtained from the shortest path, in opposite directions but by about the same order of magnitude. The modified phase-transition pressures are Pz⫽0.710Px⫹3.3 GPa and
Pz⫽0.570Px⫹4.9 GPa for the maximum and minimum
⌬¯W, respectively. The hydrostatic transition pressure
re-mained unchanged as expected. Considering the phase tran-sition macroscopically, it is physically reasonable to expect the path-dependent ⌬¯W to take the shortest path in the diamond-to--tin phase transition. We envisage the phase change starting somewhere in the crystal and spreading through the specimen as it transforms from the diamond structure to the -tin structure. We may expect the
dimen-sions to vary approximately as lx⫽(1⫺␣)ax,dia⫹␣ax, and lz⫽(1⫺␣)az,dia⫹␣az, with ␣ increasing from zero to
unity. This uniform transformation corresponds to the short-est path considered above.
All of the above calculations were also carried out for the similar phase transition of Ge. It was found that, except for small quantitative differences in the phase-transition pres-sures, the transition properties are all similar to those of Si and follow the relation Pz⫽0.737Px⫹2.5 GPa.
The effective reduction of the transition pressure for Si and Ge from diamond to -tin phases can be understood intuitively with the following consideration. The structure of the -tin phase can be seen as the z(az)-compressed and
xy(axay)-expanded diamond structure. Uniaxial compres-sion is therefore expected to lower the transition pressure as it is forcing diamond phase towards the -tin structure. However, this way of thinking should not be pushed too far as the phase transition is first order and there is still a dis-continuity in energy, and in ax, az, etc. The structures do not transform continuously with respect to the applied uniaxial pressures.
The proposed method of identifying the phase-transition pressure of diamond to -tin phase at uniaxial compression can be extended to phase transitions of more complicated systems. However, the possible paths for ⌬¯W have to be chosen carefully as there might be no obvious way of select-ing the related lxand lz for the two phases considered. This
would be particularly true if the orientation of the crystal axes of the two phases with respect to each other is more complicated than in the diamond to -tin transition. Model investigations along this line are presently under consideration.20
The diamond-anvil cell21has been used for high-pressure experiments for a long time. Although the possible phase transition of diamond under hydrostatic pressure has been proposed and studied using the ab initio method,22 the pres-sure limit of diamond is still under investigation. It will be interesting to see how uniaxial compression can change the transition pressure of the diamond carbon into other phases. Calculations along these lines are in process.
In conclusion, we have extended the thermodynamics treatment of a first-order phase transition under hydrostatic pressure, to the case of a transition under uniaxial stress. We discuss how the latter is in principle not a reversible equilib-rium process but depends on the path taken. The method is applied to the diamond-to--tin transition in Si and Ge whose total energies were evaluated with an ab initio method. Large reductions of the transition pressure were found compared to hydrostatic conditions.
We gratefully acknowledge V. Heine for invaluable dis-cussions and his careful and critical reading of the manu-script, and R. J. Needs for helpful discussion. C. C. and H. J. L. thank the support of the National Science Council in Tai-wan.
BRIEF REPORTS PHYSICAL REVIEW B 63 153202
*Email: [email protected] †Email: [email protected]
1J. C. Jamieson, Science 139, 762共1963兲. 2
M. C. Gupta and A. L. Ruoff, J. Appl. Phys. 51, 1072共1980兲. 3For examples, M. T. Yin, Phys. Rev. B 30, 1773共1984兲; R. J.
Needs and R. M. Martin, ibid. 30, 5390共1984兲; R. Biswas, R. M. Martin, R. J. Needs, and O. H. Nielsen, ibid. 35, 9559 共1987兲; A. Mujica and R. J. Needs, ibid. 48, 17 010 共1993兲. 4I.-H. Lee, J.-W. Jeong, and K. J. Chang, Phys. Rev. B 55, 5689
共1997兲.
5L. D. Landau and E. M. Lifshitz, Statistical Physics共Pergamon Press, New York, 1980兲.
6J. Wang, J. Li, S. Yip, S. Phillpot, and D. Wolf, Phys. Rev. B 52, 12 627共1995兲.
7H. Libotte and J.-P. Gaspard, Phys. Rev. B 62, 7110共2000兲. 8P. Hohenberg and W. Kohn, Phys. Rev. 136, B864共1964兲. 9J. P. Perdew, in Electronic Structure of Solids ’91, edited by P.
Ziesche and H. Eschrig共Akademie-Verlag, Berlin, 1991兲; J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh, and C. Fiolhais, Phys. Rev. B 46, 6671 共1992兲.
10G. Kresse and J. Hafner, Phys. Rev. B 47, 558共1993兲; 49, 14 251 共1994兲.
11G. Kresse and J. Furthmu¨ller, Comput. Mater. Sci. 6, 15共1996兲. 12G. Kresse and J. Furthmu¨ller, Phys. Rev. B 54, 11 169共1996兲. 13D. Vanderbilt, Phys. Rev. B 41, 7892共1990兲; K. Laasonen, R.
Car, C. Lee, and D. Vanderbilt, ibid. 43, 6796共1991兲; K. Laa-sonen, A. Pasquarello, R. Car, C. Lee, and D. Vanderbilt, ibid.
47, 10 142共1993兲.
14J. Furthmuu¨ller, J. Hafner, and G. Kresse, Phys. Rev. B 50, 15 606共1994兲.
15W. Kohn and L. J. Sham, Phys. Rev. 140, A1133共1965兲. 16
H. J. Monkhorst and J. D. Pack, Phys. Rev. B 13, 5188共1976兲. 17
J. Z. Hu and I. L. Spain, Solid State Commun. 51, 263共1984兲; M. I. McMahon and R. J. Nelmes, Phys. Rev. B 47, 8337共1993兲. 18K. A. Asaumi and S. Minomura, J. Phys. Soc. Jpn. 45, 1061
共1978兲; S. B. Qadri, E. F. Skelton, and A. W. Webb, J. Appl. Phys. 54, 3609共1983兲; A. Werner, J. A. Sanjurjo, and M. Car-dona, Solid State Commun. 44, 155共1982兲.
19N. Moll, M. Bockstedte, M. Fuchs, E. Pehlke, and M. Scheffler, Phys. Rev. B 52, 2550共1995兲.
20W.-H. Huang and C. Cheng共unpublished兲.
21H. K. Mao and P. M. Bell, Science 203, 1004共1979兲.
22S. J. Clark, G. J. Ackland, and J. Crain, Phys. Rev. B 52, 15 035 共1995兲.
BRIEF REPORTS PHYSICAL REVIEW B 63 153202