國立臺灣大學理學院化學系研究所 碩士論文
Department of Chemistry College of Science National Taiwan University
Master Thesis
釕金屬誘發含異原子有機烯炔化合物之 環化反應的研究
Study on Intramolecular Cyclization of Organic Hetero-Containing Enynes Induced by Ruthenium Metal
Complexes
羅濟賢 Ji-Xian Lo
指導教授:林英智 博士 Advisor: Prof. Dr. Ying-Chih Lin
中華民國 102 年 7 月
July, 2013
I
CONTENTS
Contents I
Numbering and Structure of Compounds II
Reaction Scheme IV
中文摘要 1
Abstract 2
Introduction 4
Results and Discussion 11
Conclusions 33
Experimental Section 34
References 55
Appendix I: X-ray Crystallographic Data 61
Appendix II: Spectra Data 78
II
Numbering and Structure of Compounds [Ru]=CpRu(PPh 3 ) 2
1a 2a-2b
3 4
5a 5b
6 7a-7c
III
8 9
10 11a
11b 12
13
IV
Reaction Scheme
1
中文摘要
本篇論文探討藉由釕金屬錯合物誘發含異原子 1,8-烯炔化合物進行分子內環
化反應而形成含氧雜環產物。利用釕金屬錯合物[Ru]Cl ([Ru] = Cp(PPh
3
)2
Ru)與尾端烯類官能基有甲基取代的化合物 1 反應,可得到含氧七環的釕金屬亞乙烯基錯合 物 3。此反應經由亞丙二烯中間體接著由親電性加成環化在 Cγ 上,並且是一溫和 的反應過程。因為在乙烯上有甲基取代,因此環化所形成的中間體有三級碳陽離
子的生成而穩定此過程。利用金屬錯合物 3 在乙腈中加熱或將[Ru]NCCH
3 +
和化合物 1 在氯仿下加熱,皆可產生環化烯炔產物 6。此外,相同的反應在氯仿及甲醇的 共溶劑環境下,經由甲氧基親核加成而得到二次環化有機產物 7a。另一方面,使 用末端烯類官能基有兩甲基取代的化合物 2a 與[Ru]Cl 進行反應,得到以反式為主
要產物的錯合物 8,。將[Ru]NCCH
3 +
和化合物 2a 在氯仿加熱可生成 11a (反式:順式=2/1)。推測此反應機制是經由六元環椅式過渡態,而使得立體障礙為主要的選 擇性因素。因此,使用含有較大的五環取代基化合物 2b,可使得異構體比例大大 的增加為 10:1.8。在溶劑為甲醇的環境下,化合物 2a 可產生相對應的雙環化合 物 12 和錯合物 13。錯合物 3 和 8 加入甲醇鈉可分別得到相對應的乙炔基錯合物 4 和 9。此外,將錯合物 3 和 8 加入烷基化試劑可分別得到相對應的亞乙烯基錯合物
5a-b 和 10.
關鍵字:釕金屬丶催化丶雜環丶烯炔丶亞乙烯基
2
Abstract
The intramolecular cyclization of 1,8-enyne containing hetero-atom mediated by ruthenium complex, leading to the formation of the oxygen heterocycle is described. In the [Ru]Cl-induced ([Ru] = Cp(PPh
3
)2
Ru) reactions of enyne 1, with an internal methyl group on the olefinic group, the vinylidene complex 3 containing an oxepane moiety, bonded at Cβ is isolated. The reaction proceeds via the formation of an allenylidene intermediate followed by a cyclization at Cγ under mild condition. Stabilization of the cationic charge by the presence of methyl substituents clearly controls the reaction pathway. The reaction of 3 with NaOMe produces the acetylide complex 4. The alkylation of 4 by allyl bromide and methyl iodide as alkylation reagents induced the formation of 5a and 5b, respectively. Heating complex 3 in CH3
CN or treatment of 1 with [Ru]NCCH3 +
in CHCl
3
at 50 °C gave the organic enyne 6 as the cyclized product.Furthermore, the same reaction in CHCl
3
/MeOH leads to the organic product 7a by tandem cyclization, formed possibly via sequential allenylidene/vinylidene cyclization followed by a nucleophilic addition of a methoxide.In the reaction of [Ru]Cl with enyne 2a, containing two terminal methyl groups on the olefinic parts, the vinylidene complex 8 with a new oxane ring is obtained as a mixture of two diastereoisomers and the anti-isomer is the major product. Mild thermolysis of 2a in CHCl
3
at 50 °C in the presence of [Ru]NCCH3 +
gave 11a
3
(anti-11a/syn-11a = 2/1). The proposed mechanism is via the formation of a six-membered ring chair-like transition state with the most bulkiest group in the pseudoequatorial position to reduce the 1,3 diaxial interactions. Hence, in the reaction of propargylic alcohol 2b, the isomer ratio was greatly improved to 10:1.8 due to the steric effect of the bulkier cyclopentyl tether. Treatment of 2a with [Ru]NCCH
3 +
in MeOH afforded two products, the hexahydro isochromene compound 12 with a methoxide group and the carbene complex 13, both containing a newly formed bicyclic ring. The same reaction of 2b in MeOH afforded no corresponding bicyclic ring products, but 11b was obtained. Similarly, the reaction of 8 with NaOMe produces the acetylide complex 9 and alkylation of 9 by allyl bromide as alkylation reagent gives 10.
Keywords: Ruthenium, catalysis, heterocycle, enyne, vinylidene.
4
Introduction:
Metal Vinylidene Complexes
The chemistry of transition metal containing unsaturated carbenes such as vinylidene and allenylidene complexes has attracted a great deal of attention because the possibility of development of new types of organometallic intermediates as emphasized in several publications.
1
During the past decade, the direct simple formation of metal vinylidene intermediates from terminal alkynes has possible the transfer from stoichiometric to catalytic reactions.
Since the first mononuclear metal vinylidene complex reported in 1972,
2
there are two major methods for the preparation, such as (1) the activation of a terminal alkyne to give an initial η2
-coordinated alkyne intermediate followed by a direct 1,2-hydrogen migration over the carbon-carbon triple bond; (2) by oxidative addition of the terminal alkynyl C-H bond to the metal center with subsequent 1,3-hydride shift to the alkynyl lignad.3
(Scheme1).Scheme 1
5
The coordinated vinylidene is considered as an electron-withdrawing ligand stabilized by electron-rich metal fragments
5
, and the reactivity of the metal vinylidene complexes largely depends on the nature of the metal. The resulting complexes exhibit a variety of reactivities, which are rationalized by taking electrophilicity of-vinylidene carbon,
nucleophilicity of-vinylidene carbon. These features boost C-C coupling and
C-heteroatom coupling of vinylidene complexes into various metal complexes (Scheme 1-2).Scheme 1-2
Metal Allenylidene Complexes
Allenylidene ligands are also cumulogous, more extended versions of carbenes and consist of three cumulated carbon atoms. Just like carbenes, these attach to metal via a formal M=C double bond. Further experimental
6
and theoretical evidence7
shows that the carbon atoms of the allenylidene ligands are alternatively electron-deficient and6
electron-rich when moving along the unsaturated chain starting from the metal center.
This behavior is easily rationalized by the contribution of three resonance forms, namely, allenylidene, propargylium and allenylium structure.
The most useful and general method for the synthesis of metal-allenylidene complexes was reported by Selegue in 1982
8
for the preparation of [Cp(PMe3
)Ru=C=C=CPh2
][PF6
]. After η2
-coordinated of the 2-propyn-1-ol to a 16 electron metal center, the spontaneous dehydration of propargylic alcohol, via the hydroxyl vinylidnen intermediate occurs (Scheme 1-3). However, other metals such as Os9a
, Mo9b
, W9c
have also been reported to give similar complexes.Scheme 1-3
Heterocycles
The development of effective strategies for the synthesis of heterocyclic compounds containing oxygen, sulfur and nitrogen is a very important challenge for modern organic and natural products synthesis.
10
For example, chromane, thiochromane, and 1,2,3,4-tetrahydroquinoline derivatives are widely found in many natural and7
biologically active compounds.
11
Some derivatives of chromane ring are well-known to exhibit extensive range of biological activities including anticancer,12
antioxidative,13
and significant pharmacological potential.14
The catalytic construction of heterocyclic skeletions is classified into two major processes, as ahown in Scheme 1-4: (1) C-C bond formation from the corresponding acyclic precursors and (2) C-Y bond formation from the corresponding acyclic precursors.
15
Scheme 1-4 Two major processes of heterocycle synthesis.
Transition-Metal-catalyzed Reaction in Heterocyclic Synthesis
Recently, metal-catalyzed cycloisomerization of enynes represents an efficient synthetic strategy for a variety of carbo-
16
and heterocycles.17
Because of the presence of metal stabilized “nonclassical” cationic intermediate, the metal-catalyzed cycloisomerization of enynes often leads to various skeleton rearrangements.18
Additionally, a variety of Ru-based transition metal have been proposed as the key intermediate in catalyzed intra and intermolecular carbon-carbon bond forming8
reactions between alkynes and alkenes.
19
Besides, a variety of preparative methods have been reported including its asymmetric version for the optically active heterocycles.20
More recently, Nishibayashi demonstrated that a thiolate-bridged diruthenium complex catalyzed cyclization of the enyne compounds via allenylidene-ene pathway to form chromane deravitives with high enantioselectivity. (Scheme 1-5, eq1)
20a-b
In addition, Tanaka and his co-workers have found that asymmetric synthesis of phenol- or naphthol-linked 1,7-enynes by the cationic rhodium(I)/(R)-BINAP complex catalyzed olefin isomerization/enantioselective intramolecular reaction giving substituted dihydrobenzofurans and dihydronaphthofurans. (Scheme 1-5, eq2)21
Also, Liu et al have developed Pt-catalyzed cycloisomerization of linear 1,8-enynes via ally-rearrangement to produce dihydrofuryl alcohols and epoxide product (Scheme 1-5, eq3).22
Therefore, the development of new, rapid, methods to generate heterocyclic products as a precursor of natural products would be a useful contribution to the synthetic community.Furthermore, the construction of more complicated ring systems, such as fused or bridged rings, is also useful for synthesizing natural products.
23
To build these fused rings, photo-rearrangement24
/thermal25
rearrangement and the use of Lewis acids,26
and transition metals27
have been developed. These exhaustive efforts have resulted in elegant methods for intricate bicyclic systems.9
Scheme 1-5. Synthesis of O-containing heteocyclic compounds.
Motivation of This Thesis
Previously, we developed the ruthenium-mediated of 1,5-enynes, which was proposed via a intramolecular cyclization, affording the vinylidene complex with a five-membered ring containing an unsaturated methylene group on the ring.
28a
(Scheme 1-6, eq1) Potentially, this vinylidene ligand may undergo further cyclization yet to observe. In a recent study, we found a ruthenium-catalyzed tandem cyclization reaction of 1,n-enynes (n=7~8) with a methyl group at the terminal vinyl group produced tricyclic products in MeOH/CHCl3
.28b
(Schene 1-6, eq2) As an extension of our previous study, we have explored the ruthenium-mediated reactions of linear 1,8-enyne propargylic alcohols, with an allylic alkoxy moiety, affording the corresponding cycloisomerization products. Herein,the results were reported on the10
study of the reactions of [Ru]Cl with linear oxygen-containing enynes also with a propargyl alcohol group.
Scheme 1-6
11
Results and Discussion:
Formation of Vinylidene Complex Containing an Oxepane Group
Scheme 2
Treatment of [Ru]Cl ([Ru] = Cp(PPh
3
)2
Ru) with 1 in the presence of NH4
PF6
in CH2
Cl2
at room temperature for one day afforded the vinylidene complex 3 containing an oxepane moiety. The mechanism of the formation of 3 is shown in Scheme 2. The reaction presumably proceeds via the formation of the γ-hydroxyvinylidene intermediate A, followed by the formation of the allenylidene intermediate B by a dehydration at Cβ and Cγ. Subsequently, the intramolecular attack of the alkene portion onto the electrophilic Cγ of the allenylidene ligand in B, resulting in a C-C bond formation to give the alkynyl complex C bearing a cationic charge at the methyl-substituted tertiary carbon of the oxepane moiety. This is followed by a12
1,5-hydrogen shift of one of the terminal protons into the acetylide moiety to give the corresponding vinylidene complex 3. Stabilization of the tertiary carbocationic intermediate by the presence of methyl substituents could assist the cyclization process.
Alternatively, direct allenylidene-ene process
20a-b
might be the other pathway for this cyclization. The structure of complex 3 is determined by NMR spectroscopy. The31
P NMR spectrum of 3, displays two doublet resonances at δ 43.24 and 43.02 with2 J PP
= 26.35 Hz indicates the presence of a stereogenic center. In the 2D-HMBC NMR spectrum, the triplet resonance at δ 343.94 with2 J CP
= 15.21 Hz assigned to Cα, shows correlation with the multiplet1
H resonance at δ 2.35 assigned to CγH. The chemical shift of Cα is similar to many Cα resonances of other ruthenium vinylidene complexes.29
Formation of Oxepane Derivative Compound
Scheme 3-1
Heating the vinylidene complex 3 in a cosolvent of CHCl
3
/CH3
CN for a day afforded the ethynyloxepane derivatives 6 and the cationic complex [Ru](CH3
CN)[PF6
] as shown in Scheme 3-1. Formation of alkyne from a metal vinylidene complex has been reported in thermolysis of the analogous metal vinylidene complex in CH3
CN.30
13
Alternatively, direct treatment of 1 with a 30 mol% of [Ru]NCCH
3 +
in CHCl
3
at 50o
C for a day also afforded 6, but with slightly lower yield (78%). In this reaction, a small amount of 3 and other impurities are also produced. Presumably, treatment of [Ru]Cl with acetonitrile produced [Ru]NCCH3 +
in the beginning which then catalyzed the cyclization reaction. The spectroscopic data of 6 are in agreement with the proposed structures. In the
1
H NMR spectrum of 6, the characteristic acetylenic proton resonance appears as a doublet at δ 2.10 with4 J HH
= 2.5 Hz. Two multiplet resonances at δ 4.92 and δ 4.83 are assigned to two olefinic methylene protons bonded to the oxepane moiety.Catalytic cyclization of analogous compound has been reported on an intramolecular Nicholas reaction via the acid treatment of exo-Co
2
(CO)6
-propargyl alcohols, leading to the formation of ethynyloxepane derivatives31
. However, their preparation required 3 steps and mixtures of diastereomers were produced. In our case, formation of 6 requires only one step with [Ru]NCCH3 +
as a catalyst under mild condition.
14
Scheme 3-2 Catalytic Cycle for the Formation of The Oxepane Derivative.
The proposed mechanism for the cyclization of compound 1 to give 6 is illustrated in Scheme 3-2. Cyclization of 1, catalyzed by [Ru]NCCH
3 +
, proceeds by the production of the allenylidene intermediate B followed by the cyclization to yield the vinylidene complex 3. Finally, an isomerization of the vinylidene ligand gives the π-coordinated alkyne which is replaced by CH
3
CN to produce 6, finishing the catalytic cycle.Complex [Ru]NCCH
3 +
is therefore successfully used to catalyze the cyclization of 1 affording enyne 6.
15
Formation of Vinylidene Complex by Alkylation
Scheme 4
Treatment of 3 with NaOMe/MeOH for 10 mins afforded the acetylide complex 4 by deprotonation and color of the solution changed from light orange to yellow.
Complex 4 could be converted back to the vinylidene complex 3 by protonation with HBF
4
in diethyl ether. The structure of 4 is confirmed by1
H NMR and31
P NMR spectra.The
31
P NMR spectrum of 4, with a stereogenic center, yet displays a broad resonance at δ 51.94. This may be due to the distant stereogenic center away from the phosphine ligands or the rotation of the C-C single bond. The resonance of CβH at δ 4.57 in the1
H NMR spectrum of 3 disappears in the1
H NMR spectrum of 4.Then, treatment of 4 separately with allyl bromide and methyl iodide as alkylation reagents in the presence of KPF
6
afforded the corresponding cationic vinylidene complexes 5a and 5b as shown in Scheme 4. Complexes 5a and 5b are stable under thermolytic condition, and show the similar NMR data. Fortunately, single crystals of5a, tethering an ally group at Cβ, are obtained at ambient temperature in diethyl
ether/CH
2
Cl2
solution, and the structure is determined by a single crystal X-ray16
diffraction study. An ORTEP type view of the cationic complex 5a is shown in Figure 1, with selected bond distances and angles. Complex 5a has distorted three-legged piano-stool coordination geometry around the ruthenium center which bound to a Cp, two PPh
3
and the vinylidene ligand with a oxepane moiety. The bond lengths of Ru(1)-C(1) and C(1)-C(2) of 1.864 (3) and 1.308 (5) Å , respectively, show a typical vinylidene bond skeletal. The bond length of C(3)-C(4) of 1.539 (6) Å attests the C-C bond formation and the C(5)-C(9) of 1.367 (9) Å corresponds to a double bond. From the crystal structure, the presence of the seven-membered oxepane ring in the vinylidene ligand is confirmed.Figure 1. An ORTEP drawing of the cationic complex 5a. For clarity, PF 6 -
and phenyl groups of the triphenylphosphine ligands on Ru except the ipso carbons are omitted (thermal ellipsoid is set at the 25% probability level). Selected bond distances (Å ) and angles (deg): Ru(1)-C(1), 1.864 (3); C(1)-C(2), 1.308 (5); C(2)-C(12), 1.536 (5);C(3)-C(4), 1.539 (6); C(5)-C(9), 1.367 (9); C(13)-C(14), 1.241 (7); Ru(1)-C(1)-C(2), 168.0 (3); C(4)-C(3)-C(8), 115.4 (3); C(6)-C(5)-C(4), 117.0 (6); C(6)-O(1)-C(7), 115.5 (6).
17
Scheme 5-1 Formation of the Cascade Cyclization Compound
As mentioned above, in the conversion of 1 to 3 or 6, two unsaturated groups are retained after the first cyclization reaction. The enyne groups in 6 are in closer proximity for cyclization than those in 1. Therefore, we have tried to explore further cyclization reaction. Thermolysis of 3 in CHCl
3
/MeOH at 50o
C generates the 1-methoxy- 5,5-dimethyl-3-oxabicyclo[4.3.1]dec-7-ene product, 7a with 90% yield shown in Scheme 5-1. In addition, a mixture of 6 and 30 mol% of [Ru]NCCH3 +
in CHCl
3
/MeOH at 50o
C could also afford 7a in high yield. Compound 7a is obtained from the treatment of 1 and [Ru]NCCH3 +
in CHCl
3
/MeOH as well, but with a lower isolated yield (70%) together with side products. In the1
H NMR spectrum of the crude product mixtures of new olefinic multiplet resonances at δ 5.81, 5.70 and a methoxy singlet resonance at δ 3.28 appeared, at the cost of the characteristic terminal olefinic resonances at δ 4.87, 4.91 of 1. The structure of 7a is determined by spectroscopic methods. In the newly formed six-membered ring, the methylene protons near the double bond display two multiplet resonances at δ 2.12 and 1.92 because of the newly formed stereogenic center at the bridged head carbon with the OMe group. Treatment of1 with [Ru]NCCH 3 +
in two other alcohols ROH (R = Et, iPr) also serve as nucleophiles
18
afforded 7b and 7c, respectively. The yield of 7c decreased to about 30% because of bulkier i-Pr group. No cyclization product was observed in tert-butyl alcohol, propargylic amine or acetone. Furthermore, treatment of disubstituted vinylidene complexes 5a-b,in CHCl
3
/MeOH at 50o
C generated no tandem cyclization products which confirms the less reactive characteristics of disubstituted vinylidene complexes.32
The seven-membered oxacycle is a novel structural feature of natural products in their molecular architecture. Structures range from fused structures janoxepin, oxepinamide C, and isoprelaurefucin to the functionalized monocycles such as lobatrienetriol and armatol A.
33
Recently, Stončius et al.34
investigated the formation of the oxabicyclo[4.3.1]decane skeleton via the Baeyer–Villiger oxidation of bicyclooctane diketones. Besides, the reaction using SmI2
as a reagent for promoting intermolecular reaction to make bicyclic product was reported.35
Herein, we provided an accessible method under mild condition to generate the oxabicyclo[4.3.1]decane moiety by intramolecular cyclization with moderate yields.19
Scheme 5-2
In order to further understand the mechanism of the formation of 7a, we carried out the cyclization of 1 with [Ru]NCCH
3 +
in CDCl
3
/CD3
OD yielding 7a-D, where both olefinic protons and the methoxy group are deuterated. According to these results, a plausible mechanism of the tandem cyclization for 1 is shown in Scheme 5-2. The tandem cyclization of 1 first yields the vinylidene complex 3. Then, nucleophilic addition of the unsaturated double bond to Cα gives the intermediate E, with a bicyclic ring containing a cationic charge on the tertiary carbon. This is followed by a methanol attack to this carbon to give F. Finally, the ligand exchange reaction between F and 1 occurs to give the corresponding fused product 7a accompanied by regeneration of the ruthenium catalyst.20
Scheme 6-1 Formation of Vinylidene Complex Containing an Oxane Group
To extent our previous study, reaction of the propargyl alcohol 2a bearing two methyl groups on the terminal carbon of the tethering alkyl group as the starting compound is investigated. Treatment of [Ru]Cl with 2a in the presence of NH
4
PF6
in CH2
Cl2
at room temperature for one day afforded the vinylidene complex 8 containing a newly formed oxane moiety as a mixture of two diastereoisomers. The ratio of major and minor isomers is 1:0.5. The structure and configuration of 8 is determined by NMR spectroscopy. The31
P NMR spectrum of 8, with two stereogenic centers, displays two sets of two doublets at δ 43.75, 43.12 (2 J PP
=26.4 Hz) for the major complex and at δ 45.22, 43.50 (2 J PP
=26.7 Hz) for the minor complex. In the13
C NMR spectrum of 8, two triplet resonances at δ 343.71 (2 J CP
=14.86 Hz) and 342.67 (2 J CP
=15.08 Hz) are assigned to the Cα for the major and minor complexes, respectively. In the1
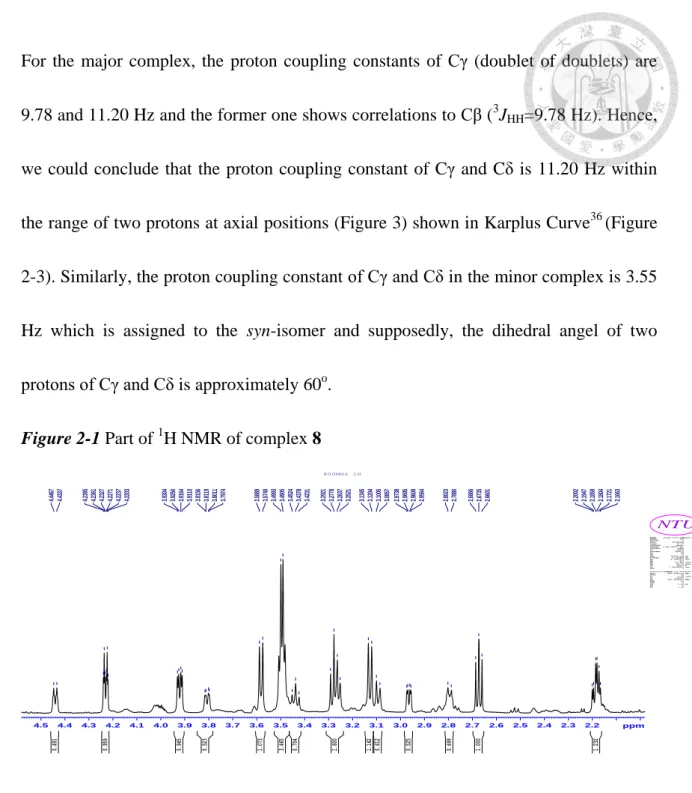
H NMR spectrum asshown in Figure 2-1, resonances at δ 4.23, 2.67 and 2.18 are assigned to the protons at Cβ, Cγ and Cδ for the major complex and resonances at δ 4.44, 2.96 and 2.79 for the
minor one, respectively. In the COSY NMR spectrum (Figure 2-2), the proton of Cγ correlates only with protons of Cβ and Cδ in both diastereomers as shown in Figure 2-2.
21
For the major complex, the proton coupling constants of Cγ (doublet of doublets) are 9.78 and 11.20 Hz and the former one shows correlations to Cβ (
3 J HH
=9.78 Hz). Hence, we could conclude that the proton coupling constant of Cγ and Cδ is 11.20 Hz within the range of two protons at axial positions (Figure 3) shown in Karplus Curve36
(Figure 2-3). Similarly, the proton coupling constant of Cγ and Cδ in the minor complex is 3.55Hz which is assigned to the syn-isomer and supposedly, the dihedral angel of two protons of Cγ and Cδ is approximately 60
o
.Figure 2-1 Part of 1
H NMR of complex 82.2 2.3 2.4 2.5 2.6 2.7 2.8 2.9 3.0 3.1 3.2 3.3 3.4 3.5 3.6 3.7 3.8 3.9 4.0 4.1 4.2 4.3 4.4
4.5 ppm
2.1663
2.17212.18042.1859
2.19472.2002
2.66012.67352.6866
2.78802.8023
2.9564
2.96092.9695
2.9738
3.0857
3.10053.12043.1345
3.25213.2637
3.27783.2921
3.4231
3.43783.45243.4905
3.49933.5749
3.5889
3.79743.80113.8119
3.81563.9110
3.91643.92503.9304
4.22034.2237
4.22714.23274.2361
4.2395
4.4337
4.4467 1.2321.0000.6990.5250.6121.1421.6000.7043.4651.0730.5230.9450.9590.491
NAME linyc-lojx-13May08-cd2cl2 EXPNO 100 PROCNO 1 Date_ 20130508 Time 16.57 INSTRUM spect PROBHD 5 mm CPTCI 1H- PULPROG zg30 TD 65536 SOLVENT CD2Cl2 NS 80 DS 0 SWH 12019.230 Hz FIDRES 0.183399 Hz AQ 2.7263477 sec RG 101 DW 41.600 usec DE 25.00 usec TE 298.0 K D1 1.00000000 sec TD0 1
======== CHANNEL f1 ========
SFO1 800.2040010 MHz NUC1 1H P1 8.00 usec SI 131072 SF 800.2000000 MHz WDW no SSB 0 LB 0.00 Hz GB 0 PC 1.00
800MHz 1H
NTU
22
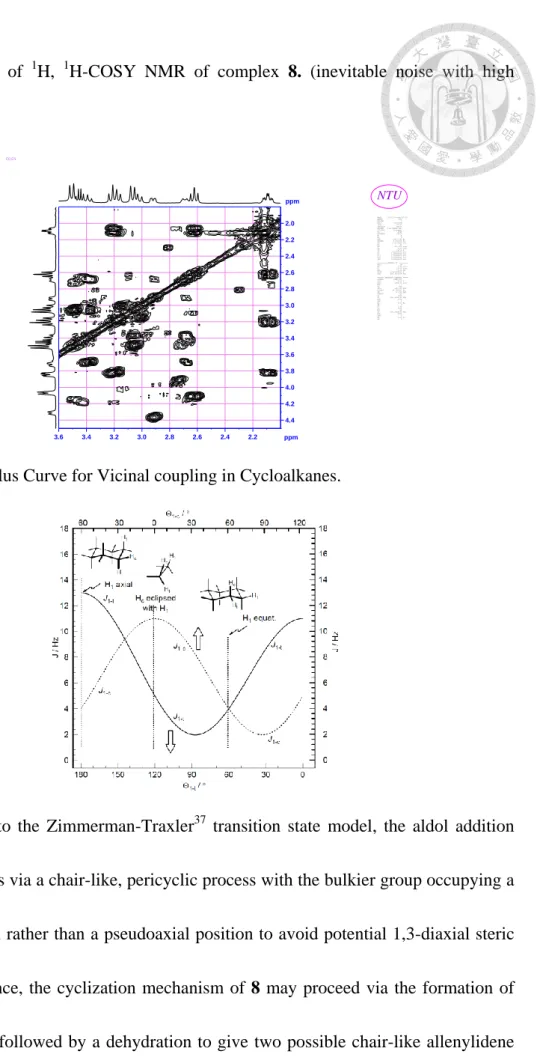
Figure 2-2 Part of 1
H,1
H-COSY NMR of complex 8. (inevitable noise with high concentration)ppm
2.2 2.4 2.6 2.8 3.0 3.2 3.4
3.6 ppm
2.0 2.2 2.4 2.6 2.8 3.0 3.2 3.4 3.6 3.8 4.0 4.2 4.4
NAME Lo121228-complx7-2d EXPNO 102 PROCNO 1 Date_ 20121228 Time 22.02 INSTRUM spect PROBHD 5 mm PABBO BB- PULPROG cosygpqf TD 2048 SOLVENT CDCl3 NS 4 DS 0 SWH 4807.692 Hz FIDRES 2.347506 Hz AQ 0.2130420 sec RG 64 DW 104.000 usec DE 6.50 usec TE 297.6 K D0 0.00000300 sec D1 1.00000000 sec D13 0.00000400 sec D16 0.00020000 sec IN0 0.00020800 sec
======== CHANNEL f1 ========
NUC1 1H P0 6.50 usec P1 13.00 usec PL1 -1.00 dB PL1W 13.43968010 W SFO1 400.1520008 MHz
====== GRADIENT CHANNEL =====
GPNAM1 SINE.100 GPZ1 10.00 % P16 1000.00 usec ND0 1 TD 256 SFO1 400.152 MHz FIDRES 18.780027 Hz SW 12.015 ppm FnMODE QF SI 1024 SF 400.1499730 MHz WDW SINE SSB 0 LB 0.00 Hz GB 0 PC 1.40 SI 1024 MC2 QF SF 400.1499729 MHz WDW SINE SSB 0 LB 0.00 Hz GB 0
400MHz COSY
NTU
Figure 2-3 Karplus Curve for Vicinal coupling in Cycloalkanes.
According to the Zimmerman-Traxler
37
transition state model, the aldol addition reaction proceeds via a chair-like, pericyclic process with the bulkier group occupying a pseudoequatorial rather than a pseudoaxial position to avoid potential 1,3-diaxial steric interactions. Hence, the cyclization mechanism of 8 may proceed via the formation of intermediate A’ followed by a dehydration to give two possible chair-like allenylidene23
transition states B’ and B’’. (Scheme 6-2). The highly demanding bulkier ruthenium complex group is thus located in a equatorial position after cyclization from a steric viewpoint.
38
In path 1, the intramolecular addition of alkene to Cγ in B’ gives the acetylide intermediate C’ with the tertiary cationic substituent at the equatorial position.This is followed by a 1,5-hydrogen shift of one of the methyl protons to Cβ of the acetylide ligand to give a less hindered product anti-8. In path 2, the intramolecular addition of the olefinic moiety to Cγ in B’’ similarly affords the corresponding C’’ but with the cationic group in the axial position. The larger steric repulsion of the axial groups of the transition state elevates the energy of intermediate C’’. As a result, complex anti-8 was obtained as the major product.
Scheme 6-2
24
Scheme 7
Treatment of 8 with NaOMe/MeOH for 10 mins afforded the acetylide complex 9 by deprotonation and color of the solution changed from deep yellow to light yellow with similar isomer ratio (anti:syn=1:0.5). Complex 9 could be converted back to the vinylidene complex 8 by protonation with HBF
4
in diethyl ether. The structure of 9 is confirmed by NMR spectra. The31
P NMR spectrum of 9, with two stereogenic centers, displays a resonance at δ 51.22 and doublets resonances at δ 52.16, 50.34 with2 J PP
= 37.9 Hz, for the anti- and syn-isomer respectively. We can see two sets of resonances in all spectra attributed to two diastereomers in a ratio of 1:0.5 and only spectroscopic dataof the major product is described below. The proton resonance of CβH at δ 4.07 in the
1
H NMR spectrum of 8 disappears in the1
H NMR spectrum of 9. In the1
H NMRspectrum of 9, the proton coupling constant of Cγ and Cδ of the anti-isomer is 11.36 Hz.
The
13
C NMR spectrum of 9 shows the triplet resonance at δ 92.19 with2 J CP
=24.98 Hz for Cα and the singlet resonance at δ 110.58 for Cβ.Then, treatment of 9 with allyl bromide as alkylation reagents, as shown in Scheme 6, afforded the cationic vinylidene complex 10 (anti:syn=1:0.3). Color of the solution
25
changed from yellow to brown. The structure of 10 is determined by NMR spectra. The
31
P NMR spectrum of anti-10, displays a broad resonance at δ 40.56. In the1
H NMRspectrum, multiplet resonance at δ 6.09 is assigned to one of the vinyl hydrogen of the allyl group and the proton coupling constant of CγH and CδH is 10.9 Hz. Resonances in
the
13
C NMR spectrum of 10 at δ 349.91 and 94.36 are assigned to Cα as a triplet with3 J CP
=12.18 Hz, and to Cp as a singlet, respectively.Isomers of the complex 10 are stable under thermolytic condition. Fortunately, single crystals of anti-isomer were obtained at ambient temperature in toluene/CH
2
Cl2
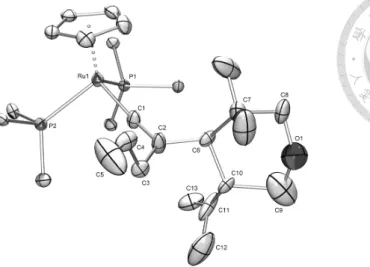
solution. The structure of anti-10 is determined by a single crystal X-ray diffraction study. An ORTEP type view of the cationic complex anti-10 is shown in Figure 3, with selected bond distances and angles. The anti-10 has distorted three-legged piano-stool coordination geometry around the ruthenium center which bound to a Cp, two PPh
3
and the vinylidene ligand with a oxane moiety. From the crystal structure, two hydrogen atoms at Cγ and Cδ are at axial position. The bond lengths of Ru(1)-C(1) and C(1)-C(2) show a typical vinylidene bond skeletal. The bond length of C(6)-C(10) of 1.578 (12) Å attests the C-C bond formation and the C(11)-C(13) of 1.376 (8) Å corresponds to a double bond.26
Figure 3. An ORTEP drawing of the cationic complex anti-10. For clarity, phenyl
groups of the triphenylphosphine ligands on Ru except the ipso carbons and PF6 -
are omitted (thermal ellipsoid is set at the 25% probability level). Selected bond distances (Å ) and angles (deg): Ru(1)-C(1), 1.886 (3); C(1)-C(2), 1.301 (5); C(6)-C(10), 1.578 (12); C(11)-C(13), 1.376 (8); C11-C12, 1.488 (11); Ru(1)-C(1)-C(2), 168.6 (4);C(7)-C(6)-C(10), 108.9 (6); C(6)-C(10)-C(9), 117.0 (7); C(10)-C(11)-C(12), 99.2 (6);
C(8)-O(1)-C(9), 140.2 (8).
27
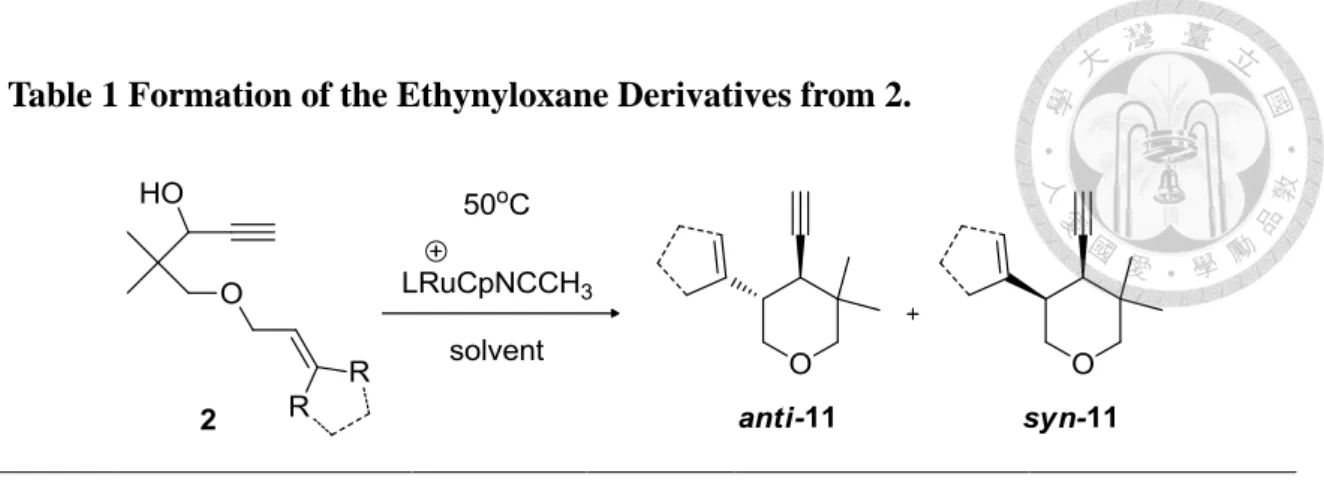
Table 1 Formation of the Ethynyloxane Derivatives from 2.
entry substrate
L (ligand)
solvent yields of 11 (%)
b
ratio of isomers (anti:syn)
c
1
2a, R=CH 3
2 PPh3
CHCl3 11a, 80
66:332
2a, R=CH 3 S-BINAP CHCl 3 11a, 80
66:333
2a, R=CH 3 R-BINAP CHCl 3 11a, 80
66:334
2b, 2R= -(CH 2
)4
- 2 PPh3
CHCl3 11b, 45
85:155
2a, R=CH 3
2 PPh3
MeOH -d
-6 2b, 2R= -(CH
2
)4
- 2 PPh3
MeOH11b, 45
85:15a All of the reactions were carried out at 50 °C for 12 h in the presence of 30 mol % LRuCpNCCH 3 +
except for entry 5 with 50 mol%. b Isolated yields as a mixture of syn- and anti-isomers. c The ratio of two stereoisomers was determined by 1 H NMR. d Products 12 and 13 were obtained in Scheme 9.
Heating 2a in CHCl
3
at 50 °C for 12 h in the presence of 30 mol % of [Ru]NCCH3 +
complex gave a mixture of diastereoisomers of 5,5-dimethyl-4-ethynyl- 3-(1-methylethenyl)-oxane (11a) in 80% isolated yield. The anti-isomer is the major product with the ratio of anti-11a:syn-11a = 2:1. (Table 1, entry1)Alternatively, use of the complex bearing optically active phosphine ligands such as S-BINAP and R-BINAP
28
did not afford satisfactory results in isomer selectivity (Table 1, entry 2-3). According to the chair-like transition state model, the diastereoselectives of product may relate to the steric hindrance by minimizing 1,3-diaxial interactions. Hence, in the reaction of the propargylic alcohol 2b with bulkier cyclopentyl tether, the isomer ratio was greatly improved to 17:3 due to the steric effect (Table 1, entry 4). The structures of 11a and
11b are determined by NMR spectra. In the 1
H NMR spectrum of 11a and 11b, the characteristic acetylenic proton resonances appear both as a doublet at δ 2.08 with4 J HH
= 2.4 Hz and 2.04 with
4 J HH
= 2.1 Hz, respectively. Two singlet resonances at δ 4.93 and δ 4.84 are assigned to two olefinic methylene protons in 11a and a singlet resonance at δ 5.52 is assigned to the olefinic proton of cyclopentene.Scheme 8 Catalytic Cycle for the Formation of the anti-11a.
29
The proposed mechanism for the cyclization of the anti-11a is illustrated in Scheme 8. The reaction may, firstly, proceed by the production of the allenylidene intermediate B’ and the intramolecular attack to give the corresponding alkynyl complex C’ which is followed by a 1,5-hydrogen shift to give the corresponding anti-8.
Finally, an isomerization of the vinylidene ligand gives the π-coordinated alkyne D’ and replaced by CH
3
CN to produce anti-11a, finishing the catalytic cycle.Scheme 9-1 Formation of the Bicyclic Ring Products
Interestingly, treatment of 2a with [Ru]NCCH
3 +
in MeOH (Table 1, entry 5) afforded two products, the hexahydro isochromene compound 12 with a OMe group and the carbene complex 13, both containing a newly formed bicyclic ring shown in Scheme 9-1. About 50 mol% of [Ru]NCCH
3 +
is required to completely consume 2a. Otherwise, treatment of 2b with a bulkier substituent with [Ru]NCCH
3 +
in MeOH afforded no corresponding product with bicyclic ring, but compound 11b with the oxane moiety was obtained. (Table 1, entry 6). The ratio of 13 and 12 is 2: 1 as determined by the
1
H NMR spectrum. The structures of 13 and 12 are determined by various 2D NMR spectra.For 13, with two stereogenic centers, there are two sets of resonances in all spectra
30
attributed to two diastereomers in a ratio of 1:0.3. The
31
P NMR spectrum displays two doublets at δ 45.26, 45.02 with2 J PP
= 29.1 Hz. In the HMBC NMR spectrum, the singlet resonance at δ 6.73 assigned to =CH shows correlations with three13
C resonances at δ 59.66, 41.11 and 20.62 assigned to methylene, the bridgehead CH and methyl carbon atoms. In the1
H NMR spectrum, two multiplet resonances at δ 1.71 and 1.34 areassigned to the two bridgehead CH. The
13
C NMR spectrum shows triplet resonance at δ 317.61 with2 J CP
=9.66 Hz for Cα. For 12, the multiplet resonance at δ 5.62, assignedto two =CH, shows correlations with the neighboring methylene group in the COSY NMR spectrum. And in the
1
H NMR spectrum, the singlet resonances at δ 3.20 and 1.10 are assigned to the methoxy and methyl groups, respectively. These correlations clearly revealing the C-C bond formation.Scheme 9-2
Complex 13 and compound 12 are formed by a tandem cyclization of 2a possibly via 8. The second cyclization presumably takes place between the terminal double bond
31
and Cα of the vinylidene ligand of 8. In order to better understand the mechanism of the
formation of 13 and 12, we treated 2a with [Ru]Cl and KPF
6
in CDCl3
/CD3
OD yielding13-D, where two hydrogens at C 1
are deuterated and 12-D where both olefinic protons and methoxy group are deuterated. According previous evidence, the reaction presumably proceeds via formation of the vinylidene complex 8 and then the nucleophilic addition of the unsaturated double bond to Cα gives the acetylide intermediate H, forming a newly bicyclic skeleton, with methyl substituted tertiary carbocation. However, there are two different path ways from here on, i.e. the catalytic cycle and the formation of 13, as shown in Scheme 9-2. In the catalytic path way, a methanol attack to the carbocation forms the intermediate I, which is followed by protonation to give 12 and [Ru]NCCH3 +
. In the formation of 13, proton migration
possibly assisted by MeOH and the presence of Ru yields the carbene complex 13. The dehydrogenation reaction of the intermediate H of the methylene group proceeded to form the more stable intermediate J with two conjugated double bonds which may cause a higher yield of 13. Finally, the reaction proceeds the protonation by methanol to form the carbene complex 13.
39
Similar reaction for the formation of fused polycyclic compounds was also proposed by Nishibayashi et al.
21a-b
However, in their case, an optically active thiolate-bridged diruthenium complex promoted catalytic cyclization process and32
afforded syn-enynes as the major product in the first step. Then Pt complex was required to catalyze cycloisomerization of the enyne product in the second step. But, in our case, the first cyclization produces the anti-isomer as the major product and the tandem cyclization readily take place in methanol.
33
Conclusion
In summary, treatment of [Ru]Cl with enyne 1, containing propargylic alcohol and allylic alkoxy moiety, in CH
2
Cl2
in the presence of NH4
PF6
affords the vinylidene complex 3 with an oxepane mioety. Furthermore, from the same reaction of 2a, the vinylidene complex 8 with a newly oxane ring is obtained as a mixture of two diastereoisomers and the anti-isomer is the major product. Additionally, the ruthenium- catalyzed cyclization of 1 generates organic enyne 6 in chloroform and in alcohol yields the organic product 7 by a tandem cyclization. Otherwise in the same reaction of 2a-b gives 11a and 11b, respectively. And the isomer ratio of 11b was greatly improved to 10:1.8 by the steric effect. Alternatively, the same reaction with 2a in methanol affords the hexahydro isochromene compound 12 with a methoxy group and the carbene complex 13, both containing a newly formed bicyclic ring. However, no corresponding bicyclic ring product was isolated from the reaction of 2b.34
Experimental Section:
General procedures: Manipulations were performed under an atmosphere of dry
nitrogen by using vacuum-line and standard Schlenk techniques unless mentioned otherwise. All reagents were obtained from commercial suppliers and were used without further purification. Solvents were dried by standard methods and were distilled under nitrogen before use. NMR spectra were recorded on Brucker DPX-400, AVIII-400, DMX-500 or on AVIII-800 FT-NMR spectrometer at room temperature and were reported in units of δ with residual protons in the solvents as a standard. Electrospray ionization mass spectrometry and X-ray diffraction studies were carried out at the Regional Center of Analytical Instrument located at National Taiwan University. The ruthenium complex Cp(PPh
3
)2
RuCl40
and compound 123
were prepared by following the method reported in the literature.Synthesis of Compound 2a.
At room temperature, to a suspension of NaH (1.48 g, 37.2 mmol) in THF (10 mL) was added dropwise to a THF solution (30 mL) of 2,2-dimethyl-1,3-propandiol (5.00 g, 33.8 mmol) with stirring for 30 min, and the resulting mixture was then heated to reflux for 1h. After cooling to room temperature, to this solution was added
35
1-bromo-3-methyl-2-butene (4.7 ml, 40.5 mmol) slowly in 1 h, and the mixture was refluxed for 8 h. The resulting solution was treated with water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated under reduced pressure to afford crude product 2a-1 (3.49 g, 60 %) that was purified by chromatography through a silica column (hexane/EA: 4/1).
Spectroscopic data of 2a-1:
1
H NMR (δ, CDCl3
): 5.28 (t, 1H,3 J HH
= 6.79 Hz, HC=);3.91 (d, 2H,
3 J HH
= 6.79 Hz, OCH2
); 3.42 (d, 2H,3 J HH
= 5.47 Hz, OCH2
); 3.24 (s, 2H, OCH2
); 2.81 (t, 1H,3 J HH
= 5.47 Hz, OH); 1.71, 1.63, 0.88 (s, 12H, 4 CH3
).13
C NMR (δ, CDCl3
): 136.97 (C=); 120.93 (HC=); 79.67, 72.19, 68.00 (OCH2
); 36.02 (C); 25.71, 21.92, 18.03 (4 CH3
). MS (ESI+
) m/z: 195.1506 (M+Na)+
.Compound 2a-1 (3.49 g, 20.3 mmol) was dissolved in CH
2
Cl2
(50 mL) and to this solution was slowly added PCC/celite (1:1 w/w, 6.51 g, 30.4 mmol). The resulting mixture was stirred for 3 h, and the resulting solution was diluted with hexane (50 mL)and purified by flash column to give 2a-2 (3.28 g, 95 %.) Spectroscopic data of 2a-2:
1
H NMR (δ, CDCl3
): 9.51 (s, 1H, CHO); 5.24 (m, 1H,HC=); 3.90 (d, 2H,3 J HH
= 7.07 Hz,OCH
2
); 3.36 (s, 2H, OCH2
); 1.69, 1.60, 1.03 (s, 12H, 4 CH3
).13
C NMR (δ, CDCl3
):205.52 (CH=O); 136.85 (C=); 120.92 (CH=); 74.89, 67.92 (2 OCH
2
); 47.03 (C); 25.69, 19.03, 17.99 (4 CH3
). MS (ESI+
) m/z: 193.1305 (M+Na)+
.To a solution of compound 2a-2 (3.28 g, 19.3 mmol) in THF (30 mL), was added
36
ethynylmagnesium bromide (46.2 ml, 23.1 mmol) at room temperature and under nitrogen. The solution was stirred for 14h. After quenching by aqueous NH
4
Cl solution (30 mL), the solution was extracted with ether (3x20 mL), then dried over sodium sulfate and concentrated under reduced pressure and eluted through a silica column(hexane/EA: 4/1) to give compound 2a (3.39 g, 90 % yields). Spectroscopic data of 2a:
1
H NMR (δ, CDCl3
): 5.29 (m, 1H, HC=); 4.14 (dd, 1H,3 J HH
= 7.11 Hz,4 J HH
= 1.96 Hz,CH); 3.94 (m, 2H, OCH
2
); 3.77 (d, 1H,3 J HH
= 7.11 Hz, OH); 3.58, 3.20 (2d, 2H,2 J HH
= 8.93 Hz, OCH2
); 2.41 (d, 1H,4 J HH
= 1.96 Hz, ≡CH); 1.71, 1.64, 1.06, 0.94 (s, 12H, 4CH3
).13
C NMR (δ, CDCl3
): 137.39 (=C); 120.61 (HC=); 83.48 (≡C); 77.85 (HC≡);73.26, 70.84 (2 OCH
2
); 67.96 (CH); 38.70 (C); 25.69, 22.19, 20.99, 18.03 (4 CH3
). MS (ESI+
) m/z: 219.1359 (M+Na)+
.Synthesis of Compound 2c
At room temperature, to a suspension of NaH (1.18 g, 18.6 mmol) in THF (10 mL) was added dropwise to a THF solution (30 mL) of 2,2-dimethyl-1,3-propandiol (2.79 g, 26.8 mmol) with stirring for 30 min, and the resulting mixture was then heated to reflux for 1h. After cooling to room temperature, to this solution was added (2-Bromoethylidene)cyclopentane (3.5 ml, 29.5 mmol) slowly in 1 h, and the mixture
37
was refluxed for 8 h. The resulting solution was treated with water and extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate and concentrated under reduced pressure to afford crude product 2b-1 (3.18 g, 60 %) that was purified by chromatography through a silica column (hexane/EA: 4/1).
Spectroscopic data of 2b-1:
1
H NMR (δ, CDCl3
): 5.38 (m, 1H, HC=); 3.91 (m, 2H, OCH2
); 3.47 (s, 1H, OH); 3.41 (d, 2H,2 J HH
= 5.84 Hz, OCH2
); 3.25 (s, 2H, OCH2
); 2.22, 1.62 (m, 8H, 4 CH2
); 0.89 (s, 6H, 2 CH3
).13
C NMR (δ, CDCl3
): 139.70 (=C); 119.77 (HC=); 77.52, 72.15, 69.27 (3 OCH2
); 39.58 (C); 33.49, 26.10, 25.82 (4 CH2
); 22.57 (2 CH3
). MS (ESI+
) m/z: 221.1652 (M+Na)+
.Compound 2b-1 (3.18 g, 16.1 mmol) was dissolved in CH
2
Cl2
(50 mL) and to this solution was slowly added PCC/celite (1:1 w/w, 6.91 g, 32.1 mmol). The resulting mixture was stirred for 3 h, and the resulting solution was diluted with hexane (50 mL)and purified by flash column to give 2b-2 (2.99 g, 95 %.) Spectroscopic data of 2b-2:
1
H NMR (δ, CDCl3
): 9.53 (s, 1H, CHO); 5.36 (m, 1H, HC=); 3.90 (m, 2H, OCH2
): 3.38(s, 2H, OCH
2
); 2.22, 1.61 (m, 8H, 4 CH2
); 1.05 (s, 6H, 2 CH3
).13
C NMR (δ, CDCl3
):206.42 (CHO); 142.36 (=C); 123.54 (HC=); 77.52, 68.15 (2 OCH
2
); 42.13 (C); 33.49, 28.62, 25.82 (4 CH2
); 18.62 (2 CH3
). MS (ESI+
) m/z: 219.1562 (M+Na)+
.To a solution of compound 2b-2 (2.99 g, 15.2 mmol) in THF (30 mL), was added ethynylmagnesium bromide (38.4 ml, 19.2 mmol) at room temperature and under
38
nitrogen. The solution was stirred for 14h. After quenching by aqueous NH
4
Cl solution (30 mL), the solution was extracted with ether (3x20 mL), then dried over sodium sulfate and concentrated under reduced pressure and eluted through a silica column (hexane/EA: 4/1) to give compound 2b (3.04 g, 90 % yields). Spectroscopic data of 2b:1
H NMR (δ, CDCl3
): 5.36-5.42 (m, 1H, HC=); 4.15 (dd, 1H,3 J HH
= 7.10 Hz,4 J HH
=2.00 Hz, CH); 3.93 (m, 2H, OCH
2
); 3.74 (d, 1H,3 J HH
= 7.10 Hz, OH); 3.58, 3.21 (2d, 2H,2 J HH
= 9.09 Hz, OCH2
); 4.15 (d, 1H,4 J HH
= 2.00 Hz, ≡CH); 2.22 (dt, 4H,3 J HH
= 19.12 Hz,4 J HH
= 6.67 Hz, 2 CH2
); 1.63 (m, 4H, 2 CH2
).13
C NMR (δ, CDCl3
): 148.40 (=C); 115.97 (HC=); 83.33 (≡C); 77.52, 69.27 (2 OCH2
); 73.13 (≡CH); 70.25 (CH);38.58 (C); 33.49, 28.63, 26.10, 25.82 (4 CH
2
); 21.92, 20.70 (2 CH3
). MS (ESI+
) m/z:245.1508 (M+Na)
+
.Synthesis of Complex 3. A mixture of [Ru]Cl (250 mg, 0.34 mmol), 1 (61 mg, 0.34
mmol), and NH
4
PF6
(81 mg, 0.50 mmol), in CH2
Cl2
(20 mL) was stirred at ambient temperature for one day. The resulting brown solution was filtered through a bed of Celite to remove the insoluble salts, and the pad was eluted with CH2
Cl2
until the eluent was colorless, then the solvent of the filtrate were removed under vacuum and the solid39
residue was extracted with a small volume of CH
2
Cl2
followed by re-precipitation by a 50 mL of stirred diethyl ether. Precipitates thus formed were collected in a glass frit, washed with diethyl ether/hexane 1:1 and dried under vacuum. The final product can be obtained as a light orange powder identified as 3 (210 mg, 70% yields). Spectroscopic data of 3:1
H NMR (δ, CDCl3
): 6.94-7.66 (m, 34H, Ph); 5.13 (s, 5H, Cp); 4.57 (d, 1H,3 J HH
= 10.47 Hz, CβH); 4.76, 4.54 (s, 2H, =CH2
); 4.12 (t, 2H,3 J HH
=15.11 Hz, CH2
);3.30, 3.16 (2d, 2H,
2 J HH
=12.71 Hz, CH2
); 2.35, 2.15 (m, 2H, CH2
); 0.99, 0.97 (s, 6H, 2CH3
).13
C NMR (δ, CDCl3
): 343.94 (t,3 J CP
=15.21 Hz, Cα); 147.04 (C=);128.21-134.61 (Ph); 115.64 (Cβ); 112.92 (CH
2
=); 94.32 (Cp); 76.80, 74.43, 38.69 (3 CH2
); 44.23 (CH); 38.16, 26.09 (2 CH3
); 21.90 (CH3
).31
P NMR (δ, CDCl3
): 43.24, 43.02 (2d,2 J PP
=26.35 Hz, PPh3
). MS (ESI+
) m/z: 855.2453 (M)+
.Synthesis of Complex 4. The mixture of 3 (85 mg, 0.099 mmol) and NaOMe (6.0 mg,
0.11 mmol) in MeOH (30 mL) was stirred for 5 m at room temperature. After that, solvent of the solution was removed under vacuum and then 20 mL of diethyl ether was added and the mixture was stirred using an ultrasonic cleaner. The solution was filtered through neutral Al
2
O3
to remove the insoluble salts, and then solvent of the filtrate was removed under vacuum. The final product can be obtained as a yellow solid identified40
as 4 (77 mg, 90% yield). Spectroscopic data of 4:
1
H NMR (δ, C6
D6
): 7.69-7.73 (m, 12H, Ph); 6.92-6.98 (m, 18H, Ph); 5.00, 4.79 (s, 2H, =CH2
); 4.39 (s, 5H, Cp); 4.59, 4.26 (2d, 2H,2 J HH
=14.17 Hz, OCH2
); 3.90, 3.33 (2d, 2H,2 J HH
=11.81 Hz, OCH2
); 2.86 (m, 2H, CH2
); 2.76 (m, 1H, CγH); 1.51 (s, 3H, CH3
); 1.22 (s, 3H, CH3
).13
C NMR (δ, C6
D6
):127.39-140.12 (Ph); 113.45 (Cβ); 92.69 (t,
2 J CP
=24.43 Hz, Cα); 85.44 (Cp); 110.72 (CH2
=), 151.45 (C=); 78.96, 75.42, 39.64 (3 CH2
); 26.63, 21.74 (2 CH3
); 39.59 (C).31
P NMR (δ, C6
D6
): 51.94 (s, PPh3
). MS (ESI+
) m/z: 855.2469 (M+1)+
.Synthesis of Complex 5a. Complex 4 (110 mg, 0.13 mmol) and KPF 6
(26 mg, 0.14 mmol) were added into a Schlenk flask, and CH2
Cl2
(20 mL) was added under nitrogen atmosphere. Allyl bromide (17 mg, 0.14 mmol) was added to the resulting solution which was stirred for 8 h. After that, the solution was filtered through a bed of Celite to remove the insoluble salts, then the solvent of the filtrate was removed under vacuum and the solid residue was extracted with a small volume of CH2
Cl2
followed by re-precipitation by adding to a 50 mL of stirred ethyl ether. Precipitates thus formed were collected in a glass frit, washed with ethyl ether/hexane 1:1 and dried under41
vacuum. The final product was obtained as a deep yellow powder identified as 5a (100 mg, 90% yield). Spectroscopic data of 5a:
1
H NMR (δ, CDCl3
): 7.74-6.79 (m, 49H, Ph);5.89 (m, 1H, =C(C)H); 5.11 (s, 5H, Cp); 5.11 (m, 2H, =CH
2
); 4.63, 4.55 (s, 2H, =CH2
);4.16, 3.75 (2d, 2H,
2 J HH
=14.25 Hz, OCH2
); 3.26, 2.72 (2d, 2H,2 J HH
=12.21 Hz, OCH2
);3.13, 2.77 (dd, 2H,
2 J HH
=16.79 Hz,3 J HH
=6.11 Hz, CH2
); 2.65, 2.25 (m, 2H, CH2
); 2.22(d,
3 J HH
=6.11 Hz, 1H, CH) 1.04, 0.83 (s, 6H, 2CH3
).13
C NMR (δ, CDCl3
): 349.67 (t,3 J CP
=14.04 Hz, Cα); 148.16-111.99 (Cβ, Ph, =C(CH2
)2
, =CH2
); 94.02 (Cp); 81.98(OCH
2
); 74.50 (OCH2
); 4.77 (CH); 39.97 (CH2
); 38.33 (C); 27.56 (CH2
); 26.46 (CH3
);21.00 (CH
3
).31
P NMR (δ, CDCl3
): 41.38, 40.68 (2d,2 J PP
=27.68 Hz, PPh3
). MS (ESI+
) m/z: 895.2813 (M)+
.Synthesis of Complex 5b. Complex 5b (120 mg, 93% yield) was similarly prepared
from 4 (130 mg, 0.15 mmol), KPF
6
(31 mg, 0.18 mmol) and methyl iodide (26 mg, 0.18 mmol). Spectroscopic data of 5b:1
H NMR (δ, CDCl3
):7.77-6.93 (m, 42H, Ph); 5.14 (s, 5H, Cp); 4.63, 4.41 (s, 2H, =CH2
); 4.18, 3.87 (2d, 2H,2 J HH
=14.18 Hz, OCH2
); 3.34, 2.92 (2d, 2H,2 J HH
=12.02 Hz, OCH2
); 2.49 (t, 1H,3 J HH
=12.91 Hz, CH); 2.20, 1.86 (2d, 2H,2 J HH
=11.57 Hz, CH2
); 1.76, 1.04, 0.93 (s, 9H, 3CH3
).13
C NMR (δ, CDCl3
): 350.8142
(t,
3 J CP
=15.19 Hz, Cα); 148.22 (=C(CH2
)2
); 135.23-128.42 (Cβ, Ph); 112.23 (=CH2
);93.93 (Cp); 81.00 (OCH
2
); 74.59 (OCH2
); 43.85 (CH2
); 40.15 (CH2
); 36.34 (C); 27.12 (CH3
); 21.65 (CH3
); 7.98 (CH3
).31
P NMR (δ, CDCl3
): 42.43, 41.44 (2d,2 J PP
=26.42 Hz, PPh3
). MS (ESI+
) m/z: 869.2641 (M)+
.Synthesis of Compound 6. The solution of complex 3 (150 mg, 0.17 mmol) in CDCl 3
(1.5 mL) and CH
3
CN (121 mg, 3.0 mmol, 0.15 mL) in an NMR tube was heated at 60o
C for 24 h. Then CDCl3
and CH3
CN were removed in vacuo and CH2
Cl2
(1.0 mL) was used to extract the product and diethyl ether (6.0 mL) was then added. The pale-orange precipitates thus formed was filtered and washed with diethyl ether and dried under vacuum to give [Ru]NCCH3 +
. The filtrate was evaporated to dryness under vacuum and the crude product purified by flash chromatography (silica gel, hexanes/EtOAc = 10/1) to afford 6 (26 mg, 92%). Spectroscopic data for 6:
1
H NMR (δ, CDCl3
): 4.92, 4.83 (s, 2H, =CH2
); 4.23, 4.13 (2d, 2H,2 J HH
=14.51 Hz, OCH2
); 3.35, 3.13 (2d, 2H,2 J HH
=12.51 Hz, OCH2
); 2.48 (m, 2H, CH2
); 2.23 (dt, 1H,3 J HH
=9.54 Hz,4 J HH
= 2.50 Hz, CH); 2.10 (d, 1H,4 J HH
=2.50 Hz, HC≡); 1.03, 0.94 (s, 6H, 2CH3
).13
C NMR (δ, CDCl3
): 147.49 (=C); 112.67 (H2
C=); 85.72 (≡C); 77.28, 74.59, 36.23 (3CH2
); 70.97 (HC≡); 34.21 (≡C);43
37.74 (C); 25.31, 20.92 (3 CH
3
). MS (ESI+
) m/z: 165.1217(M+1).Synthesis of Compound 7a. The solution of 1 (65 mg, 0.36 mmol) and [Ru]NCCH 3 +
(78 mg, 0.11 mmol) in a 2:1 cosolvent of CHCl
3
/MeOH was heated to 50o
C for 1 day.Then the solvent was removed under vacuum and 1 mL of CH
2
Cl2
was used to extract the crude product. This is followed by re-precipitation by a 10 mL of stirred diethyl ether. The pale-orange precipitates thus formed were filtered and washed with diethyl ether and dried under vacuum to give [Ru]NCCH3 +
. The filtrate was evaporated to dryness under vacuum and the crude product purified by flash chromatography (silica
gel, hexanes/EtOAc = 10/1) to afford 7a (55 mg, 77%). Spectroscopic data for 7a:
1
H NMR (δ, CDCl3
): 5.81 (m, 1H, =C(C)H); 5.70 (m, 1H, =C(C)H); 4.12, 3.19 (2d, 2H,2 J HH
=14.07 Hz, OCH2
); 3.44, 3.11 (2d, 2H,2 J HH
=12.27 Hz, OCH2
); 3.28 (s, 3H, OCH3
);2.62 (2d, 1H,
2 J HH
=12.82 Hz, CH2
); 2.12, 1.92 (m, 2H, CH2
); 2.05 (br, 1H, CH); 1.43 (dd, 1H,2 J HH
=12.82 Hz,3 J HH
=7.31 Hz, CH2
); 1.14, 0.81 (s, 6H, 2CH3
).13
C NMR (δ, CDCl3
): 128.96, 126.25 (2C=); 83.09, 81.60 (2OCH2
); 78.42 (C); 49.29 (OCH3
); 44.46 (CH); 38.01 (C); 36.57, 28.91 (2CH2
); 26.67, 23.75 (2CH3
). MS (ESI+
) m/z: 197.810244
(M+H)
+
.Synthesis of Compound 7b. Compound 7b (49 mg, 70% yield) was similarly prepared
from 1 (61 mg, 0.33 mmol), [Ru]NCCH
3 +
(73 mg, 0.10 mmol) in a 2:1 cosolvent of
CHCl
3
/EtOH, and the solution was heated to 50o
C for 1 day. Spectroscopic data of 7b:1
H NMR (δ, CDCl3
): 5.80 (m, 1H, =C(C)H); 5.70 (m, 1H, =C(C)H); 4.11, 3.22 (2d, 2H,2 J HH
=13.96 Hz, OCH2
); 3.53 (m, 2H, OCH2
); 3.43, 3.11 (2d,2 J HH
=12.16 Hz, 2H, OCH2
);2.61 (d, 1H,
2 J HH
=12.77 Hz, CH2
); 2.14, 1.93 (m, 2H, CH2
); 2.04 (br, 1H, CH); 1.47 (dd, 1H,2 J HH
=13.30 Hz,3 J HH
=7.48 Hz, CH2
); 1.15 (t, 3H,3 J HH
=7.06 Hz, CH3
); 1.13 (s, 3H, CH3
); 0.81 (s, 3H, CH3
).13
C NMR (δ, CDCl3
): 128.94, 126.34 (2C=); 83.08, 81.60 (2 OCH2
); 78.42 (C); 49.29 (OCH3
); 44.46 (CH); 38.01 (C); 36.57, 28.91 (2 CH2
); 26.67, 23.75 (2 CH3
). MS (ESI+
) m/z: 211.8102 (M+H)+
.Synthesis of Compound 7c. Compound 7c (30 mg, 43% yield) was similarly prepared
from 1 (57 mg, 0.31 mmol), [Ru]NCCH
3 +
(68 mg, 0.093 mmol) in a 2:1 cosolvent of
CHCl
3
/i-PrOH, and the solution was heated to 50o
C for 1 day. Spectroscopic data of 7c:1
H NMR (δ, CDCl3
): 5.78 (m, 1H, =C(C)H); 5.68 (m, 1H, =C(C)H); 4.11, 3.23 (2d, 2H,2 J HH
=13.95 Hz, OCH2
); 3.94 (Septet, 1H,3 J HH
=6.18 Hz, CH); 3.44, 3.12 (2d, 2H,2 J HH
=12.12 Hz, OCH2
); 2.58 (d, 1H,2 J HH
=12.68 Hz, CH2
); 2.12, 1.91 (m, 2H, CH2
);2.03 (br, 1H, CH); 1.51 (dd, 1H,
2 J HH
=12.51 Hz,3 J HH
=7.33 Hz, CH2
); 1.14(s, 3H,45
CH
3
);0.81 (s, 3H, CH3
); 1.13 (s, 6H, 2 CH3
).13
C NMR (δ, CDCl3
): 128.86, 126.38 (2C=); 83.16, 83.07 (2 OCH2
); 79.03 (C); 64.23 (OCH); 44.53 (CH); 37.98 (C); 37.64, 31.07 (2 CH2
); 26.70, 25.54, 25.33, 23.76 (4 CH3
). MS (ESI+
) m/z: 247.1661 (M+Na)+
.Synthesis of Complex 8. A mixture of [Ru]Cl (148 mg, 0.21 mmol), 2a (50 mg, 0.25
mmol), and NH
4
PF6
(85 mg, 0.32 mmol), in CH2
Cl2
(20 mL) was stirred at ambient temperature for one day. The resulting brown solution was filtered through a bed of Celite to remove the insoluble salts, and the pad was eluted with CH2
Cl2
until the eluent was colorless, then the solvent of the filtrate were removed under vacuum and the solid residue was extracted with a small volume of CH2
Cl2
followed by re-precipitation by a 50 mL of stirred diethyl ether. Precipitates thus formed were collected in a glass frit, washed with diethyl ether/hexane 1:1 and dried under vacuum. The final product can be obtained as a deep yellow powder identified as 8 (147 mg, 68% yields).The ratio of anti- and syn- isomers is 1:0.5.
Spectroscopic data for the anti-isomers:
1
H NMR (δ, CD2
Cl2
): 7.93- 6.99 (m, 70H, Ph);5.12 (s, 5H, Cp); 5.10, 4.89 (s, 2H, 2 HC=); 4.23 (dt, 1H,