Structure and magnetic properties of the Fe
3O
4„001… surface: Ab initio studies
C. Cheng*
Department of Physics, National Cheng Kung University, Tainan, Taiwan, Republic of China 共Received 1 August 2004; published 10 February 2005兲
First-principles calculations are employed to study four frequently proposed surface models for the Fe3O4共001兲 surface, i.e., the A-terminated surface 共A model兲, the A-terminated surface with Fe vacancy 共A-vac model兲, the B-terminated surface 共B model兲, and the B-terminated surface with O vacancy 共B-vac model兲.
However, calculations have revealed that the outmost surface Fe atoms of the four surface models are all from the B-type layers, i.e., the A-site Fe atoms which were originally situated at the surface, according to the ideal bulk-terminated positions for the A and A-vac models moved underneath the next B-type layer following relaxation processes. Magnetic degradation of the four surface models is demonstrated to be mainly due to atomic relaxations. The half-metallic property remains in the B and B-vac models but destroyed in the A and A-vac models. The relative stability of the four surface models is also discussed.
DOI: 10.1103/PhysRevB.71.052401 PACS number共s兲: 75.50.Gg, 75.70.Rf, 68.47.Gh Thin films of ferrites have technological importance as
catalysts, anticorrosives, and magnetic devices. In particular, magnetite, as a half-metallic material, is an attractive candi- date for applications in spin electronics and magneto record- ing. In applications of magnetite in thin magnetic films the morphology of the layers as well as the structure and com- position of the surface are crucial factors for the functional- ity. In this respect, knowledge of the surface structure on an atomic scale is important for understanding the electromag- netic behavior of Fe3O4 thin films. Despite this fact there is still a lot of controversy on the termination and the structure of the 共001兲 surface of Fe3O4.1–3 We have used ab initio methods to study the共001兲 surface properties of Fe3O4. Cal- culations show interesting results of atomic relaxations near surfaces which are beyond the conventional conjectures and also provide information on how the degradation of magnetic properties on the surface arises.
Bulk Fe3O4 has a cubic inverse-spinel structure at room temperature where the O2− anions form a fcc lattice, one- third of the Fe ions共Fe3+兲 occupy the tetrahedral interstices 共A sites兲, and the other two-thirds of the Fe ions 共half Fe3+
and half Fe2+兲 are located in the octahedral interstices 共B sites兲. Seen from the 共001兲 face of the crystal, the bulk unit cell can be described by four pairs of alternating atomic sublayers, which are shifted in the plane of the layer with respect to each other. Within a pair, one sublayer is com- posed of the A-site Fe3+ cations and the other sublayer is composed of the B-site Fe3+, Fe2+ cations, and O2− anions.
Up to date, a significant number of attempts has been under- taken to understand the structural changes taking place at the Fe3O4共001兲 surface. However, the experimental results until now have not led to a consistent determination of the atomic arrangement of the surface layer.
In the present study, we performed ab initio calculations to investigate the structural and magnetic properties of four frequently proposed Fe3O4共001兲 surface models, i.e., the A-terminated surface共A model兲, the B-terminated surface 共B model兲, and the A-vac and B-vac models. The A-vac model is the A model with an ordered array of tetrahedral Fe vacan- cies 共half monolayer of Fe兲 and the B-vac model is the B model with one O vacancy per共
冑
2⫻冑
2兲 surface.The total energy of systems is determined in the frame-
work of spin-polarized density functional theory.4 The pro- posed generalized gradient approximation by Perdew and Wang5 is used for the nonlocal correction to a purely local treatment of the exchange-correlation potential and energy.
The single-particle Kohn-Sham equations6 are solved using the plane-wave-based Vienna ab initio simulation program 共VASP兲 developed at the Institut für Material Physik of the Universität Wien.7The interactions between the ions and va- lence electrons are described by the projector augmented- wave method8in the implementation of Kresse and Joubert.9 The numbers of treated valence electrons are 8 and 6 for Fe and O atoms, respectively. The energy cutoff for the plane- wave basis is 400 eV in all calculations. The Monkhorst-Pack10 method of sampling k points is used for the Brillouin-zone integration.
The calculated lattice constants 共a0兲 for the cubic bulk Fe3O4 is 8.308 Å and the u parameter is 0.0044; they com- pare well with the experimental values11 of 8.394 Å and 0.0048. Here the k-point set used corresponds to the Monkhost-Pack parameters 共6 6 6兲 for the cubic unit cell which consists of 24 Fe atoms and 32 O atoms. The calcu- lated local moments for the A-site and B-site Fe atoms in the bulk Fe3O4are 3.38B共minority spin兲 and 3.50B共majority spin兲, repsectively, which compare well with the previous calculations.12
All surface systems are simulated by periodic slabs sepa- rated by vacuum. A thickness of at least 11.4 Å, i.e., 11 interlayer spacings between atomic layers along the Fe3O4共001兲 direction, for vacuum is used in the simulations.
A slab consisting of nine atomic planes was used in the ini- tial siumlation and a thicker slab consisting of 17 atomic planes was used to check the convergence in the thickness of the slabs. Exactly the same sizes of supercells were used for the four surface models in the two sets of calculations. The tests showed that the atomic displacments for atoms near the surface can be described quite well using the thin slabs.
However, the atomic relaxation actually goes down quite deep into the slab, i.e., at least six atomic layers when counted from the surface 共see Fig. 3兲. The relative energy changes, as due to using slabs of different thickness, are not similar for the four models either; they are much larger for the A and A-vac models than for the B and B-vac models.
PHYSICAL REVIEW B 71, 052401共2005兲
1098-0121/2005/71共5兲/052401共4兲/$23.00 052401-1 ©2005 The American Physical Society
Unless otherwise specified, the presented results are from the calculations using the thick slabs. The k-point set used in the supercells is defined by the Monkhorst-Pack parameters 共6 6 2兲 corresponding to the same density of k points in the lateral direction as in the bulk and somewhat higher density in the film-growing direction. Relaxation processes are ac- complished by moving atoms to the positions at which all atomic forces are smaller than 0.02 eV/Å.
The calculated atomic structures of the A-vac and B-vac models are presented in Figs. 1 and 2. Only atoms from the first two共B-vac兲 or three 共A-vac兲 atomic layers 共atomic lay- ers according to the bulk structure, i.e., the A-type and B-type layers兲 are shown in the figures. The lateral positions of the atoms are plotted according to the calculated relaxed structures and the coordinates perpendicular to the surface 共hereafter denoted as the z direction兲 are written explicitly in the figures and schematically displayed by using different sizes of symbols for A-site Fe and different degrees of dark- ness for oxygen atoms. The zero of the z-direction coordinate is chosen to be at the position of the outmost surface atoms.
The squares with dashed borders correspond to the 共
冑
2⫻
冑
2兲R45° surface cell.The most surprising results in the relaxation calculations are that the outmost Fe atoms in the four considered surface models were all found to be the B-site ones. This possibility has never been conjectured in the previous studies of Fe3O4共001兲 surface structure. In fact, the A-type Fe atoms in the A and A-vac models which were originally, according to the ideal bulk-terminated positions, situated at the outmost positions moved underneath the next B-type layer following relaxation processes. Simulations using slabs of both thick- nesses have led to similar results in this respect. The surface B-type layer which consists of both B-site Fe and O atoms further splits into three layers and some of the O atoms be- come the outmost surface atoms. For the four surface models considered here, the outmost surface atoms are always found to be O atoms. This might be due to the fact that being the outmost atoms allows O anions, which attract more valence electrons than the Fe cations, to spread out electron clouds into the vacuum to reduce kinetic energy and accordingly lower the systems’ energy. The interlayer distance between the surface O layer and the second 共counted from the sur-
face兲 A-type layer is around 1.2 Å. This is much smaller than 2.08 Å, the interlayer distance of the nearest-neighbor A-type layers in the bulk.
In the lateral direction the distances between O atoms near the surface in the A and A-vac models can deviate from that of the bulk value, i.e., 2.94 Å, as shown in Fig. 1. In the A model, the O-O distance of 1-2 and 7-8 is 3.4 Å while that of 4-5 is 3.2 Å. In the A-vac model the O-O distances, when compared to those in the A model, can be even larger, e.g., that of 7-8, due to the submerged A-site Fe, or smaller, e.g., that of 1-2, due to the Fe vacancy. In general, the O-O dis- tances across the atomic chains of A-site Fe were found larger than that in the bulk while those across B-site Fe were smaller. Similar conclusions can be drawn for the B and B-vac models.
In the B-vac model, one notices that the interlayer dis- tance between the surface B-site Fe layer and the next A-site Fe layer is only 0.8 Å. The same result was also found in the B model. Similar to the A and A-vac models, the surface B-type layer also split into three layers with O atoms being the outmost surface atoms. In the lateral direction the dis- tances between O atoms can deviate much from that in the bulk due to the presence of O vacancy, e.g., the O-O dis- tances of 1-2 and 7-1U in Fig. 2. In the B model, the O-O distance of 1-2 and 7-8共see Fig. 1 for the whereabouts of the O atom denoated as 8兲 is about 3.5 Å. The O vacancy in the B-vac model can be chosen to be different from the present one, e.g., the vacancy can be due to the absence of the O atom 3 in Fig. 2 instead. However, it was found that this alternative choice is about 共0.5 eV兲/共
冑
2⫻冑
2兲 higher in en- ergy than the present one.In the present study we have found that the atomic relax- ation plays an important role in degradation of the surface magnetic properties. To demonstrate this, we shall present the calculated results of the four surface models in the ideal structures, i.e., the bulk-terminated structures without atomic relaxation, and compare them with those of the relaxed ones.
However, first we shall make a guess at how the surface magnetic properties of the ideal surface structures can be different from the bulk by assuming that the O atoms in the systems have a strong tendency to stay as O2−anions. In bulk Fe3O4 every A-site Fe3+ cation provides 43 electrons to each FIG. 1. The calculated relaxed structure of the A-vac model. For details please see the text.
BRIEF REPORTS PHYSICAL REVIEW B 71, 052401共2005兲
052401-2
of the four O atoms bonded tetrahedrally to it while every B-site Fe cation, which is half Fe3+ and half Fe2+, provides
5
2⫻16=125 electrons to each of the six O atoms bonded octa- hedrally to it. Every O atom in the bulk Fe3O4is bonded to three B-site Fe and one A-site Fe which, according to the previous counting, makes it an O2− anion.
In the A model, considering a共
冑
2⫻冑
2兲R45° surface cell henceforth, the missing bonds for atoms near the surface can be easily identified from the ideal A-terminated surface. They include four O atoms for the two surface A-site Fe atoms 共4⫻34兲, four O atoms for the four surface B-site Fe atoms 共4⫻125兲, and four B-site Fe atoms for the four surface O atoms 共4⫻125兲. To keep the oxygen in the system as O2−anions, there are still three electrons to be distributed among the surface Fe atoms. If the three electrons are distributed only on the two surface A-site Fe atoms, the spin moment of the minority-spin electrons is reduced共as the d orbital for the A-site Fe3+cations is already half filled兲 and the surface has more magnetic moments than that of the bulk. Another pos- sibility is that the three electrons are distributed not only on the two surface A-site Fe atoms but also on the four surface B-site Fe atoms. The resulting effect will depend on how the three electrons are distributed among these six Fe atoms and the enhancement of the magnetic moment is lowered when it is compared to the previously discussed case. A similar dis- cussion can be applied to the other three surface models and all are found to have larger magnetic moments than that of the bulk.
Qualitatively similar results were indeed obtained in our calculations for the four surface models in the ideal struc- tures as listed in Table I. The magnetic moments of the ideal surface structures are enhanced due to surface effects and for
the A-vac, B, and B-vac models, even the surface O atoms were observed to have non-negligible local moments of around 0.4B. However, the enhancement of surface mag- netic moments was either reversed to degradation or much reduced when atomic relaxation was allowed to take place 共Table I兲.
In the relaxed A and A-vac models, the local moments for the submerged A-site Fe atoms are about 0.3B and 0.2B
less than that in the bulk. However, the local moments for the surface B-site Fe are also reduced which results in a total of slight magnetic degradation on the surface for the A model and almost no surface magnetic degradation for the A-vac model. The lateral-plane averaged electron distribution for the A-vac model is shown in Fig. 3 with the indicated z-direction positions of the ideal共bulklike兲 and the relaxed A-site and B-site atoms. The magnetic polarizations of the outmost surface layer for the four relaxed models were all found to be corresponding to the majority spin as in the relaxed A and A-vac models the outmost surface Fe atoms are the B-site Fe atoms. In the relaxed B-vac model, the local
FIG. 3. The lateral-plane averaged electron distribution for the relaxed A-vac model. “up” and “dn” represent the majority-spin and minority-spin electrons, respectively. The ideal and relaxed atomic positions for the A-site and B-site Fe are also indicated.
FIG. 2. The calculated relaxed structure of the B-vac model. For details please see the text.
TABLE I. The changes in magnetic moments共positive for in- crease and negative for decrease兲 of the four surface models in the ideal and relaxed structures with respect to the calculated bulk mag- netic moments. The units areBper共
冑
2⫻冑
2兲 surface area.A A-vac B B-vac
Ideal ⫹0.4 ⫹2.0 ⫹2.9 ⫹2.4
Relaxed ⫺1.1 ⫹0.1 ⫺3.6 ⫺1.8
BRIEF REPORTS PHYSICAL REVIEW B 71, 052401共2005兲
052401-3
moments for the surface B-site Fe atoms which are not around the O vacancy 共numbers 1 and 4 in Fig. 2兲 were found to be about 0.9B lower than that in the bulk while those for the other two surface B-site Fe atoms共numbers 2 and 3 in Fig. 2兲 remain roughly the same as that in the bulk.
In the relaxed B model, all four surface B-site Fe atoms have moments about 0.8B lower than that of the bulk B-site Fe.
Overall, the relaxation effect is the major reason why the magnetic properties of these four surface models are de- graded.
The half-metallic property of the bulk Fe3O4 remains for the B and B-vac models. For the A-vac model, there is no longer a gap in the density of states for the majority-spin electrons and the system is transferred to a normal metal due to the surface structure. For the A model, although the gap in the density of states for the majority-spin electrons is still there, the Fermi level is located at about 0.1 eV below the lower edge of the gap, which makes it also a normal metal.
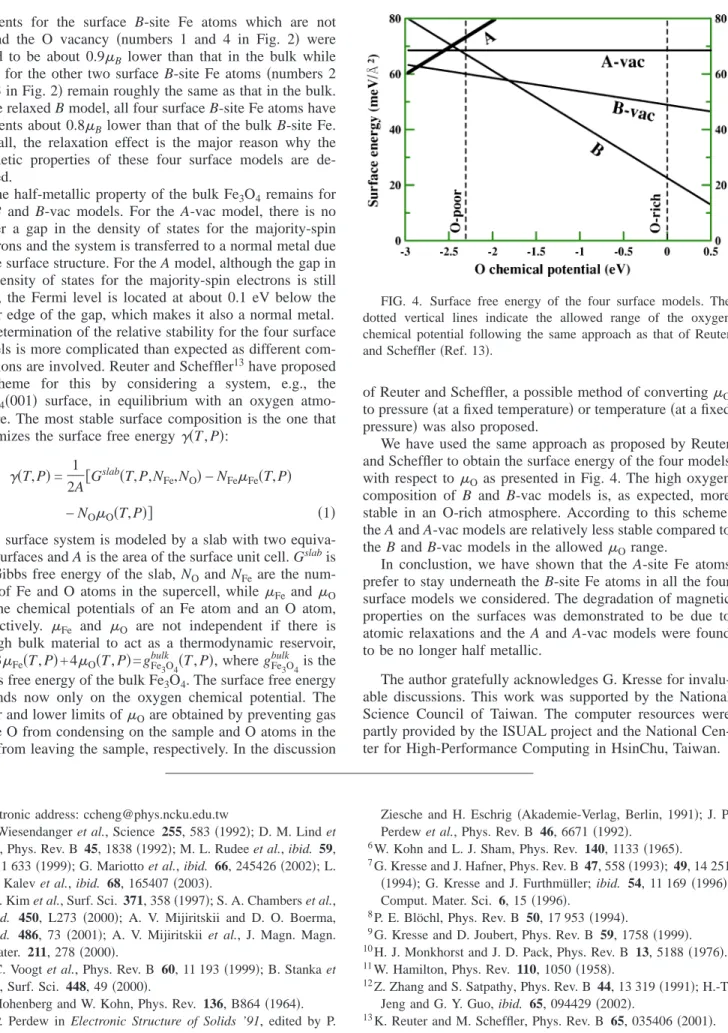
Determination of the relative stability for the four surface models is more complicated than expected as different com- positions are involved. Reuter and Scheffler13have proposed a scheme for this by considering a system, e.g., the Fe3O4共001兲 surface, in equilibrium with an oxygen atmo- sphere. The most stable surface composition is the one that minimizes the surface free energy␥共T, P兲:
␥共T,P兲 = 1
2A关Gslab共T,P,NFe,NO兲 − NFeFe共T,P兲
− NOO共T,P兲兴 共1兲
if the surface system is modeled by a slab with two equiva- lent surfaces and A is the area of the surface unit cell. Gslabis the Gibbs free energy of the slab, NOand NFe are the num- bers of Fe and O atoms in the supercell, whileFe andO
are the chemical potentials of an Fe atom and an O atom, respectively. Fe and O are not independent if there is enough bulk material to act as a thermodynamic reservoir, i.e., 3Fe共T, P兲+4O共T, P兲=gFe3O4
bulk 共T, P兲, where gFe3O4 bulk is the Gibbs free energy of the bulk Fe3O4. The surface free energy depends now only on the oxygen chemical potential. The upper and lower limits ofOare obtained by preventing gas phase O from condensing on the sample and O atoms in the slab from leaving the sample, respectively. In the discussion
of Reuter and Scheffler, a possible method of convertingO
to pressure共at a fixed temperature兲 or temperature 共at a fixed pressure兲 was also proposed.
We have used the same approach as proposed by Reuter and Scheffler to obtain the surface energy of the four models with respect to Oas presented in Fig. 4. The high oxygen composition of B and B-vac models is, as expected, more stable in an O-rich atmosphere. According to this scheme, the A and A-vac models are relatively less stable compared to the B and B-vac models in the allowedOrange.
In conclustion, we have shown that the A-site Fe atoms prefer to stay underneath the B-site Fe atoms in all the four surface models we considered. The degradation of magnetic properties on the surfaces was demonstrated to be due to atomic relaxations and the A and A-vac models were found to be no longer half metallic.
The author gratefully acknowledges G. Kresse for invalu- able discussions. This work was supported by the National Science Council of Taiwan. The computer resources were partly provided by the ISUAL project and the National Cen- ter for High-Performance Computing in HsinChu, Taiwan.
*Electronic address: [email protected]
1R. Wiesendanger et al., Science 255, 583共1992兲; D. M. Lind et al., Phys. Rev. B 45, 1838共1992兲; M. L. Rudee et al., ibid. 59, R11 633共1999兲; G. Mariotto et al., ibid. 66, 245426 共2002兲; L.
A. Kalev et al., ibid. 68, 165407共2003兲.
2Y. J. Kim et al., Surf. Sci. 371, 358共1997兲; S. A. Chambers et al., ibid. 450, L273 共2000兲; A. V. Mijiritskii and D. O. Boerma, ibid. 486, 73共2001兲; A. V. Mijiritskii et al., J. Magn. Magn.
Mater. 211, 278共2000兲.
3F. C. Voogt et al., Phys. Rev. B 60, 11 193共1999兲; B. Stanka et al., Surf. Sci. 448, 49共2000兲.
4P. Hohenberg and W. Kohn, Phys. Rev. 136, B864共1964兲.
5J. P. Perdew in Electronic Structure of Solids ’91, edited by P.
Ziesche and H. Eschrig共Akademie-Verlag, Berlin, 1991兲; J. P.
Perdew et al., Phys. Rev. B 46, 6671共1992兲.
6W. Kohn and L. J. Sham, Phys. Rev. 140, 1133共1965兲.
7G. Kresse and J. Hafner, Phys. Rev. B 47, 558共1993兲; 49, 14 251 共1994兲; G. Kresse and J. Furthmüller; ibid. 54, 11 169 共1996兲;
Comput. Mater. Sci. 6, 15共1996兲.
8P. E. Blöchl, Phys. Rev. B 50, 17 953共1994兲.
9G. Kresse and D. Joubert, Phys. Rev. B 59, 1758共1999兲.
10H. J. Monkhorst and J. D. Pack, Phys. Rev. B 13, 5188共1976兲.
11W. Hamilton, Phys. Rev. 110, 1050共1958兲.
12Z. Zhang and S. Satpathy, Phys. Rev. B 44, 13 319共1991兲; H.-T.
Jeng and G. Y. Guo, ibid. 65, 094429共2002兲.
13K. Reuter and M. Scheffler, Phys. Rev. B 65, 035406共2001兲.
FIG. 4. Surface free energy of the four surface models. The dotted vertical lines indicate the allowed range of the oxygen chemical potential following the same approach as that of Reuter and Scheffler共Ref. 13兲.
BRIEF REPORTS PHYSICAL REVIEW B 71, 052401共2005兲
052401-4