I
國立交通大學
物理研究所
碩士論文
N-甲基乙酰胺分子上的能量耗散途徑
Energy relaxation pathways in
N-methylacetamide molecules
研究生:翁瑞辰
指導教授:張正宏 副教授
中華民國一百零二年五月
II
N-甲基乙酰胺分子上的能量耗散途徑
學生:翁瑞辰 指導教授:張正宏
國立交通大學物理研究所碩士班
中文摘要
為了了解生物相關分子上的能量耗散途徑,我們使用 GROMACS 軟體架構出 N-methylacetamide molecules 模型,計算原子的速度與位置組態,由速度組態計 算出功率譜,找出譜上特定化學鍵峰值隨時間的變化,以及不同振動模態隨時間 的消長,分析能量傳遞路徑和遲豫時間。在幾種不同的操作方式下,我們得到很 穩定一致的遲豫時間,約為 4.5~5.0 ps,這和最近實驗觀察到的遲豫時間 4.9 ps 及瞬時正則模數值模擬的總耗散時間 11.7 ps 很相近。「時變功率譜」分析成為另 一種描述分子內能量耗散的可用工具,有別於 anti-Stokes Raman 分析及瞬時正則 模分析。III
Energy relaxation pathways in N-methylacetamide molecules
Student: Ruei-Chen Weng Advisor: Dr. Cheng-Hung Chang
Institute of physics
National Chiao Tung University
Abstract
In order to understand the energy transduction path way in biologically relevant molecules, we use GROMACS to construct N-methylacetamide molecule models, calculate their atomic speeds and configurations, convert these information to power spectral density, extract the evolution of the frequency peaks of specific chemical bonds, find out the relations between peak variations, and analyze the paths of energy transfer and relaxation time. Under various protocols, we obtain fairly stable and consistent relaxation time around 5.0ps, which is close to the experimentally measured relaxation time 4.9ps and the total relaxation time 11.7ps calculated by the instantaneous normal mode analysis. The time-dependent power spectrum analysis turns out to be another feasible tool for analyzing intra molecular energy relaxations, beyond the anti-Stokes Raman analysis and the instantaneous normal mode analysis.
IV
誌謝
首先要謝謝張正宏老師不辭辛勞且耐心的指導,不管是論文的撰寫或是研究上的 討論真的讓我受益匪淺,對於能被張老師指導感到非常幸運。謝謝邦杰學長教我 程式與軟體技能讓我在研究問題上能夠更順利更方便的進行,且對於解決問題開 始感到有趣,碰到困難時給予許多研究上的資源幫我渡過許多難關。也要感謝爸 爸媽媽還有姊姊、姊夫們,在我讀碩班的期間給予支持與肯定,讓我更有信心的 讀完碩士班。 在讀碩士班的期間謝謝我的同學翌、彥勳、烜任、郃諺、郁竣、閔琨說說笑笑的 319 解決了不少研究以及修課上帶來的壓力。博士班的唐老大與陳琳元學長,在 唐老大身上總可以挖出私囊行程外出走走增添不少生活色彩與陳琳元討論許多 電腦上的知識增加不少觸角。同實驗室的德明學長以及宏慶學長總是適時的給與 建議點出研究上的漏洞,讓我能及時的修正。感謝袁珮宏這個當了七年的學長, 給我許多幫忙讓我能快速的適應碩士班生活。還有當然少不了碩士班的學弟賜得 與冠智,那些一起吃吃喝喝一起奮鬥量力的日子快樂又充實,為我的碩士班生活 帶來了許多驚喜。 另外要感謝交大羽球隊的廖老師以及王老師,一圓加入校隊夢想之於還點出許多 態度上需要改進以及注意的缺點,還有交大羽球隊的大家互相扶持,讓我除了在 課業之外能精進我的球技,感謝那些一起流汗一起努力一起挨罵的時光。 特別感謝斑馬、承翰、雅欣、胖子那些在我焦躁不安的時候給我許多的鼓勵的朋 友,謝謝你們讓我有力氣與勇氣繼續面對困境。還有在碩士班生涯裡關心我照顧 我的人,謝謝你們,讓我能夠為我的碩士班畫下完美的句點。V
目錄
第一章研究背景 ... 1
第二章理論基礎 ... 5
2.1 速度相關函數(Velocity Auto-correlation function) ... 5
2.2 傅立葉變換( Fourier Transformation) ... 5 2.3 功率譜(Power spectrum) ... 8 第三章模型建構與模擬計算 ... 10 3.1 系統模型建構 ... 11 3.1.1 NMA200 模型建構 ... 12 3.1.2 NMA_water 模型建構... 14 3.2 功率譜計算(Power spectrum) ... 15 第四章結果與討論 ... 20 4.1 系統期望值 ... 20 4.1.1 速度相關函數的期望值 ... 21 4.1.2 功率譜的期望值 ... 22 4.2 不同時間區間下的功率譜 ... 24 4.3 不同溫度下的遲豫時間 ... 27 4.4 結論 ... 30 第五章未來工作 ... 31 參考文獻 ... 32
1
第一章研究背景
大自然是一個不斷趨向平衡的系統,大到宇宙小到基本粒子,當一個區域有過多 的能量,它就會以不同方式將能量傳遞出去,讓系統趨向平衡。但是能量是怎麼 耗散出去的,是個有趣的問題。在此論文我們探討在分子尺度微小世界的能量耗 散行為,包含能量經由什麼模態轉換傳遞出去,耗掉多少時間,以及傳遞過程和 熱庫溫度的關係。 分子以分子間作用力與它的周圍環境相耦合,當分子有過多能量時,會藉由這些 耦合將能量釋放到周圍環境,在以下討論中的周圍環境即為熱庫(heat bath)。1993 年A. Tokmakoff等人發現當一個溶質分子周圍液體的溫度改變時,液體密度、振 動聲子數量將隨之改變[1]。液體密度升高會提升非諧(anharmonic)交互作用的貢 獻,而聲子數量的改變會影響振動鬆弛(vibration relaxation)。在此工作,作者將兩 種溶質W(CO)6和Cr(CO)6溶於兩種溶劑四氯化碳(CCl4)和氯仿(CHCL3 chloroform)當中,形成四種溶液(a)W(CO)6/(CCl4),(b)W(CO)6/CHCL3,(c)Cr(CO)6/(CCl4),以及
(d)Cr(CO)6/CHCL3。利用2 picoseconds pulse的雷射去激發溶質的CO鍵,觀察它們
的振動生命周期(vibration lifetime)τ。研究發現,溶液(a)(c)(d)的τ隨熱庫溫度增 高而減小,也就是溫度高能量傳遞效率會變好。然而,溶液(b)的τ會隨溫度增高 上升,有別於其他溶液。為闡述此現象,讓我們觀察跟振動生命周期τ成反比的 振動鬆弛常數(vibrational relaxation constant) K = π
ℏ ρ 〈V
( )〉 (n + 1) ,其中ρ
是聲子態密度,np是聲子佔據數(phonon occupation number),<V>是和系統位能有
關的非諧偶和矩陣。CCL4與CHCL3是兩個很相似的溶劑,彼此有著相近的ρ。隨
著溫度升高,兩液體的液體密度會下降,造成分子間交互作用下降,使得Debye frequency的減小,因此遠離Debye frequency的聲子態密度ρ會隨溫度升高而下降
2
加幅度,然而溶劑CHCL3的ρ下降幅度會蓋過np的增加幅度,因為CHCL3的np增
加較緩和。後者是人們認為(b)的τ為何隨溫度增高上升的原因[2]。
1999 年 C.W.Rella 等人使用自由電子雷射(free electron laser,FEL)觀察血清肌紅素 (Myoglobin)的醯胺(Amid I)鍵,FEL 的脈衝半高寬大約是 1ps,樣品是結晶狀的 Myoglobin,溶劑是 D2O 與甘油(glycer(ol-d3))以 1:4 體積比混合而成。實驗指出
在不同溫度下,傅立葉轉換紅外光譜儀吸收光譜(FTIR absorption spectra)的形狀寬 度差不多,且醯胺鍵振動頻率位置並沒有太大的改變,暗示著醯胺鍵結對溫度相 依性低。在此系統熱庫的頻率處在約 500[cm ]以下的低頻區,高於此區被稱為 高頻區。理論上溫度升高,熱庫模態(bath mode)增多,振動的傳遞加快,能量 relax 的速率會增加。但既然溫度對於鍵結吸收光譜沒有影響,顯示高頻 modes 不是直 接轉成熱庫模態,也就是該鍵的振動能量會先在高頻 modes 之間傳遞[3]。 2009年Shigeto等人以三階段模型來描述分子能量耗散過程經過的途徑。第一個階 段是高頻振動模態(vibration mode)的能量傳至中、低頻的振動模態。第二階段為 中頻振動模態能量傳至與低頻振動模態與熱庫。第三階段為低頻振動模態能量傳 至熱庫。為驗證此模型,實驗上使用超快雷射脈衝,將一道紅外脈衝分別打在甘 胺酸(glycine, GLY)、N-甲基乙酰胺(N-methylacetamide, NMA)、及苯甲酸離子 (Benzoate anion, BZ)樣品上,激發碳氫鏈(chain)並測量anti-Stokes Raman頻譜以觀察 能量在不同振盪模態間的轉移。實驗發現GLY與BZ高頻振盪模態的能量直接耗散 到熱庫的量遠高於耗散到其他低頻振盪模態量。反之,NMA的能量是間接耗散
到熱庫的,其中NMA能量由CH3模態間接傳遞到熱庫模態的總生命週期為4.9
3
2010 年 Bastida 進一步利用非線性分子模擬來驗證分子上能量的傳輸途徑。在平 衡態下瞬間置換 NMA 的甲基振動模態來模擬碳氫鍵被激發,觀察計算出的遲豫 時間第一階段為 0─0.14ps,第二階段為 0.14─1.84ps,第三階段為 1.84ps─10.6ps。 在模擬中考慮了三種不同熱庫,分別為 semirigid D2O、rigid D2O、及 vacuum 環境。
在實際的 D2O 分子,氫氧鍵長能微量伸縮,氫-氧-氫的鍵角能小幅擺動。在 semirigid 模型,鍵長是固定的,但鍵角依舊可以擺動。在 rigid 模型,鍵角與長度 皆固定。在 vacuum 則是無熱庫的真空狀態。當能量傳遞進行到第一階段,三個 環境的遲豫時間皆與熱庫為彈性(flexible)D2O 時的計算結果相同,依此判斷在第 一階段的能量只在模態與模態之間傳遞,沒有耗散到溶劑熱庫中。在第二階段三 種水分子模型皆有相同的遲豫時間,而真空熱庫模型的遲豫時間則有明顯的上升, 這表示第二階段的能量已經不再單純傳遞於兩個模態之間,其中有部分的能量被 傳到熱庫當中。第三階段主要是低頻模態到熱庫模態的能量傳遞,計算得到 flexible 和 semirigid D2O 熱庫環境下的遲豫時間相近。如果進一步將鍵長固定,遲 豫時間將大大增加,可見熱庫分子振動方式對於 NMA 能量耗散的重要性。這結 果也顯示 NMA 分子的特殊性,有別於一般分子,NMA 有複雜的內部振動模態重 組(intramolecular vibrational redistribution)與能量耗散路徑[5]。
以上能量耗散過程的模態消長資訊,在實驗上得自於時變 infrard-Raman 光譜,在 數值模擬上來自於由瞬時正則模態(Instantaneous Normal-Mode)分析得到的態密度 頻譜。在此論文我們使用另一種工具「時變功率譜」來探討這種問題。功率譜可 以用來做訊號分析,在物理中它常用於電磁波、聲波、以及力學波等的研究,在 化學上它可用來訂定分子的內部成份。在本論文,我們使用時變功率譜來觀察 NMA 分子能量耗散時的非平衡態過程,其精神類似於超快雷射中的時變光譜。 兩者隨時間變小的峰值都代表著振盪族群(population)的減少。在此工作,首先我 們使用 GROMACS 軟體架構出系統模型,並計算系統的速度與位置組態,由速度
4 組態計算出功率譜,找出譜上特定鍵結峰值隨時間的變化,以及不同振動模態隨 時間的消長,分析能量傳遞路徑和遲豫時間。在幾種不同的操作方式下,我們得 到很穩定一致的遲豫時間,約為 4.5~5.0 ps,和實驗裏的遲豫時間 4.9 ps[4]及瞬時 正則模模擬的總耗散時間 11.7 ps [5]很相近。「時變功率譜」分析成為描述分子內 能量耗散的工具,和最近實驗的 anti-Stokes Raman 及理論的瞬時正則模分析相輔 相成。
5
第二章理論背景
2.1 速度相關函數(Velocity Auto-correlation function)
速度相關函數可用來探討原子與分子的振動及擴散性質。如果將速度向量函數寫 成V t( ),速度相關函數(autocorrelation function)定義成 0 1 ( ) lim T ( ) ( ) x C V t V t dt T
eq(2.1) 或 / 2 / 2 1 ( ) lim T ( ) ( ) T x C V t V t dt T
eq(2.2) 其中 ※C( ) 是時間間隔為 的兩個點之間的自洽,它相當於兩個向量的內積 ( ) ( ) V t V t 。 ※當C( ) 愈大時函數V t( )與函數V t( )之間有愈大的相關性。 ※C( ) 是實數偶函數C( ) C()。 ※C( ) 的最大值是C(0)。 ※C(0)是函數V t( )平方的平均。 ※如果V t( )具有週期性,則C( ) 也具有周期性。若周期為T,C(0)以及C nT( ), n 皆為最大值。2.2 傅立葉變換( Fourier Transformation)
傅立葉變換是一種線性的積分變換。它可將時間域(time domain)函數轉換成方便 分析的頻率域(frequency domain)函數。任何周期函數都可以用正弦函數和餘弦函 數構成的無窮級數來表示,它就是所謂的傅立葉級數。假設訊號 f x( )為一連續6 函數,則其傅立葉級數為 1 ( ) 2 ikx k k f x f e
, ( ) ikx k f f x e dx
eq(2.3) 其中 fk ,k ,可以視為 f x( )在整數頻率波上的振幅。 函數 f x( )的傅立葉轉換為 ( ) ( ) ikx F f x e dx
eq(2.4) 為了在電腦中處理離散點的傅立葉變換,我們需要使用離散傅立葉變換(Discrete Fourier Transform),簡稱 DFT。對於 N 個不連續的數據點x n[ ], 0nN,它的離 散傅立葉變換與傅立葉反變換為 2 1 0 1 { [ ]} [ ] [ ] N i nk N n F x n x k e x n N
k0,1...N1 eq(2.5) 2 1 1 0 1 { [ ]} [ ] [ ] N i nk N n F x k x n e x k N
n0,1...N1 eq(2.6) 由於離散傅立葉變換的計算效率過低,因此人們發展了快速傅立葉變換(Fast Fourier Transformation),簡稱 FFT。最常見的快傅立葉變換演算法是 1965 年 Cooley 及 Tukey 共同提出的,此方法將一個序列長度為 N 的離散傅立葉序列拆成兩個長 度各為 N/2 的離散傅立葉序列,此一作法可以將運算的複雜度由 2 N 降為Nln( )N 。 然而離散傅立葉轉換有幾個條件限制: (1)取樣頻率(Sampling rate)必須符合取樣定理:取樣頻率為最高成分頻率的兩倍或 以上。 (2)取樣周期必須為量測數據周期的整數倍。 (3)量測數據必須有周期性。 (4)取樣數必為2k個,K ∈ 。 其中取樣頻率是每單位時間從連續訊號擷取資料的次數。舉例來說,假設我們的7 系統每 0.2fs 做一個改變,那麼成分頻率為 50[1 ps]⁄ 。如果我們每 2fs 記錄一次系 統的位置與速度,那我們取樣周期為 2fs,取樣頻率為 500[1 ps]⁄ 。如此,取樣頻 率為成分頻率的 10 倍。假使我們的主要觀察對象是 CH3L 頻率為 3000[cm ]的 振盪,它相當於 94[1 ps⁄ ]。在取樣訊號還原成連續訊號時,為了不讓不同頻率的 訊號彼此交疊在一起造成訊號失真,因此有條件(1)的限制。 假設W = e π,則 WNnk:=(WN)nk。快速傅立葉轉換之所以效率高,是因為利用了 以下(a)與(b)兩條件: (a)對稱性WNn k WNnk eq(2.7) (b)週期性WNnk WNn k N( )WN(n k k ) eq(2.8) 快速傅立葉轉換實際操作如下: 將 N 個數據點x n[ ], 0nN 拆為兩部分,一為 偶數列x n[ ]n2i,一為奇數列x n[ ]n2 1i , 0,1, 2...( 1) 2 N i 。此兩個長度 2 N 序列的 離散傅立葉變換為 4 / 2 1 ( ) 0 4 / 2 1 ( ) 0 2 [ ] [2 ] , 0,1, 2...( 1) 2 2 [ ] [2 1] N i n k N even n N i n k N odd n x k e x n N N k x k e x n N
eq(2.9) 或等價於 ( / 2) 1 / 2 0 ( / 2) 1 / 2 0 1 ( ) (2 ) / 2 , 0,1, 2...( 1) 2 1 ( ) (2 1) / 2 N n k even N n N n k odd N n F k f n W N N k F k f n W N
eq(2.10) 由此可以得到一個 N 點序列的離散傅立葉變換為 2 2 1 { } { ( ) ( )} 2 1 { } { ( ) ( )} 2 2 i k N even odd i k N even odd F k F n e F n N F k F n e F n eq(2.11)8
2.3 功率譜(Power spectrum)
譜分析是描述振盪的基本工具。根據傅立葉定理,對於周期 T 的函數x t( ),我們 可將它表成傅立葉級數 ( ) n inwt n x t a e
, / 2 / 2 1 ( ) T inwt n T a x t e dt T
eq(2.12) 由於x t( )是週期連續函數,它自然符合 / 2 / 2 ( ) T T x t dt
eq(2.13) 假如x t( )是一個非周期(或周期無限大的)函數,則級數由積分取代: ( ) ( ) iwt x t F w e dw
, ( ) 1 ( ) 2 iwt F w x t e dt
eq(2.14) 其存在條件為 x t dt( )
eq.(2.15) 對於只存在於有限時間區間 T 內的實際信號,F(w)將被取代為: 0 ( ) T ( ) iwt T F w
x t e dt 對於這種非週期運動,頻率譜是連續譜。 對於一般平穩隨機過程(stationary process),x(t)的平均值(甚至其均方值等)不隨時 間變化,其自變量(時間 t)將延伸到(-∞,∞),例如正弦波或理想白噪聲。對於這 種函數,條件 eq(2.15)不成立,甚至|x(t)| 的積分也是發散的, 即: 2 2 0 0 lim T ( ) ( ) T x t dt F w dw
eq(2.16) 因此我們不可能利用傅立葉變換式 eq(2.14)求出其振福-頻率譜。即使有的可以求 出,也不能反映運動真實特性。為了用譜分析來了解平穩隨機過程必須採取新的9 辦法。 在 eq(2.16)式左邊通常代表系統的總能量(如在電路中,x(t)代表電流強度,x (t)代 表通過單位電阻的功率)。假如x(t)是一個平穩過程,雖然 eq(2.14)左邊的積分不 是有限的,但條件: 2 0 1 lim T ( ) TT
x t dt eq(2.17) 仍能成立。此式子的意義是功率不發散。即如果令 2 1 ( ) T( ) S w F T 則有 2 0 0 1 ( ) lim T T( ) T S w dw F d T
eq(2.18)其中可看出 S(w)為x(t)的功率譜密度(power spectral density)。很明顯,用功率譜密 度 S(w)取代振幅進行譜分析比較合適。
另外 Wiener-Khintchine theorem 告訴我們,廣義平穩(wide-sense stationary)隨機過程 的功率譜密度是 2.1 節裡自相關函數的傅立葉轉換: 1 ( ) ( ) 2 iw S w C e d
eq(2.19) / ( ) ( ) iw C S w e dw
其中C( ) 來自 eq(2.2.1)及 eq(2.2.2)。當 =0 時即回到 eq(2.18)的結果
/ 2 2 / 2 1 lim T ( ) (0) ( ) T T T x t dt C S w dw
10
第三章模型建構與模擬計算
在這邊簡單的介紹 N-甲基乙酰胺(N-methylacetamide)分子,N-甲基乙酰胺分子是 由十二顆原子組成的分子,圖 3.1 可以看到在 N-甲基乙酰胺分子的兩側各有一個 甲基(CH3),為了方便描述,這邊將靠近氮的甲基(CH3)命名為 CH3L,靠近氧的 甲基(CH3)命名為 CH3R。在一般環境下,它會形成白色針狀結晶。熔點 30.55℃ (28℃),沸點 206℃,可溶於水、乙醇、苯、醚、氯仿。它是在合成藥劑時的中 間產物,可用作溶劑也可製藥。NMA 擁有一個胜肽鍵,是一個生物相關的簡單 分子,常被拿來當 Model,探討分子尺度的生物相關問題。 圖 3.1 N-甲基乙酰胺結構分子示意圖,綠色的碳(C)、藍色是氮(N)、紅色是氧(O), 灰色是氫(H)。11
3.1 系統模型建構
GROMACS 是 Groningen Machine for Chemical Simulations 的縮寫,是一套研究生物 分子系統與分子動力學的集合封包,可以模擬聚合物、晶體、蛋白質等分子,並 有多種的能量檔案對於分子模擬分析非常方便。GROMACS 是免費軟體可以在 www.gromacs.org 下載。 本研究的系統模型分為兩種,一種是將 N-甲基乙酰胺(N-methylacetamide)分子填 滿整個盒子,如圖 3.2,此系統被稱為 NMA200,另一種是在充滿水的盒子中放 一顆 NMA 分子,如圖 3.2,此系統被稱為 NMA_water。 圖 3.2:NMA200 是將兩百個 NMA 分子丟入盒子中。
12 圖 3.3:NMA_water 將一顆 NMA 分子丟入 2nm*2nm*2nm 充滿水的盒子中。
3.1.1 NMA200 模型建構
建構NMA200模型,需要準備系統結構資訊(.gro)、力場資訊(.itp)、環境條件 (.mdp)。.gro告訴GROMACS系統起始的分子位置、分子結構。.itp記錄分子間作用 力函數、鍵結角度、鍵結長度、鍵結強度。這裡我們用的力場是OPLSAA[6]。mdp 設定模型模擬時的邊界條件、系統溫度、系統壓力、模擬時間、cutoff等等,當 所有資訊導入GROMACS之後會產生拓譜檔(.top)記錄系統資訊,下面將介紹模擬 的運算流程。 步驟一:創造平衡系統13
grompp_d -f md1.mdp -c em.gro -p topol.top -o md1.tpr mdrun_d -nt 4 -v -deffnm md1
指令 grompp 將所有資訊導入拓譜檔(topol.top),值得注意的是如果 GROMACS 是 倍精度版本(double verson)就必須在指令後加上”_d”才能執行,在上面的指令中, -f 導入系統環境條件(.mdp),-c 導入系統結構(.gro),-p 產生拓譜檔(.top),-o 生成 GROMACS 看得懂的系統檔案(.tpr)。-nt 指定 cpu 運算核心數目,-deffnm 運算完 模擬之後生成檔案的檔案名稱,模擬結束之後會產生各種紀錄系統資訊的檔案, 包括系統位置與速度檔案(.trr)、系統能量(.edr)、系統結構(.gro)、系統運算資訊(.log)、 系統狀態(.cpt)。系統能量包括動能、位能、總能、庫倫作用力、系統溫度、系統 壓力、鍵能等等。系統運算資訊包括系統運算速率、系統運算效率、系統各種參 數、系統各種初始值、總運算時間,直接當作文件打開就可以取得資訊。 make_ndx –f md1.gro make_ndx 指令可以標識出系統原子或是將不同分子結合成群組,若加上-o 可指 定生成檔名,預設生成檔案為 index.ndx。 步驟二:將所有的 NMA 的 CH3L 加熱至 1000K
grompp_df – md3.mdp -c md1.gro -t md1.cpt -n index.ndx -p topol.top -o md3.tpr mdrun_d -nt 4 -v -deffnm md3
在 GROMACS 裡可以將模擬分段,若想要將已計算出的結果做為起始條件,只需 要將前一個系統結構(.gro)、系統狀態(.cpt)利用 grompp 指令引入 tpr 即可。
14 步驟三:加熱 NMA 後的 relaxation
grompp_df – md4.mdp -c md3.gro -t md3.cpt -n index.ndx -p topol.top -o md4.tpr mdrun_d -nt 4 -v -deffnm md4
步驟四:平衡態
grompp_df – md2.mdp -c md3.gro -t md1.cpt -n index.ndx -p topol.top -o md1.tpr mdrun_d -nt 4 -v -deffnm md2 指令 g_energy 可以將先前提到系統能量(.edr)內的資訊提出,方便觀察整個系統變 化,唯一美中不足的是 g_energy 指令只能取得各種整個系統的平均能量,並不能 對單一的局域部分做測量。
3.1.2 NMA_water 模型建構
步驟一:製造模型結構editconf_d -f nmlgopls.gro -o box.gro -box 2.089 2.085 2.08 -center 1.0 1.0 1.0 genbox_d -cp box.gro -cs spc216.gro -o solv1.gro -p topol.top
editconf_d -f solv1.gro -o solv.gro -box 2.0 2.0 2.0 -center 1.0 1.0 1.0
指令ediconf製作結構檔,將原有的NMA分子結構引入,-box定義盒子大小,-center 定義分子位置。指令genbox可以將給定的分子填滿給定的空間,在我們的系統就 是利用這個指令將水填滿整個系統空間模來擬體內環境。spc216.gro在GROMACS
15 裡是採用TIP3P[7]的水分子模型。
步驟二:能量最小化
grompp_d -f min.mdp -c solv.gro -p topol.top -o em.tpr -maxwarn 5 mdrun_d -nt 4 -v -deffnm em 能量最小化並不是一個力學計算,通常初始條件(粒子的位置與速度)都是亂數給 定,為了讓系統有適當的位能、較完整的結構,因此需要執行能量最小化,讓系 統有一個趨近於真實的起始條件。 步驟三到六則重複 NMA200 的步驟一到四。
3.2 功率譜計算(Power spectrum)
為了之後方便講述,先來定義一些符號: T:表示 MD 模擬到某一個時刻。 dt:表示速度相關函數的運算區間範圍。為了分析取得的模擬資料,需先讀取系 統記錄分子位置與速度的.trr 檔。圖 3.4(上)為在 CH3L 記錄到的速度對時間的函數。 每一個藍色的小方框代表一個時刻 T 到時刻 T+dt 的速度函數區間。利用 g_velacc 計算時刻 T 到時刻 T+dt 的速度相關函數(velocity autocorrelation function),圖 3.4(中) 為這些藍色區間內的速度相關函數。若將圖 3.4(中)各區間個別做傅立葉轉換 (Fourier transform),將得到不同時刻的功率譜(power spectrum),如圖 3.4(下)。16 圖 3.4:取出 CH3L 的速度函數(上圖),將 T→T+dt 的速度函數定義為時刻 T 的速 度函數。計算時刻 T 的速度相關函數(中圖)。最後將每時刻 T 的速度相關函數做 傅立葉轉換得到瞬時功率譜(下圖)。 由功率譜上看出在頻率 W=3000.9982[ 1 cm ]的地方有一峰值(peak),將每一時刻峰 值記錄得到峰值與時間的對應關係如圖 3.5,其中峰值定義如下: 假設在時刻 T 所記錄到的峰值高度為P T( ),例如時刻 T=0[ps]的峰值記為P(0)、 時刻 T=1[ps]的峰值記為P(1),其中P T( )可為達平衡或未達平衡的峰值高度。假 如系統已達平衡,平衡時的平均峰值為 0 0 1 [ (0) (1).... ( )] ( ) N i P P P P N N P i N
17 圖 3.5:趨向平衡過程峰值隨時間的變化圖 圖 3.6:達平衡後峰值隨時間的漲落平均值即為P 。 0 由圖 3.5 及 3.6 得到P T( )與P 。利用 0 y (T)=P(T)-P0 0 eq(3.1) 算得圖 3.7 的紅色圓點。利用 -x y=A exp( ) eq(3.2) P(T) P0
18 對紅色圓點做指數(exponential)擬合(fitting),得圖 3.7 的紅色實線。所得的 即為 CH3L 的遲豫時間(relaxation time)。 圖 3.7:W=3000.9982[ 1 cm ]CH3L 峰值擬合。黑點為模擬數據點,紅線為擬合曲線。 有別於峰值高度,另外一種作法為觀察峰值底下面積變化,首先找出功率譜上 P(T)/2 的頻率,也就是圖 3.8 上看到功率譜與紅線相交的兩點以定義出計算面積 的邊界圖中藍線,並計算出曲線下面積即為黑色斜線部分。
19 圖 3.8:功率譜取線下面積取法示意圖‧
重新定義觀察峰值底下面積變化的面積值,將 A(T)定義為非平衡態下隨時間變的
功率譜取線下面積值對比於 eq(3.1)的 P(T),A0定義為平衡狀態下 A(T)值的平均
對比於 eq(3.1)的 P0。利用此方法觀察遲豫時間時 eq(3.1)可以改寫成
y (T)=A(T)-A0 0 eq(3.3)
20
第四章結果與討論
4.1 系統功率譜平均
我們有兩種系統,對於 NMA200 系統,在計算速度相關函數時已經做過 200 個分 子的平均,結果相當於系綜(ensemble)平均,因此遲豫傾向明顯,我們可以利用 第三章敘述的方法很簡單的將遲豫時間計算出來。但是對於 NMA_water 系統, 觀察的 CH3L 只來自單一 NMA 分子,因此雜訊很多,使得頻率譜上峰值隨時間 變化的數據(如圖 4.1)非常雜亂,無法估計甲基的遲豫時間。為了消除這些雜訊。 需要做系綜平均。以下採用兩種取得系統系綜平均的方法,一種是先將速度相關 函數取平均後再做功率譜分析,另一種是取得功率譜後再做平均。 圖 4.1:NMA_water 系統峰值衰變圖,平衡溫度為 298k,在時間區間 dt=1ps。圖 中點為模擬計算數據點,實線數據點為擬合取線。21
4.1.1 速度相關函數的平均
利用不同亂數種子給定不同粒子位置與速度做為系統初始條件,建構一個系綜。 經過模擬,算出系綜內各系統裏唯一的 CH3L 的速度變化,算出數個時間區間(如 圖 3.4 的藍色區間)的速度相關函數,並將每個區間的速度相關函數做系綜平均, 並做傅立葉轉換得到功率譜。圖 4.2 由上而下依序為 10、30、50、100、200 組速 度相關函數做平均的結果,圖中可以發現當組數越多的時候,數據點與指數衰減 的遲豫曲線越來越穩合。由表 4.1 可以看到隨著組數的增加,峰值數據點與指數 衰減的標準差會下降。 圖 4.2:系統平衡溫度為 298K 的情況下,CH3L 時變速度相關函數的系綜平均, (a)-(e)分別是組數為 10、30、50、100、200 的結果。黑點為計算值,紅線為擬合 函數。22 組數 遲豫時間[ps] 峰值標準差 平均峰值 標準差/平均 1 0.200 0.073 0.113 0.648 10 6.076 0.035 0.117 0.3 30 4.522 0.03 0.121 0.244 50 3.242 0.028 0.121 0.234 100 2.898 0.026 0.128 0.204 200 3.347 0.026 0.124 0.208 表 4.1:對速度相關函數做不同組數系綜平均後擷取 CH3L 振盪 mode 的遲豫時間
4.1.2 功率譜的平均
如同上面 4.1.1 所提到的系綜平均方法,只是在這邊將平均的對象從速度相關函 數變成功率譜,簡單的將功率譜利用上面的方法求得平均。圖 4.3 由上而下依序 為 10、30、50、100、200 組的功率譜做系綜平均。分析圖中遲豫時間發現其結 果與利用速度相關函數取系綜平均的結果非常雷同,組數越多一樣可以達到與擬 合曲線越穩合的效果。表 4.2 列出不同組數下的遲豫時間,與達到平衡後 CH3峰 值平均高度,與標準差。標準差標示著平衡狀態的漲落。23 圖 4.3:系統平衡溫度為 298K 的情況下,時變頻譜圖的 CH3L 峰值系綜平均,(a)-(e) 分別是組數為 10、30、50、100、200 的結果。黑點為計算值,紅線為擬合函數。 組數 遲豫時間[ps] 峰值標準差 平均峰值 標準差/平均 1 0.222 0.035 0.12 0.291 10 6.022 0.03 0.124 0.242 30 4.605 0.029 0.124 0.23 50 3.200 0.027 0.131 0.203 100 2.834 0.027 0.131 0.203 200 3.293 0.026 0.128 0.207 表 4.2:對功率譜做不同組數系綜平均後擷取 CH3L 振盪 mode 的遲豫時間
24 圖 4.4:比較表 4.1 與 4.2 對速度相關函數與功率譜取系綜平均的分析方法,兩者 間遲豫時間(上圖)、峰值標準差(中圖)、平均峰值的差異(下圖)。 比較表 4.1 與 4.2 對遲豫時間兩種不同的分析方法得到圖 4.4。很明顯可看出兩種 方法在不同組數下遲豫時間的估計幾乎完全吻合,而在標準差的估計發現利用功 率譜取平均的標準差會稍微大於利用速度相關函數取平均的標準差。因此不論是 利用速度相關函數系綜平均計算遲豫時間亦或是功率譜系綜平均計算遲豫時間 都可得到相近的結果。
4.2 不同時間區間下的功率譜
要利用功率譜分析 mode 的消長,必須先計算速度相關函數。然而計算速度相關 函數有幾個決定性的參數,其中一個就是時間區間,當我們取不同的時間區間(如25 下圖 4.5)計算速度相關函數時,不僅該函數會隨區間長度有所不同,頻率譜的解 析度也會隨區間長度而變。較短的區間所包含的振盪周期較少,解析度也會較差, 這個效應對低頻 mode 的影響尤其劇烈,因為低頻的振盪周期較長,所以在較短 區間內振盪次數較少,因此縮短時間區間後振盪次數減少的比例會遠遠大出高頻。 以圖 4.5 為例,觀察 4 種時間區間內的速度相關函數,以四種顏色表達,各區間 都從時間零開始算,沒從零開始的顏色是因為被其它顏色蓋過去。對這 4 區間求 功率譜,發現它們在 800 1 cm 到 1700cm1之間的變化如圖 4.6,由該圖可明顯看 出短時間區間只能看到一個峰值而長時間區間則可以看到較多的峰值,另外高頻 峰值較不受影響。表 4.3 列出不同功率頻率對應的振盪頻率。而不同時間區間長 度的速度相關函數,經由傅立葉轉換後也會有所不同。在小時間區間的速度相關 函數(dt 較小)的功率譜會有較大的曲線下面積,以及較大的峰值。如圖 4.6 可以 看出不同的時間區間功率譜的峰值以及曲線下面積的變化。 [cm ] [ps ] CH3L 3000.998 94.24778 CH3R 3000.998 94.24778 Coa 1533.844 48.17109 Cob 1733.910 54.45427 NH 3334.442 104.7198 表 4.3:不同模態頻率的振盪次數的對照表
26
圖 4.5:不同時間區間的速度相關函數,橫軸為時間區間 dt[ps],縱軸為速度相關 函數的值。藍線 dt=0.2ps、綠線 dt=0.4ps、紅線 dt=1ps、黑線 dt=4ps。
圖 4.6:不同時間區間下的速度相關函數算出來的功率譜,橫軸為功率頻率,縱 軸為功率值。藍線 dt=0.2ps、綠線 dt=0.4ps、紅線 dt=1ps、黑線 dt=4ps。(圖的 y 軸)
27 不同的區間在速度相關函數及功率譜上會有所不同,但由圖 4.7 可以看出遲豫時 間(relaxation time)在不同速度相關函數的時間區間下雖然有所差異,但差異並不 顯著。 圖 4.7:不同時間區間的遲豫時間(relaxation time),實線為擬合曲線點為模擬數值, 系統的平衡溫度為 298K。
4.3 不同溫度下的遲豫時間
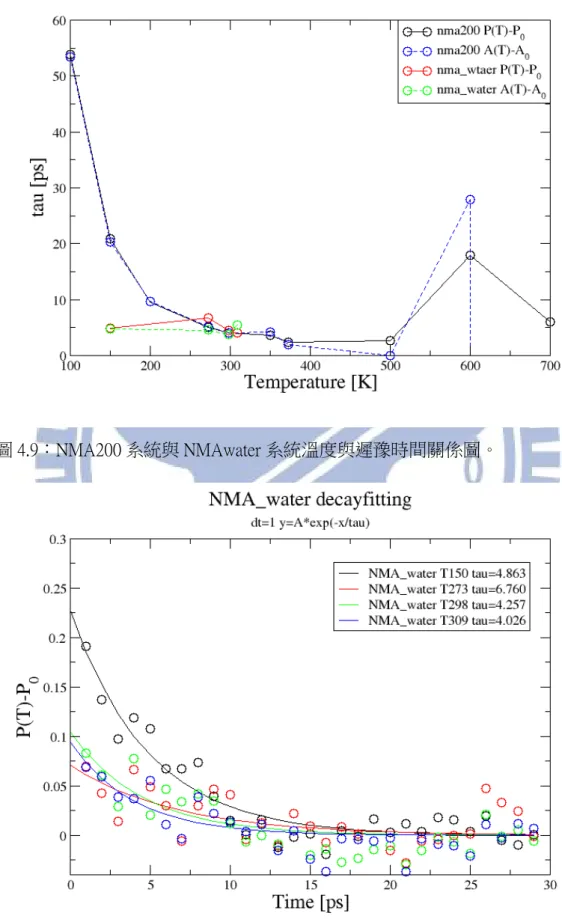
不管是什麼樣的蛋白質,都會有其適應或者存活的溫度,溫度不同蛋白質的結構 會產生變化,遲豫時間也會隨之改變,遲豫時間隨時間如何改變是另一個我們感 興趣的問題。在 NMA200 的系統裡,對 NMA 的 CH3L 局部加熱並給予不同的平 衡溫度來觀察在不同溫度下高頻 mode 的遲豫現象與遲豫時間。28 圖 4.8:NMA200 系統下不同溫度下的遲豫時間變化。橫軸為模擬時間[ps],縱軸 為峰值比值。 由圖 4.8 為各個不同溫度衰變曲線,點為模擬數值點,實線為擬合曲線。很明顯 的可以看出,當外界溫度越低會有越大的遲豫時間,NMA200 系統遲豫時間與溫 度關係畫在圖 4.9。
29
圖 4.9:NMA200 系統與 NMAwater 系統溫度與遲豫時間關係圖。
30 縱軸為峰值比值。 在圖 4.9 裡實線表示 P(T)-P(0)做出的遲豫時間與溫度關係,曲線分為三個區段, 第一部分黑色實線溫度小於 0 0 C,第二部分紅色實線較接近液態水溫度 0℃ ~100℃ 的區間,第三部分黑色實線為溫度大於 100℃ 。虛線表示 A(T)-A(0) 做出的遲豫時間與溫度關係。在 NMA_water 系統下結果如圖 4.10,我們可以看 到它跟圖 4.8 有相同的趨勢。圖 4.10 的遲豫時間也畫在圖 4.9。其中點線及點虛 線分別代表 P(T)-P(0)與 A(T)-A(0)做出的遲豫時間與溫度關係。由圖 4.8,圖 4.9 及圖 4.10 可以看出當溫度越低會有越大的遲豫時間,這與先前在第一章裡提到 的當溫度越高會有越短的遲豫時間是相吻合的。
4.4 結論
我們利用功率譜分析碳氫鏈的遲豫時間得到幾個結論: (1) 在功率譜的分析,不管是利用速度相關函數系綜平均計算遲豫時間,亦或是 利用功率譜系綜平均計算遲豫時間都得到非常相近的 5 ps。 (2) 在不同溫度下,溫度越高遲豫時間越短,這與期刊文獻吻合。31
第五章未來工作
以下為幾個可繼續探討的問題:
(1) 改變熱庫分子,可以進一步驗證 power spectrum 在 Three-Stage Model[4]下是否 得到相似的結果。
(2) 在 NMA200 與 NMA_water 兩種系統當中遲豫時間隨著溫度的上升而下降的幅 度為何有所不同?
32
參考文獻
[1] H. Graener, T. Q. Ye, and A. Laubereau. Ultrafast dynamics of hydrogen bonds directly observed by time-resolvedinfrared spectroscopy. J. Chem. Phys. 90, 3413 (1989)
[2]A. Tokmakoff, B. Sauter, M. D. Fayer. Temperature‐dependent vibrational relaxation in polyatomic liquids Picosecond infrared pump–probe experiments.J. Chem. Phys. 100, 9035 (1994).
[3]C. W. Rella, J. R. Engholm, H. A. Schwettma. Ultrafast Vibrational Dynamics of the Myoglobin Amide I Band. J. Phys. Chem. B 1999, 103
[4]Ying Fang, Shinsuke Shigeto, Nak-Hyun Seong, and Dana D. Dlott. Vibrational Energy Dynamics of Glycine, N-Methylacetamide, and Benzoate Anion inAqueous (D2O)
Solution. J. Phys. Chem. A 2009, 113, 75–84
[5]Adolfo Bastida,Miguel A. Soler,Jose´ Zu´n iga,Alberto Requena,Adria´n Kalstein,andSebastian Ferna´ndez-Alberti.Molecular Dynamics Simulations and Instantaneous Normal-Mode Analysis of the C-H Stretching Modes of
N-methylacetamide-d in Liquid Deuterated Water.pdf.J. Phys. Chem. A 2010, 114, 11450-11461
[6] Jorgensen, W. L., Tirado-Rives, J. The OPLS potential functions for proteins. energy minimizationsfor crystals of cyclic peptides and crambin. J. Am. Chem. Soc.
33
[7]Jorgensen,W. L., Chandrasekhar, J., Madura, J. D., Impey, R.W., Klein, M. L. Comparisonof simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935, 1983
![圖 4.5:不同時間區間的速度相關函數,橫軸為時間區間 dt[ps],縱軸為速度相關 函數的值。藍線 dt=0.2ps、綠線 dt=0.4ps、紅線 dt=1ps、黑線 dt=4ps。](https://thumb-ap.123doks.com/thumbv2/9libinfo/7685928.142682/31.892.144.812.105.1028/時間區間速度相關函數橫軸為時間區間縱軸為速度相函數值藍綠線.webp)