行政院國家科學委員會專題研究計畫 期中精簡報告
酚亞硫酸基轉移酵素催化機制之研究與修改(1/2)
計畫類別: 個別型計畫 計畫編號: NSC91-2311-B-009-002- 執行期間: 91 年 08 月 01 日至 92 年 07 月 31 日 執行單位: 國立交通大學生物科技學系 計畫主持人: 楊裕雄 計畫參與人員: 蘇天木, 陳偉迪, 邱靜玟 報告類型: 精簡報告 處理方式: 本計畫可公開查詢中 華 民 國 92 年 5 月 12 日
中文摘要 關鍵詞:亞硫酸基轉移酵素;分子模擬;定點突變; 基因選殖與表達;酵素反應 機制。 亞硫酸基轉移酵催化亞硫酸基轉移至生物系統中各種親和受體的反應。生物 體中藉由硫酸化來進行調控的物質有蛋白質、醣蛋白、醣類、荷爾蒙、神經傳導 物質及包括藥物及致癌物等化合物;因此,亞硫酸基轉移參與生物訊息之分辨與 傳遞、荷爾蒙之調節、解毒作用及調節神經傳 導物質等重要生物反應。然而我們 對於此酵素反應機制的了解卻非常有限。針對 酚亞硫酸基轉移酵素催化反應管制 與機制之研究,我們預計進行的相關實驗包括:核酸與酵素之親和性標記,酵素 結構之分子模擬,氧化還 原對於此酵素生理與轉移反應之不同影響,決定此酵素 之雙硫鍵位置,及定點突變以改變酵素特性。上述各種實驗方法,將可探討此酵 素如何應用其結構上的變化來催化與調控亞硫酸基轉移反應,並進一步設計出使 用不同核酸的新酵素。目前 我們的研究發現亞硫酸基轉移酵素中有一形態不穩定 區(主要包括 63-Leu-Glu-Lys-Cys-Gly-Arg-68),與核酸之催化反應無直接相關, 但可被核酸與酵素之親和性標記。由分子模擬看出,此形態不穩定區位於核酸進 出口,而定點突變此區的氨基酸後,嚴重影響到核酸與酵素的結合與釋放。其中 C66 形成酵素內之雙硫鍵後,生理反應活性 (Vmax)逐漸增加,亞硫酸基轉移反應 (非生理反應)則僅 Km 增加。據此,我們可設計出抗氧化之酚亞硫酸基轉移酵素。 酵素分子模擬顯示 C66,C82 及 C232 這三個胺基酸位置相當接近,可形成雙硫 鍵;此外,K65 與 R68 經過定點突變後,也合乎根據酵素與反應物結合之三度 空間立體結構所提出之反應機制。目前我們已建立催化亞硫酸基轉移機制及其管 制的模式。
Keywords: Sulfotransferase, molecular modeling, site-directed mutagenesis, gene expression, enzyme mechanism.
Sulfotransferase catalyzes sulfuryl group transfer between nucleotide and a variety of nucleophiles that may be sugar, protein, xenobiotics and other small molecules. Nucleotides may serve as cosubstrate, cofactor, inhibitor or regulator in an enzyme catalyzed sulfuryl group transfer reaction. We are trying to understand how nucleotide regulates the activity of phenol sulfotransferase (PST) through the expression of two enzyme forms. The homogeneous rat recombinant PST was obtained from E. coli and the nucleotide co-purified was examined. The nucleotide was completely removed from inactive PST in high salt and oxidative condition. Total enzyme activity was recovered following incubation in reductive environment. Many nucleotides are kno wn t o t ight ly bind t o PST but o nly o ne nucleot ide, 3’-phosphoadenosine 5’-phosphate (PAP), was identified to combine with PST by ion-pair RP-HPLC,
UV-visible spectra, 31P NMR and ESI-MS and MS-MS spectrometry. In addition to the presence or absence of PAP, oxidation following reduction of PST was required to completely inter-convert the two forms of PST. According to the experimental results, a mechanism for the formation of the two enzyme forms was proposed.
成果報告內容
The following report funded by NSC 91-2311-B-009-002 is to be published in the June 24, 2003 issuse of Biochemistry (24).
Mechanism of Post-Translational Regulation of Phenol Sulfotransferase: Expression of Two Enzyme Forms through Redox Modification and Nucleotide Binding†
Tian-Mu Su and Yuh-Shyong Yang*
Department of Biological Science and Technology National Chiao Tung University, Hsinchu, Taiwan, ROC Running title: Post-Translational Regulation of Phenol Sulfotransferase *Correspondence to:
Dr. Yuh -Shyong Yang
Department of Biological Science and Technology National Chiao Tung University
75 Po-Ai Street
Hsinchu, Taiwan, ROC Tel: 886-3-5731983 Fax: 886-3-5729288
E-mail: [email protected]
†
This research is supported by a grant from the National Science Council (NSC 91-2311-B-009-002), Taiwan.
Abbreviations used: Bis-tris propane, 1,3-bis[tris(hydroxymethyl) methylamino] propane; CD, circular dichroism; DTT, dithiothreitol; EDTA, (ethylenedinitrilo) tetraacetic acid; ESI-MS, electrospray ionization mass spectrometry; FAD, Flavin adenine dinucleotide; GSH, glutathione, reduced form; GSSG, glutathione, oxidized form; MS-MS, collision-induced dissociation tandem mass spectrometry; m/z, mass-to-charge ratio; -NAD, -nicotinamide adenine dinucleotide; -NADH, -nicotinamide adenine dinucleotide, reduced; NADP, nicotinamide adenine
dinucleotide 3’-phosphate; NADPH, nicotinamide adenine dinucleotide 3’-phosphate, reduced; PAP, 3 ’-phosphoadenosine 5’-phosphate; PAPS, 3’-phosphoadenosine 5’-phosphosulfate; PMSF, phe nylmethylsulfonyl fluoride; PNP, 4-nitrophenol; PNPS, 4-nitrophenyl sulfate; SDS, sodium dodecyl sulfate; TCEP, Tris(2-carboxyethyl) phosphine; Tris-HCl, tris(hydroxymethyl) aminomethane hydrochloride
It has been known for a long time that sulfation occurs in a biological system (1). Sulfotransferases (EC 2.8.2), which catalyze sulfuryl group transfer between nucleotide and a variety of nucleophiles, are responsible for all the known biological sulfation. The nucleotide, 3’-phosphoadenosine-5’-phosphosulfate (PAPS), is a biologically active form of inorganic sulfate that serves as the sulfate donor in various biological processes (2). Macromolecular substrates (such as proteins (3) and polysaccharides (4)) are metabolized by membrane-bound sulfotransferases. Small molecules, like xenobiotics (5) or endogenous compounds (such as hormones (6) and neurotransmitters (7) are metabolized by cytosolic enzymes. Sulfotransferases have been classified into several subfamilies according to the degree of the similarities of the deduced amino acid sequences (8, 9) or substrate specificities (10).
The regulation of the activity of sulfotransferase has not been well studied. Four phenol sulfotransferases (PST) have been separated and purified from rat liver (11,
12). PST has been cloned and expressed in E. coli (13). During the growth of the
bacterial cells, the - and -forms of the sulfotransferase, are produced and both active. The ratio of two forms PST express is dependent on cell culture condition (14). The two forms are separable from each other by hydroxyapatite (13) and PAP-agarose chromatography (15). It has been shown that the interaction of a nucleotide with PST is important for the formation of the two different enzyme forms, but is not the only factor (16). Both forms of the recombinant enzyme are similar when compared by circular dichroism (CD) spectroscopy below 240 nm. Above 240 nm, the maximal difference is at about 260 nm with additional peaks at 280 and 290 nm. Addition of PAP with -form PST gives similar CD spectrum to that of -form PST (16).
The postulated nucleotide that pre-exist in -form PST before purification has never been chemically and biochemically identified. The activity of -form PST shows a hyperbolic dependence on PAP concentration in the nano mo lar range, whereas the activity of -form is not stimulated by the addition of nucleotide (16). The two forms of PST can also be distinguished by the PST assays and separated by chromatography (13, 16). PST activity can be measured by the physiological or the transfer reactions. The former reaction represents the transfer of sulfuryl group from PAPS to another co-substrate. The transfer reaction is the two-stage transfer of the sulfuryl group, for example, from PNPS to PAP and the subsequent transfers from PAPS to 2-naphthol (16). Only -form PST is active toward both the physiological and the transfer reactions. The -form PST is found unable to catalyze the physiological reaction, which requires the release of PAP to complete a turnover of a sulfuryl group transfer (16). Routine analysis of the activity of sulfotransferase generally measures only the physiological activity (11, 12) catalyzed by -form PST
forms in physiological condition is yet to be studied.
Complete conversion of the two enzyme forms has not been achieved previously
in vitro. Part of the physiological activity (catalyzed only with -form PST) can be
generated following partial oxidation of -form PST with glutathione, oxidized form (GSSG). However, prolong incubation with GSSG resulted in the loss of PST activity and incubation of PAP with -form PST did not give complete -form (16, 17). Factors involved in the transformation of the two forms of PST are the subject of this study. There are many nucleotides that bind tightly to PST. Some of the dissociation constants are in the nanomolar range (15). In addition to PAP, several other nucleotides are also found to be the cofactors or substrates of PST (15). PAP analogs, coenzyme A and its derivatives, are strong inhibitors for PST (18, 19). Cells contain numerous nucleotides. It is intriguing to learn what is the nucleotide irreversibly attached to PST and what is the mechanism responsible for the formation of the two PST forms from the expression of a single cDNA. Here we present a series of chemical and biochemical methods that identify the naturally produced nucleotide and study its effect on the formation of two PST forms.
EXPERIMENTAL PROCEDURE S
Materials— 4-nitrophenyl sulfate (PNPS), 4-nitrophenol (PNP), 3’-phosphoadenosine
5’-phosphate a g a r o s e g e l ( PA P-agarose), 3’-phosphoadenosine 5’-phosphate (adenosine 3’, 5’-diphosphate; PAP) and other nucleotide analogs were purchased from Sigma (USA). Tris(2-carboxyethyl) phosphine (TCEP) was obtained from Pierce (USA). DEAE sepharose fast flow and HiTrap desalting column were obtained from Amersham pharmacia biotech Asia pacific (Hong Kong). Hydroxyapatite ultrogel® was purchase from Biosepra (USA). All other chemicals were obtained commercially at the highest purity possible.
Enzyme assay— PST activity was determined by the change of absorbency at 400 nm
due to 4-nitrophenol (=10,500 cm-1M-1 at pH 7.0) as described previously (16). Standard assay is a transfer reaction that consists of 2-mercaptoethanol (5 mM), 4-nitrophenyl sulfate (5 mM), 2-naphthol (50 M), PAP (2 M) and bis-tris propane (100 mM at pH 7.0). The -form activity was determined in the absent of PAP. The -form activity was the difference between standard assay and -form activities. Protein concentration of homogeneous form of PST was estimated on the basis of absorbency at 280 nm (=58,350 cm-1
M-1 or 1.7 ml/mg cm-1) (20).
Preparation of nucleotide from homogeneous PST— Recombinant rat phenol
sulfotransferase was cloned into expression vector pET3c and transformed into
Escherichia coli BL21 (DE3). PST was extracted by sonication, and purified by
DEAE and hydroxyapatite liquid chromatography. Detailed methods were described
previously (13, 16) e xc e p t t ha t a ne w h yd r o x ya p a t it e c hr o ma t o g r a p h y (Hydroxyapatite ultrogel® from Biosepra) was used. The purity of PST used for this
study was examined by SDS polyacrylamide gel electrophoresis to be a single band and the purity was estimated over 95% or higher. Extra high purity PST was used for the extraction of nucleotide to prevent any possible contamination. Purified PST was further passed through a HiTrap column with desalting buffer (100 mM bis-tris propane at pH 7.0, 1 mM EDTA, 10% glycerol (v/v), and 125 mM sucrose) to remove
any unbound small molecules. Active fractions were pooled (a typical preparation contained 16 mg or 2.3×10-7 mole PST dimer; including 66% -form) and mixed with 70% ethanol. The denatured and precipitated PST was removed by centrifugation. The supernatant was collected and concentrated with vacuum dryer (EZ550R, FTS system, USA). According to A260 (=15.1 cm-1mM-1 for adenine) (21), 1.4×10-7 mo le of
adenine nucleotide was recovered. The yield was 92% calculated base on one nucleotide per -form PST dimer (16).
Separation of - and -form PST— The two forms of PST were separated by
PAP-agarose chromatography as described previously (15) with the fo llowing
modification. The gel was incubated overnight with capture buffer which included DTT (1 mM), sucrose (125 mM), glycerol (10%), 2-mercaptoethanol (14 mM), EDTA (1 mM) and phosphate (25 mM at pH 6.8). Capture buffer (5-fold column volume) was used to equilibrate column. Unbound protein (-form PST) was washed out with 10-fold column volume capture buffer, and bound PST (-form PST) was eluted with capture buffer containing NaCl (0.3 M). This preparation of -form PST was used as control to extract adenine nucleotide as described above and the existence of nucleotide was not found.
Ion-pair RP-HPLC analysis of nucleotides— Nucleotides were separated with a 5 m
(250mm) pre-packed LiChrospher 100 RP-18 column (Merck, USA) and were monitored at 260 nm wit h a UV-visible detector using a D-7000 HPLC system (Hitachi, Japan). The separation was achieved in an isocratic eluent at a flow-rate of 1 ml/min as previously described (15, 22) with some modification on the mobile phase
bu ffer s yst e m. T h e s e p a r a t io n b u ff e rc o n t a i n e d 1 0 m M T B H S (tetra-n-butyl-ammonium hydrogen sulfate) and 0.1 M phosphate at pH 5.8. HPLC mobile phase was 90% separation buffer and 10% acetonitrile, and they were mixed completely before running.
UV-visible spectral measurements— Nucleotides were dissolved in HPLC mobile
phase solution as described above. The absorption spectra were recorded between 340 to 220 nm o n a UV-visible spectrophotometer (U-3300, Hitachi, Japan) and the background absorption obtained by the HPLC mobile phase solution was subtracted. The scan parameters were 2.0 nm of slit, 120 nm/min of scan speed and 1 nm of sample interval.
31
P NMR spectroscopy— The 31P NMR spectra were recorded on a VARIAN
UNITYINOVA 500 NMR spectrometer (VARIAN, USA) operating at a frequency of
202.31 MHz. Samples were placed in 5 mm NMR tubes and the spectra were recorded at room temperature (25℃). A pulse of 4.7 s was used with an acquisition
time of 0.271 sec. The spectral width was set to 60.6 kHz, and 32,000 data points were recorded for each free induction decay. Chemical shifts were referenced relative to external 85% H3PO4 at 0 ppm.
Electrospray ionization mass spectrometry (ESI-MS)— ESI-MS investigations were
carried out by means of a quadrupole time-of-flight (Q-TOF) mass spectrometer (Micromass, UK) equipped with an electrospray ion source. Electrospray ionization mass spectrometry was performed in the positive-ion modes. Samples were dissolved in hydroxyapatite buffer (10 mM phosphate at pH 7.0, 1 mM EDTA, 1 mM DTT, 10% glycerol, and 125 mM sucrose), and sample volumes of 20 l/min were applied by loop injection. The liquid eluent was water/acetonitrile/1% formic acid (8/1/1, v/v). The collision energy of MS-MS spectra was 25 V. Nitrogen was used as nebulizing and drying gas. Mass spectra were acquired in scan range from m/z 200 to m/z 2500.
RESULTS & DISCUSSION
Identification of the nucleotide co-purified with PST— One of the main purposes
of this study was to chemically identify the nucleotide that trapped and co-purified with PST. Alcohol (70%) was used to denature PST and to release the nucleotide from -form PST. The adenine nucleotide recovered was equal to 0.92 PAP per PST dimer
for -form PST. None of the adenine nucleotide can be observed in a parallel experiment with -form PST. This result was consistent with previous report (16), that
fully active PST dimer require one PAP for the transfer reaction. The one PAP per PST dimer ratio is also previously determined by circular dichroism (16).
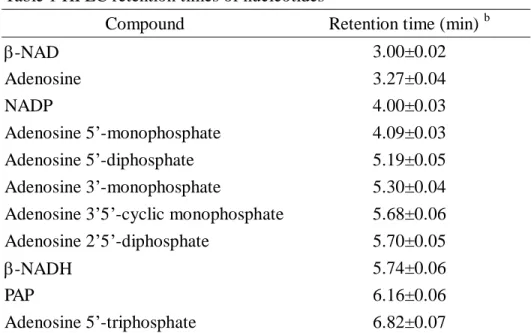
Under the selected HPLC condition, separation for a variety of nucleotides was obtained within 20 min, as shown in Figure 1 . Other nucleotides that could be separated by this method were listed in Table 1. The ion-pair RP-HPLC profile gave only one peak from the extract o f recombinant PST. The retention time of the extracted nucleotide was identical with that of commercial PAP at 6.150.04 min (Figure 1B). This result indicated that only one nucleotide was still tightly bound to purified PST after gel filtration to separate small molecules from purified PST. It has been reported previously (15) that many nucleotides bind tightly to PST at Kd in the
nanomolar range. However, to remain bound to PST through a series of protein purification procedure, the nucleotide might need to be irreversibly trapped inside the enzyme. As reported later for the conversion of the two enzyme forms, we found that partial denaturation of PST by oxidation or high salt solution was required to remove the trapped nucleotide.
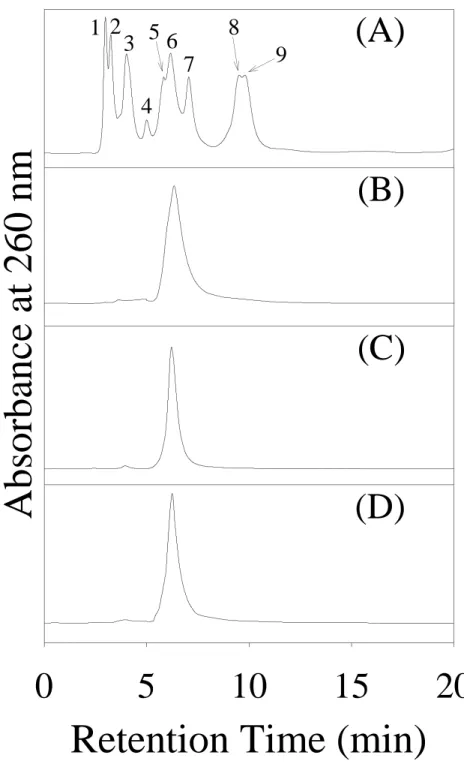
The UV-visible absorption spectrum of the isolated nucleotide was shown in Figure 2, which had the same spectrum as that of commercial PAP. In the previous study (16), the CD spectra of the two PST forms were compared. The major difference at 260 nm indicates that adenine may be involved in the binding of -form PST. Biochemical test of this isolated nucleotide was examined as cofactor of -form PST as described under “EXPERIMENTAL PROCEDURES” for standard assay. As
compared to that of commercial PAP, identical PST activity was obtained. The concentration of the nucleotide was determined by A260 and the equivalent of 2 M
PAP (o r A260 = 3.0×10-4) was used as cofactor in standard assay condit io n.
Specificities obtained were 2.010.05 and 1.900.11 mole/min/mg for commercial PAP and nucleotide isolated from PST, respectively. This was the first time that the nucleotide in -form PST was isolated and tested chemically and biochemically, and they were found to be identical.
The 31P NMR spectrum of desalted PST was shown in Figure 3A. Figure 3B was the 31P NMR spectrum of the solution containing the targeted nucleotide extracted from PST. The 31P chemical shifts of PAP can be significantly different in distinct environments (23) and the variation between Figure 3A and 3B indicated that the
phosphorus signals were sheltered or interacted strongly with PST. Crystal structures of PAP and sulfotransferase complex (24, 25) show that both of the phosphate groups are strongly coordinated by neighboring amino acids in the enzyme binding site. Denaturation of PST released PAP into solution and gave the spectral differences (shown in Figures 3A and 3B). This observation confirmed our previous proposal that the PST bound nucleotide might be trapped inside the enzyme binding pocket. The proton-decoupled 31P NMR spectrum gave three chemical shifts (Figure 3B), 3.86, 3.58 and 2.60 ppm from the extract of desalted PST. The two lower field chemical shifts were identical to those of 3’-phosphate and 5’-phosphate of PAP in Figure 3C (15, 26). Figure 3D was the mixture of the equal amount of enzyme extract and commercial PAP (determined by A260). The third chemical shift shown in Figure 3B
overlapped with that of sodium phosphate (data not shown). It is known that PAP is subjected to hydrolysis in aqueous solution (15); which may result in the presence of free phosphate in PST extract.
The positive-ion ESI-MS spectrum of purified PST dominated by different charged molecular ions was shown in Figure 4A. These signals were deconvoluted using the “MassLynx” program to yield molecular masses for the PST. PST (33,901 Da) and PST-PAP complex (34,327 Da) were appeared as shown in Figure 4B. Electrospray ionization mass spectrometry (ESI-MS) is a powerful tool to study biomolecular non-covalent interactions involving proteins with metals, ligands, peptides, proteins or oligonucleotides (27-29). This result further confirmed the strong interaction of PAP and PST. The other peaks of Figure 4B were salt clusters (30-32) with PST or PST-PAP complex. These salt cluster peaks disappeared when PST was passed through HiTrap desalting column with H2O (data not shown). However, PST is
unstable in pure water and precipitates easily. The MS-MS spectrum of m/z 428 produced two peaks, m/z 428 and m/z 136 that could be assigned to PAP and adenine, respectively (Figure 4C).
Conversion of the two PST forms— Table 2 detailed the procedures and results of
complete transformation of - to -form PST. In a reverse process, Table 3 detailed the procedures and results of complete transformation of - to -form PST.
The ratio of - to -form PST expressed in E. coli is dependent on the cell growth condition. The percentage of -PST can be as high as nearly 100% or as low as 30% and can be controlled by the growth temperature and oxygen supply (14). Incubation of PAP (20 M) with 4 mg -form PST (14 M) produces only 60% -form activity (16). Although PAP seems to help the refolding of denatured PST, only 4% of the total enzyme activity is recovered (14). In this study, 66% -form PST was expressed in E. coli at 37℃. Accumulation of PAP is found in E. coli in the
(21) and it is possible that the two enzyme forms are similar ly regulated in mammalian cells.
As proposed above, PAP could be trapped inside PST so that the release of PAP could not be achieved without partially denaturing the enzyme. It has been shown that oxidat ion releases the tight ly bound PAP easier (17) a nd high salt effects PAP-sulfotransferase interaction (23, 33). We were thus utilizing these strategies to exchange PAP and to interconvert these two enzyme forms (Tables 2 and 3). Oxidative modification at cysteines regulates PST activity and the Kd of PAP and PST
is about 30-fold higher following incubation with 1 mM GSSG for 1 hour (17). However, oxidation of PST was found unable to completely transfer -form to -form PST and prolong oxidation inactivated the enzyme activity. In our experiments the -form activity reduced to 20% (from 66% of total PST activity) with the incubation of 1 mM cystamine for 6 hours or 1 mM GSSG for 9 hours (data not shown). As shown in Table 2, a combination of oxidation and high salt treatment completely eliminated the activity of -form PST (II to IV or II to V shown in Table 2). In such condition PAP could be removed from PST and then separated by ultrafiltration or gel filtration chromatography. PST was inactive in o xidat ive st at e (IV), but was completely reactivated (V) following reduction with TCEP as shown in Table 2.
Different degrees of transformation were obtained following the incubation of -form PST (0.27 mg/ml or 4M dimer) with different PAP concentrations in reducing condition (Figure 5). The PAP to PST ratios were 1, 10, 100 and 1000 (i.e., 4 M, 40 M, 0.4 mM and 4 mM PAP, respectively). The two dissociation constants of PAP and PST are 31 nM and 152 M (15). In such conditions, nearly all the first binding site of PST was occupied by a PAP as calculated and shown in Figure 5 as solid circles. Accordingly, 3, 21, 72 and 96% of PST would contain the second PAP in such conditions (open circles in Figure 5). In our experiments, the transformations of -form to -form PST were in between the above calculated results. Repeated desalting (up to t hree t imes) did not change t he- to -form ratio in these experiments. This result confirmed our proposal that PAP was irreversibly trapped inside -form PST. It also indicated that the binding of PAP in both binding sites could facilitate the transformation of - to -form PST, i.e., to entrap one PAP inside PST.
Complete transformation (within experimental error) of -form to -form PST was achieved with the pretreatment of cystamine to inactivate PST following PAP incubation and reduction with TCEP (VII in Table 3). As has been shown previously (16), a PST2-PAP (one PAP per PST dimer) complex is required for the catalysis of
the transfer reaction. In a typical assay condition (about 20 nM PST was used) the calculated concentration of PST2-PAP complex in the solution is only 6.2 nM for the
equal amount of PAP (20 nM) using Kd =31 nM for PAP and PST determined
previously (15). That is, only 31% enzyme activity would have been obtained if equal amount of PAP and PST were present due to the tight binding but did not irreversibly trapped inside PST as proposed for -form. The treatment of PST with high salt concentration (0.3 M NaCl) did not increase the ratio of transformation significantly (VI in Table 3). It indicated that partial denaturation of PST by oxidation was required to facilitate the transformation.
Proposed model for the formation of the two forms of PST- In addition to this PST example (13, 16), expression of two protein forms from a single cDNA are found for other proteins. Rat liver malonyl-CoA decarboxylase (MCD) generates two isoforms from one cDNA (34), because it contains two potential translational start sites. However, two forms PST are indistinguishable in their sequence for they have identical first eight amino acids, molecular weight and western blot results (13). Another example is prion protein (PrP). The normal, cellular PrP (PrPc) is converted into modified protein (PrPsc) through a post-translational process whereby a portion of its -helical and coil structure is refolded into -sheet (35, 36). Their amino acid sequences and molecular weights are identical. Two forms structures can convert reversibly by oxido-reduced met ho d (3 6 , 37). PS T ma y be s u b je c t e d t o post-translational modification through redox regulation. In the absence of PAP, -form PST is active in catalyzing the transfer reaction. However, partial oxidation is required for -form PST to be able to catalyze the physiological reaction of sulfuryl group transfer (17), which requires the removal of PAP to complete a turnover of sulfuryl group transfer (16). In liver tissue, the concentrations of glutathione, reduced and oxidized form (GSH and GSSG), are in the mM ranges (39) and the concentration of PAP is in M range (21). There are plenty of redox reagents and PAP available for the transformation of the two PST forms in physiological condition.
Scheme I illustrates how the two PST forms can be converted according to the results from this and previous studies. We propose that the direct transformation between - and -form PST requires conformational change that can be facilitated by
partial denaturation and renaturation through oxidat ion and reduct ion. The intermediate, the partially oxidized PST, is inactive for both the transfer and the physiological reactions. High salt seems to help the transformation but redox is crucial for the release and binding of PAP that induces conformational change of PST (16). Only the -form PST is fully active for both the transfer and the physiological reactions. The binding of PAP does not transform the -form to - form PST directly. PAP needs to be irreversibly trapped inside PST to produce -form. This makes -form PST inactive toward the catalysis of the physiological reaction, which requires the release of PAP to complete a turnover.
A flexible loop, amino acid residue 64-68, which includes a cysteine at 66, may be accountable for the transformation of the two PST forms. The possible interaction of C66 with PST nucleotide binding site has been demonstrated by affinity labeling (40). Interestingly, crystal structures of sulfotransferases (24, 25, and 41) indicate that this highly conserved region is not part of the nucleotide-binding site. This region may be very flexible or disordered, so that it cannot be resolved in the crystal structures (24, 25). As evidence in the crystal structure, this flexible region may be in the way of nucleotide entrance and releasing and thus affect two forms transformation of PST. It has been shown that in the presence of GSSG, C66 and C232 formed a disulfide bond (17). The Kd of PST and PAP is significantly increased in oxidation
state (17). Mutation around C66, K65ER68G, also greatly affects the binding of PST and PAP (16). Detailed study concerning the effect of loop 64-68 on the formation of two PST forms is under investigation by site directed mutagenesis.
CONCLUSION
Although it has been demonstrated that many nucleotides can be tightly bound to PST and can be function as cofactor of PST for sulfuryl group transfer (15), only one nucleotide was found to trap inside PST. This nucleotide was identified as PAP according to ion-pair RP-HPLC, UV-visible spectra, 31P NMR, ESI-MS and MS-MS
spectrometry and biochemical test as cofactor of PST.
Partial denaturation of PST that results in its inactivation was required to remove the trapped PAP from -form PST. This process can be induced by oxidation. Oxidation, reduction and the binding of PAP facilitate the transformation between - and -form PST.
Acknowledgements
We thank Ms. Hui-Chi Tan for collecting 31P NMR data at Instrument Center of National Tsing Hua University. Ms. Yun-Ming Li obtained ESI-MS data at Instrument Center of National Chiao Tung University.
RFERENCE
1. Williams, R. T. (1959) Detoxication Mechanisms, second ed., Chapmam and Hall, London
2. Klaassen, C. D. and Boles, J. W. (1997) Sulfation and sulfotransferases 5: the importance of 3'-phosphoadenosine 5'-phosphosulfate (PAPS) in the regulation of sulfation, FASEB J. 11, 404-418.
3. Niehrs, C. and Huttner, W. B. (1990) Purification and characterization of tyrosylprotein sulfotransferase, EMBO J. 9, 35-42.
4. H abu c h i, O . ( 2 0 0 0 ) D ive r s it y a nd fu nc t io ns o f g lyc o sa m ino g l yc a n sulfotransferases, Biochim. Biophys. Acta 1474, 115-127.
5. Jakoby, W. B. and Ziegler, D. M. (1990) The enzymes of detoxication, J. Biol. Chem. 265, 20715-20718.
6. Strott, C. A. (1996) Steroid sulfotransferases, Endocr. Rev. 17, 670-697
7. Sakakibara, Y., Takami, Y., Zwieb, C., Nakayama, T., Suiko, M., Nakajima, H., and Liu, M. C. (1995) Purification, characterization, and molecular cloning of a novel rat liver Dopa/tyrosine sulfotransferase, J.Biol.Chem. 270, 30470-30478 8. Weinshilboum, R. M., Otterness, D. M., Aksoy, I. A., Wood, T. C., Her, C., and
Raftogianis, R. B. (1997) Sulfation and sulfotransferases 1: Sulfotransferase molecular biology: cDNAs and genes, FASEB J. 11, 3-14
9. Nagata, K., and Yamazoe, Y. (2000) Pharmacogenetics of sulfotransferase,
Annu. Rev. Pharmacol. Toxicol. 40, 159-76
10. Guo, W.-X., Yang, Y.-S., Chen, X., McPhie, P., and Jakoby, W. B. (1994) Cha nges in su bst rat e spec ific it y o f t he r ec o mbina nt fo r m o f p he no l sulfotransferase IV (tyrosine-ester sulfotransferase), Chem. Biol. Interact. 92,
25-31.
11. Sekura, R. D. and Jakoby, W. B. (1979) Phenol sulfotransferases, J. Biol. Chem.
254, 5658-5663.
12. Sekura, R. D. and Jakoby, W. B. (1981) Aryl sulfotransferase IV from rat liver,
Arch. Biochem. Biophys. 211, 352-359.
13. Chen, X., Yang, Y.-S., Zheng, Y., Martin, B. M., Duffel, M. W., and Jakoby, W. B. (1992) Tyrosine-ester sulfotransferase from rat liver: bacterial expression and identification, Protein Expr. Purif. 3, 421-426.
14. Yang, Y.-S., Tsai, S.-W., and Lin, E.-S. (1998) Effects of 3'-phosphoadenosine 5'-phosphate on the activity and folding of phenol sulfotransferase, Chem. Biol. Interact. 109, 129-135.
15. Lin, E.-S. and Yang, Y.-S. (2000) Nucleotide binding and sulfation catalyzed by phenol sulfotransferase, Biochem. Biophys. Res. Commun. 271, 818-822.
16. Yang, Y.-S., Marshall, A. D., McPhie, P., Guo, W. X., Xie, X., Chen, X., and Jakoby, W. B. (1996) Two phenol sulfotransferase species from one cDNA: nature of the differences, Protein Expr. Purif. 8, 423-429.
17. Marshall, A. D., Darbyshire, J. F., Hunter, A. P., McPhie, P., and Jakoby, W. B. (1997) Control of activity through oxidative modification at the conserved residue Cys66 of aryl sulfotransferase IV, J. Biol. Chem. 272, 9153-9160.
18. Leach, M., Cameron, E., Fite, N., Stassinopoulos, J., Palmreuter, N., and Beckmann, J. D. (1999) Inhibition and binding studies of coenzyme A and bovine phenol sulfotransferase, Biochem. Biophys. Res. Commun. 261, 815-819. 19. Tulik, G. R., Chodavarapu, S., Edgar, R., Giannunzio, L., Langland, A., Schultz,
B., and Beckmann, J. D. (2002) Inhibition of bovine phenol sulfotransferase (bSULT1A1) by CoA thioesters. Evidence for positive cooperativit y and inhibition by interaction with both the nucleotide and phenol binding sites, J.
Biol. Chem. 277, 39296-39303
20. Gill, S. C. and von Hippel, P. H. (1989) Calculation of protein extinction coefficients from amino acid sequence data, Anal. Biochem. 182, 319-326.
21. Lin, E.-S. and Yang, Y.-S. (1998) Colorimetric determination of the purity of 3'-phospho adenosine 5'- phosphosulfate and natural abundance of 3'-phospho adenosine 5'- phosphate at picomole quantities, Anal. Biochem. 264, 111-117. 22. Kanno, N., Nagahisa, E., Sato, M., and Sato, Y. (1993) Nonradioactive assay for
adenosine 5'-phosphosulfate sulfotransferase using reversed-phase ion-pair high-performance liquid chromatography, Biochem. Mol. Biol. Int. 29, 47-55. 23. Marsolais, F., Laviolette, M., Kakuta, Y., Negishi, M., Pedersen, L. C., Auger,
M., and Varin, L. (1999) 3'-Phosphoadenosine 5'-phosphosulfate binding site of flavonol 3- sulfotransferase studied by affinity chromatography and 31P NMR,
Biochemistry 38, 4066-4071.
24. Kakuta, Y., Pedersen, L. G., Carter, C. W., Negishi, M., and Pedersen, L. C. (1997) Crystal structure of estrogen sulphotransferase, Nat. Struct. Biol. 4, 904-908.
25. Bidwell, L. M., McManus, M. E., Gaedigk, A., Kakuta, Y., Negishi, M., Pedersen, L., and Martin, J. L. (1999) Crystal structure of human catecholamine sulfotransferase, J. Mol. Biol. 293, 521-530.
26. Barrio, J. R., Barrio, M. C., Leonard, N. J., England, T. E., and Uhlenbeck, O. C. (1978) Synthesis of modified nuc leo side 3',5'-bisphosphates and their incorporation into oligoribonucleotides with T4 RNA ligase, Biochemistry 17, 2077-2081
27. Cheng, X., Harms, A. C., Goudreau, P. N., Terwilliger, T. C., and Smith, R. D. (1996) Direct measurement of oligonucleotide binding stoichiometry of gene V protein by mass spectrometry, Proc. Natl. Acad. Sci. U. S. A. 93, 7022-7027. 28. Tang, X. J., Brewer, C. F., Saha, S., Chernushevich, I., Ens, W., and Standing, K.
G. (1994) Investigation of protein-protein noncovalent interactions in soybean agglutinin by electrospray ionization time-of-flight mass spectrometry, Rapid
Commun. Mass Spectrom. 8, 750-754.
29. Veenstra, T. D. (1999) Electrospray ionization mass spectrometry: a promising new technique in the study of protein/DNA noncovalent complexes, Biochem.
Biophys. Res. Commun. 257, 1-5.
30. Charles, L., Pepin, D., Gonnet, F., and Tabet, J. C. (2001) Effects of liquid phase composition on salt cluster formation in positive ion mode electrospray mass spectrometry: implications for clustering mechanism in electrospray, J. Am.
Soc. Mass Spectrom. 12, 1077-1084.
31. Hao, C., March, R. E., Croley, T. R., Smith, J. C., and Rafferty, S. P. (2001) Electrospray ionization tandem mass spectrometric study of salt cluster ions. Part 1--investigations of alkali metal chloride and sodium salt cluster ions, J.
Mass Spectrom. 36, 79-96.
32. Pramanik, B. N., Bartner, P. L., Mirza, U. A., Liu, Y. H., and Ganguly, A. K. (1998) Electrospray ionization mass spectrometry for the study of non-covalent complexes: an emerging technology, J. Mass Spectrom. 33, 911-920.
33. Marsolais, F. and Varin, L. (1995) Identification of amino acid residues critical for catalysis and cosubstrate binding in the flavonol 3-sulfotransferase, J. Biol.
Chem. 270, 30458-30463.
34. Dyck, J. R., Berthiaume, L. G., Thomas, P. D., Kantor, P. F., Barr, A. J., Barr, R., Singh, D., Hopkins, T. A., Voilley, N., Prentki, M., and Lopaschuk, G. D. (2000) Characterization of rat liver malonyl-CoA decarboxylase and the study of its role in regulating fatty acid metabolism, Biochem. J. 350 Pt 2, 599-608.
35. Pan, K. M., Baldwin, M., Nguyen, J., Gasset, M., Serban, A., Groth, D., Mehlhorn, I., Huang, Z., Fletterick, R. J., Cohen, F. E., and Prusiner, S. B. (1993)
Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins, Proc. Natl. Acad. Sci. U. S. A 90, 10962-10966.
36. Prusiner, S. B. (1998) Prions, Proc. Natl. Acad. Sci. U. S. A 95, 13363-13383. 37. Jackson, G. S., Hosszu, L. L. P., Power, A., Hill, A. F., Kenney, J., Saibil, H.,
Craven, C. J., Waltho, J. P., A. R. Clarke, and Collinge, J. (1999) Reversible conversion of monomeric human prion protein between native and fibrilogenic conformations, Science 283, 1935-1937.
38. Lu, B. Y., Beck, P. J., and Chang, J. Y. (2001) Oxidative folding of murine prion mPrP(23-231), Eur. J. Biochem. 268, 3767-3773.
39. Senft, A. P., Dalton, T. P., and Shertzer, H. G. (2000) Determining glutathione and glutathione disulfide using the fluorescence probe o-phthalaldehyde, Anal.
Biochem. 280, 80-86.
40. Zheng, Y., Bergold, A., and Duffel, M.W. (1994) Affinity labeling of aryl sulfotransferase IV. Identification of a peptide sequence at the binding site for 3'-phosphoadenosine-5'- phosphosulfate, J. Biol. Chem. 269, 30313-30319.
41. Kakuta, Y., Pedersen, L. G., Pedersen, L.C., and Negishi, M. (1998) Conserved structural motifs in the sulfotransferase family, Trends. Biochem. Sci. 23,
129-130.
Table 1 HPLC retention times of nucleotides a
Compound Retention time (min) b
-NAD 3.00±0.02 Adenosine 3.27±0.04 NADP 4.00±0.03 Adenosine 5’-monophosphate 4.09±0.03 Adenosine 5’-diphosphate 5.19±0.05 Adenosine 3’-monophosphate 5.30±0.04
Adenosine 3’5’-cyclic monophosphate 5.68±0.06
Adenosine 2’5’-diphosphate 5.70±0.05
-NADH 5.74±0.06
PAP 6.16±0.06
Adenosine 5’-tetraphosphate 9.12±0.10
NADPH 9.63±0.12
FAD 20.37±0.18
Coenzyme A 25.93±0.25
a
Separations were described under “EXPERIMENTAL PROCEDURES ”.
b The meanstandard deviation was obtained from triplicate HPLC separation.
Table 2 Complete transformation of - to -form PST a
Purified PST Desalt Cystamine Reduced Oxidized Reduced Desalted PST PST PST PST TCEP Desalt Ultrafiltration TCEP Desalt Desalt
I
II
III
IV
V
0.3 M NaCl Composition of PST (%)PST step Total specific activity (mole/min/mg) Specific activity of -form PST (mole/min/mg) I. Purified PST 1.770.03 f 1.240.02 70 30 II. Desalted b 1.700.02 1.020.02 60 40 III. Reduced c 1.850.01 1.120.02 61 39 IV. Oxidized d 0.100.01 0.040.02 (40) g (60) g V. Reduced e 1.920.03 0.0070.004 (0.4) g 99.6 a
Assays and calculations of PST composition were performed as described under “EXPERIMENTAL PROCEDURES”.
b
Purified PST (I) following DEAE column was passed through a HiTrap desalting column to remove DTT and NaCl and to exchange buffer (100 mM bis-tris propane at pH 7.0, 1 mM EDTA, 10% glycerol, and 125 mM sucrose). The PST active fractions were pooled and concentrated by ultrafiltration.
c Desalted PST (II, 1 ml, 7.8 M or 0.53 mg/ml) was incubated in TCEP (1 mM) for 6
hours at 25℃, then passed through HiTrap desalting column immediately.
d Another fraction of desalted PST (II, 5 ml, 7.8 M or 0.53 mg/ml) was incubated
with 1mM cystamine for 1 hour. This mixture was concentrated to 1 ml by ultrafiltration. PAP released from PST was removed by ultrafiltration from 10 ml to 1ml in the present of desalting buffer plus 1 mM cystamine and 0.3 M NaCl. The above ultrafiltration procedure was repeated 4 times. The inactive enzyme solution was run through a desalting column and was collected based on A280.
e
Complete reactivation of PST was achieved by incubating the enzyme solution (1 ml, 4 M or 0.27 mg/ml) with TCEP (1 mM) at 25℃ for 6 hours.
f The meanstandard deviation was obtained from triplicate activity assay. g
Table 3 Complete transformation of - to -form PST a Purified PST Desalt PAP agarose -form -form Desalted Reduced PAP added
I
II
III
IV
V
TCEP TCEP Desalt Desalt Cystamine TCEP TCEP Desalt Desalt Desalt Desalt Desalt Buffer PAP PAP PAP NaCl PAP addedVI
Desalt Desalt Buffer PAP PAP added PAP addedVII
Composition of PST (%)PST step Total specific activity (mole/min/mg) Specific activity of -form PST (mole/min/mg) I. -form PST 1.950.03 g 0.0100.005 (0.5) h 99.5 II. Desalted b 1.900.04 0.0090.005 (0.5) h 99.5 III. Reduced c 1.840.04 0.0100.005 (0.5) h 99.5
IV. PAP added d 1.890.08 1.700.08 90 10
V. PAP added d 1.860.06 1.660.03 89 11
VI. PAP added e 1.860.07 1.690.03 91 9
VII. PAP added f 1.910.04 1.890.05 99 (1) h
a
PST assay and separation of -form PST (I) were performed as described under “EXPERIMENTAL PROCEDURES”.
b
The -form PST was passed through a desalting column (HiTrap) to remove DTT that was added to stabilize PST. The resulting -form PST solution contained 100 mM bis-tris propane at pH 7.0, 1 mM EDTA, 10% glycerol, and 125 mM sucrose.
c
III was obtained by incubating the above enzyme (II, 1 ml, 4 M or 0.27 mg/ml) with TCEP (1 mM) at 25℃ for 6 hours.
d The desalted -form PST (1 ml, 10 M or 0.68 mg/ml) was incubated with desalted
buffer at 25℃ for 6 hours, and then passed through HiTrap desalting column. The active fractions were pooled (4 M) and incubated with PAP (4 mM) and without (IV) or with (V) TCEP (1 mM) at 25℃ for 6 hours.
e Alternatively, the desalted -form PST (1 ml, 10 M or 0.68 mg/ml) was incubated
with 0.3 M NaCl, first. Then the active fractions were pooled (4 M) and incubated with PAP (4 mM) and TCEP (1 mM) at 25℃ for 6 hours.
f The desalted -form PST (1 ml, 10 M or 0.68 mg/ml) incubating with cystamine (1
mM) at 25℃ for 6 hours and then with PAP (4 mM) and TCEP (1 mM) at 25℃ for 6 hours. The -form PST was obtained following by a desalting column (Hitrap) to remove free PAP.
g The meanstandard deviation was obtained from triplicate activity assay. h
Scheme 1
FIGURE LEGENDS
Figure 1. Ion-pair RP-HPLC separation of nucleotides. A 10 l sample was
injected for each run. (A) Commercial nucleotides included: 1 -NAD; 2 adenosine; 3 adenosine 5’-mo no pho sp hate; 4 adeno sine 5’-diphosphate; 5 adenosine 2’, 5’-diphosphate; 6 PAP; 7 adenosine 5’-triphosphate; 8 adenosine 5’-tetraphosphate; 9 NADPH; (B) PST extract (A260=0.44); (C) Commercial PAP (100 M or A260=1.5);
(D) Mixture of enzyme extract and commercial PAP (A260=0.22 for each)
Figure 2. UV-visible spectra of PAP and nucleotide extracted from PST. PAP (0.1
mM) was dissolved in HPLC mobile phase solution (90% separation buffer and 10% acetonitrile). The background of the solution in UV-visible spectra was subtracted. The PST extract following ion-pair RP-HPLC separation was collected at desire retention time (6.15 min) as shown in Figure 1B. The two spectra were normalized at 260 nm. The absorption of PST extract at 260 nm was 0.44.
Figure 3. 31P NMR spectra. Purified PST was passed through a HiTrap column with
sonication buffer (10 mM Tris-HCl at pH 7.0, 1 mM EDTA, 1 mM DTT, 10% glycerol, and 125 mM sucrose). Nucleotides were obtained as described under “EXPERIMENTAL PROCEDURES” and dissolved in sonication buffer. (A) Desalted PST (20 mg/ml or A280=34); (B) PST extract (A260=0.5); (C) Commercial PAP (0.1
mM or A260=1.5); (D) Mixture of enzyme extract and commercial PAP (A260=0.38 for
PAP
PAP PAP
PAP
-form PST PST-PAP complex
-form PST Oxidized PST Oxidation Reduction PAP PAP Oxidation Reduction (Conformational change required) (Fully active ) Slow
each)
Figure 4. ESI-MS spectra. (A) ESI-MS spectrum of native PST; (B) Deconvoluted
using the “MassLynx” program; (C) The MS-MS spectrum of m/z 428 peak from (A).
Figure 5. Addition of PAP partially transforms --form to -form PST. The
-form PST (4 M or 0.27 mg/ml) was used to incubate with different concentrations of PAP (4 M to 4 mM) in the presence of TCEP (1 mM). PST2-PAP ( ●) and
PST2-PAP2 (○) complexes were calculated using Kd1 =31 nM and Kd2 = 152 M.
The transformation ratio (▼ as % of -form) was determined as describe under “EXPERIMENTAL PROCEDURES”. The error bar was one standard deviation and calculated from triplicate activity assay.
Figure 1
Retention Time (min)
0
5
10
15
20
A
bsorbance at
26
0 nm
(B)
(C)
(D)
1 2
3
5
4
7
6
8
9
(A)
Figure 2 Figure 3
Wavelength (nm)
220
260
300
340
Abs
o
rbanc
e
0.0
0.5
1.0
1.5
Extract
PAP
ppm
6
5
4
3
2
1
0
(A)
(B)
(C)
(D)
PO4 3-3’ 5’Figure 5
log [PAP] (M)
-6
-5
-4
-3
-2
%
of
bi
n
di
n
g site
oc
c
up
ie
d
0
20
40
60
80
100
計畫成果自評We feel that we have accomplished the proposed research goal. The second manuscript entitled “Site-directed mutagenesis reveals the regulation of the activity of phenol sulfotransferase by a flexible loop” which is funded by this grant is now in preparation and will be submitted to Biochemistry for publication before the end of this funded period.