Transfer of Foreign Gene to Giant Freshwater
Prawn (Macrobrachium rosenbergii) by
Spermatophore-Microinjection
SI-SHEN LIANDHUAI-JEN TSAI*Institute of Fisheries Science, National Taiwan University, Taipei, Taiwan
ABSTRACT We developed a spermatophore-microinjection (SMI) technique that allows exogenous DNA fragments to be transferred easily into the giant freshwater prawn (Macrobrachium rosenbergii), an im-portant aquacultural shellfish and aquatic invertebrate model. From 28 to 1,000 ng of the circular plasmid pGL, in a total volume of 1ml, were directly microin-jected into spermatophores. Fertilization and hatching of prawns created with SMI were completed in vivo. Fertilization and hatching rates in the SMI treatments did not differ from those of the untreated control group. The genomes of free swimming, SMI-created larvae (21 days after fertilization) were analyzed using PCR and Southern blot analyses. A product with a molecular mass of 680 bp was amplified. It corresponded to amplifications of pGL, and Southern blot analysis re-vealed that the amplified band was positive. The gene transfer rate was primarily dependent on the concentra-tion of DNA during SMI. The higher the concentraconcentra-tion of pGL, the higher the rate of gene transfer. PCR and Southern blot analyses detected the existence of foreign DNA in 16 of 23 samples (70%) of genomic DNA isolated from hatched larvae in the 750 ng pGL SMI treatment. SMI, described here for the first time, is the simplest and most efficient method for mass producing transgenic giant freshwater prawns.Mol. Reprod. Dev. 56:149–154, 2000 r2000 Wiley-Liss, Inc.
Key Words: gene transfer; microinjection; prawn; spermatophore
INTRODUCTION
Transgenic animals provide a powerful system for in vivo study of gene regulation, expression, and function, and make possible the production of transgenic variet-ies having special genetic traits. Many approaches have been developed to introduce foreign DNA molecules into zygotes. In fish, methods for gene transfer include microinjection of foreign DNA into oocyte nuclei (Ozato et al., 1986; Tsai et al., 1995b), the cytoplasm of developing embryos (Chourrout et al., 1986; Dunham et al., 1987), and fertilized eggs (Fletcher et al., 1988; Dunham et al., 1992; Lu et al., 1992). Electroporation of exogenous DNA fragments into fertilized eggs (Inoue et al., 1990; Powers et al., 1992; Tsai and Tseng, 1994) is a popular technique because, compared with the
tradi-tional microinjection technique, electroporation is simple, convenient, and efficient. However, the efficacy of gene transfer by electroporation of fertilized eggs is still not high enough to treat the tremendously large number of eggs spawned within a very short time by cultured finfish and shellfish species.
Thus, for aquatic animals, interest in sperm-medi-ated gene transfer has increased because this tech-nique has several advantages over other methods of gene transfer applied to finfish and shellfish. First, sperm-mediated gene transfer can be applied to huge numbers of oocytes. Second, sperm-mediated gene trans-fer overcomes some of the disadvantages of conven-tional gene transfer systems resulting from egg charac-teristics such as opaqueness, stickiness, buoyancy, invisible pronuclei, and a tough chorion. Third, foreign DNA carried by sperm is transferred into the nucleus; with electroporation, the probability that DNA frag-ments will be transferred into the blastodisc, the vol-ume of which is extremely small in a fertilized egg, is very low. Fourth, finfish and shellfish sperm can be activated by simply adding water, and they can be cryopreserved. Thus, treated sperm is ready for use at any time.
A number of researchers have attempted to use sperm as a vector for introducing foreign DNA and producing transgenic finfish and shellfish. Chourrout and Perrot (1992) reported failure using a sperm-incubation technique with rainbow trout. However, some success has been achieved using incuba-tion with zebrafish (Khoo et al., 1992), and sperm-electroporation with common carp, catfish, tilapia (Muller et al., 1992), salmon (Symonds et al., 1994), loach (Tsai et al., 1995a), and abalone (Tsai et al., 1997). There have been few attempts to apply gene transfer techniques to an important group of shellfish: crusta-ceans. Gendreau and colleagues (1995) successfully used a ballistic technique to transfer genes into brine shrimp (Artemia). However, the facilities and DNA
Grant sponsor: National Science Council; Grant sponsor: Council of Agriculture, Republic of China.
*Correspondence to: Huai-Jen Tsai, Institute of Fisheries Science, National Taiwan University, Taipei, Taiwan, 106.
E-mail: hjtsai@ccms.ntu.edu.tw
Received 15 November 1999; Accepted 20 January 2000
preparations required for ballistic transfer are ex-tremely expensive, and the work is tedious and unsuit-able for field use. Bensheg and Khoo (1997) used microinjection to transfer a reporter gene into the fertilized eggs of freshwater shrimp, Macrobrachium
lanchesteri. Again, traditional microinjection is
labori-ous, time-consuming, and can only treat a limited number of eggs. In this communication, we describe a simple and effective way to transfer foreign DNA frag-ments into giant freshwater prawn on a massive scale using spermatophore-microinjection (SMI). SMI may be quite useful for studying gene regulation and for creating new crustacean strains having special traits.
MATERIALS AND METHODS Experimental Animals
Nine- to twelve-month-old, aquacultured, giant fresh-water prawns were purchased locally. They averaged 16.66 4.8 cm (males) and 11.5 6 2.9 cm (females) in length, 3.76 0.6 cm (males) and 2.5 6 0.4 cm (females) in width, and 61.46 11.7 g (males) and 33.6 6 7.4 g (females) in weight. Males and females were main-tained in separate aquarium tanks at a density of 1 (males) or 2 (females) per 10 liters at 28°C on a natural light cycle.
Spermatophore Extrusion and In Vitro Fertilization
Spermatophores were extruded from male prawns using electric shock (36–10 V, DC, model PS-304, Shin-Ray Electronic, Taiwan). The positive pole was attached to the fifth swimming pod and the negative pole was placed on the ventral ganglia. Thirteen ma-ture males were used to examine the degree of spermato-phore extrusion caused by voltages ranging from 0 to 10 V at 10 A.
Foreign DNA, at concentrations of 28, 250, 400, 750, and 1,000 ng/µl in a total volume of 1 µl, was directly microinjected into the extruded spermatophore. For the mock-treated control group, we microinjected 1 µl of double-distilled water into the extruded phore. For in vitro fertilization, a treated spermato-phore was placed for 2–3 min in the spermatheca, located on the thorax between the third and fifth swimming pods, of a female prawn that had just finished molting and had well-developed ovaries. In general, treated females began to fertilize their eggs in vivo 2–6 hr after spermatophores were attached. When their eggs were close to hatching, these females were moved to a 100 liter tank containing 12 ppt salt water.
DNA Preparation
pGreen Latern-1 (pGL, Gibco BRL), with a molecular mass of 5 kb, consists of an immediate early-gene promoter and enhancer of cytomegalovirus, fused with the cDNA of a green fluorescence protein (GFP) and a 38-untranslated region of SV40. They were prepared by the cesium chloride-ethidium bromide ultra-centrifuga-tion method (Sambrook et al., 1989). The circular form
of pGL was resuspended in double-distilled water (ster-ilized) at concentrations of 28, 250, 400, 750, and 1,000 ng per µl.
DNase Digestion
In order to ensure PCR detection of foreign DNA molecules in embryos, we had to avoid contamination of the DNA fragments that remained outside the embryos. First, we determined the conditions that allowed DN-ase I (Sigma Chemical Co., St. Louis, MO) to completely digest the greatest amount of DNA employed in trans-genesis. pGL of 1,000 ng was resuspended in 100 µl of digestion buffer (10 mM MgCl2and 20 mM TrisCl, pH
8), then 0.5, 0.75, 1, or 10 µg of DNase I was added, and the solution was incubated at 37°C for 1 hr. Each treatment was duplicated. One-hundredth of the reac-tion volume was analyzed by PCR to determine the concentrations of DNase capable of completely digest-ing DNA molecules.
Genomic DNA Extraction
Five embryos that had developed to the zoea-I-stage were pooled and collected in an eppendorf tube, washed three times with double-distilled water, and resus-pended in a 500 µl of digestion buffer. Then, DNase I was added until a final concentration of 100 µg/ml was achieved. The solution was incubated at 37°C for 1 hr. After the reaction, the embryos were centrifuged at 76g for 3 min. The supernatant was decanted, and samples were boiled for 10 min to halt DNase activity. A volume of 400 µl lysis solution (6 M guanidine hydrochloride and 0.1 M sodium acetate, pH 5.5) was added to the embryos, which were briefly crushed and then incu-bated at 37°C for 1 hr in a roller shaker. Then the genomic DNA of embryos was obtained by phenol-chloroform extraction and ethanol precipitation.
Twenty-three free-swimming larvae, randomly se-lected from the experimental and control groups, were examined for foreign DNA. Genomic DNA was ex-tracted from each larva and prepared as described above for embryo DNA, except that DNase was not added. Each larva was washed three times in double-distilled water, resuspended in 400 µl lysis buffer, crushed slightly, and incubated at 37°C for 1 hr in a roller shaker. The genomic DNA was obtained by phenol-chloroform extraction and ethanol precipita-tion.
PCR Analysis
Two oligonucleotide primers, EGFP-F and GFP-R, were synthesized for detection of GFP cDNA by PCR analysis. The nucleotide sequences for forward and reverse primers were ATGGTGAGCAAGGGCGAGGA (EGFP-F) and CAGCTCGTCCATGCCATGTG (GFP-R), respectively. The primers for detection of the endog-enous b-tubulin gene, which served as an internal control, were CCCTTCCCTCGTCTCCAC (forward, f6) and GCCAGTGTACCAGTGAAGGGA (reverse, tub-r7). Each PCR sample consisted of 20 µl of solution containing 10–20 ng of templates, 5–10 pmol of each
primer, 25 µM of each dNTP, 20 µg of bovine serum albumin (Giambernardi et al., 1998), and five units of ProZyme II (Protech) in a 13 PCR buffer (Protech). Amplification was performed with a DNA Thermal Cycler (Perkin-Elmer Cetus, Norwalk, CT). PCR con-sisted of 25 cycles of denaturing at 94°C for 1 min, annealing at 60°C for 1 min and extension at 72°C for 2 min, followed by 10 min extension at 72°C. Each PCR sample (10 µl) was subjected to electrophoresis on a 3% NuSieve GTG agarose gel (FMC BioProducts, Rock-land, ME).
Probe Preparation
The 0.6 kb fragment containing GFP cDNA was recovered from the agarose gel using Jetsorb Gel Extrac-tion (Genomed, Inc., Research Triangle Park, NC) after plasmid pGL was restricted by NotI and run on the gel. Three micrograms of the 0.6 kb DNA fragment were DIG-labeled by the random priming method at 37°C for 20 hr, according to the manufacturer’s recommended procedures (Boehringer Mannheim).
Southern Blot Analysis
PCR products were analyzed on an agarose gel and transferred onto a nitrocellulose membrane (Amer-sham, Pharmacia Biotech, Uppsala, Sweden). After air drying, the DNA was cross-linked to the membrane by UV irradiation and then hybridized to a probe. Hybrid-ization was carried out overnight, at 42°C, in a stan-dard buffer solution (Boehringer Mannheim) contain-ing 50% formamide and 10 ng/ml of denatured probe. After the membranes were washed, colorimetric detec-tion was carried out with nitroblue tetrazolium and 5-bromo-4-chloro-3-indolyl phosphate reagents, as rec-ommended by the manufacturer (Boehringer Mann-heim).
Fertilization Rate, Hatching Rate, and Gene Transfer Rate
The fertilization rate was calculated as the propor-tion of 40 randomly selected eggs that developed one day after spawning. The hatching rate was calculated as the proportion of 40 randomly selected eggs that hatched three weeks after spawning. The gene transfer rate was the number of positive PCR and Southern blot analyses divided by the total number of hatched larvae examined.
Green Fluorescence Detection
To detect green fluorescence, hatched larvae were examined under a microscope (Olympus BH-2, Tokyo, Japan) using an excitation filter (BP 490 nm) and a dichron mirror (DM 500 nm).
RESULTS
Effect of Voltage on Spermatophore Extrusion In Vitro
Eleven of 13 (85%) male giant freshwater prawns extruded their spermatophore in vitro when shocked with 7.4 V (Fig. 1). No spermatophores were extruded
at voltages less than 7 V, and two male prawns failed to extrude spermatophores even at voltages above 7.4 V.
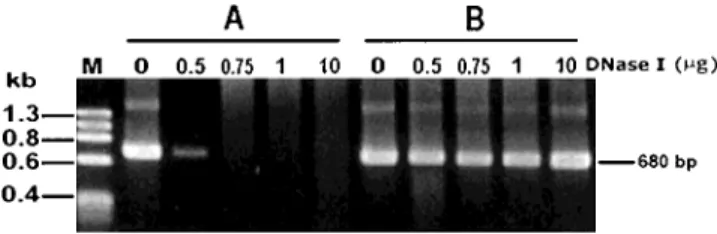
Presence of Foreign DNA in Embryos Before we could ascertain whether foreign DNA was present in the embryos in the SMI treatments, we had to determine the conditions under which PCR sensitiv-ity was sufficient to detect DNA molecules that had not been completely digested by DNase I. In the control and 0.5 µg DNase I treatments, a 680-bp band appeared on the gel, but this band did not form in treatments using 0.75 µg, or more, DNase I (Fig. 2). In the next experi-ment, we determined whether the transgene was pres-ent in prawn embryos under a more stringpres-ent condi-tion: embryos were treated with 10 µg DNase I.
After DNase I digestion, genomic DNA was extracted from four samples, each consisting of five, pooled, zoea-I-stage embryos from the 400 ng/µl pGL SMI treatment. Three of the four samples of embryonic genomes yielded a PCR product with a molecular mass of 680 bp (Fig. 3, lanes 8, 9, and 11). The positive control group, for which pGL was used as a template, also generated a PCR product with the same size (Fig. 3, lane 3). Positive signal occurrence was at least 15%, that is, at least 3 of the 20 larvae in the four samples tested positive.
Fig. 1. Effect of voltage (4–10 V at 10 A) on spermatophore
extrusion. The cumulative percentage was the cumulative number of males, out of 13, whose spermatophore was extruded at or below a given voltage.
Fig. 2. Effect of DNase concentration on pGL digestion. An aliquot
of 1 µg pGL was incubated with 0.5, 0.75, 1, or 10 µg DNase in a 100 µl solution at 37°C for 1 hr. After the DNase was denatured by boiling, samples were analyzed by PCR without (A) or with (B) an additional 1 µg of pGL. M, molecular marker of phagefX174 digested with HaeIII. The arrow indicates the 680 bp PCR-product.
Analysis of Transgenic Larva by PCR and Southern Blotting
Genomic DNA was extracted from each hatched larva from the 750 ng pGL SMI treatment. Larval genomes were analyzed by PCR and Southern blot analyses. A single, positive band, with a molecular mass of 680 bp, was observed exclusively in the experimental group (lanes 5–14 of Fig. 4). Based on positive signal appear-ance in PCR and Southern blot analyses, the SMI gene transfer success rate for prawns was about 70% (16 of 23 larvae were positive).
Effect of SMI on Fertility, Hatching, and Transgenesis
Egg fertilization and embryo hatching rates for the untreated group and the four SMI treatments (28, 250, 750, and 1,000 ng of pGL) did not differ (Fig. 5). However, transgenesis increased as the concentration of pGL used in SMI was increased, with the greatest increase occurring from 250 to 750 ng pGL.
Green Fluorescence
Green fluorescence was used to detect the expression of the GFP reporter gene in hatched larvae. Unexpect-edly, green fluorescence was not seen in any larvae from the SMI with plasmid (experimental) treatments, nor in larvae from the SMI with water (mock-treated control) treatment.
DISCUSSION
Giant freshwater prawns are one of the most valu-able aquacultural crustaceans, and they are also suit-able for indoor, laboratory-scale culture. In addition, because embryogenesis can be achieved in vitro under
controlled conditions, this valuable decapod is used as an invertebrate model. Unfortunately, it is difficult to transfer genes into the fertilized eggs of M. rosenbergii using electroporation because they are retained in a clump on the abdomen and cannot be detached until 16 hr postfertilization (Caceci et al., 1996). Separating individual eggs from the clump and placing each fertil-ized egg in a container for electroporation is very tedious work. Consequently, only a small number of fertilized prawn eggs can be treated. In contrast, SMI provides a fast, efficient, and easy method for transferring genes to fertilized, giant freshwater prawn eggs.
We demonstrated that at least 1 µg of foreign DNA fragments (pGL) was completely digested by 0.75 µg DNase kept at 37°C for at least 1 hr. However, PCR
Fig. 3. Detection of pGL in giant freshwater prawn embryos using
PCR analysis. Genomic DNAs were isolated from five pooled, zoea-I-stage embryos developed from SMI treatment using 400 ng pGL. All embryos were incubated with 10 µg DNase at 37°C for 1 hr before their genomes were extracted and analyzed. Lane 1, molecular markers, HaeIII-digestedfX-174-RF DNA; lane 2, 0.5 ng pGL as a template (positive control); lane 3, no template; lanes 4–7, from an SMI treatment using a solution lacking pGL (mock-treatment group); lanes 8–11, from an SMI treatment using a solution containing pGL (experimental group). Primers added to amplify a 680-bp fragment from transgenic GFP cDNA by using GFP primer, and a 425-bp from endogenousb-tubulin gene by using Tub primer, were indicated.
Fig. 4. PCR (A) and Southern blot (B) analyses of genomic DNA
isolated from hatched larvae using GFP cDNA as a probe. Lane 1, 0.1 ng pGL as a template (positive control); lane 2, molecular markers, HaeIII-digestedfX-174-RF DNA; lane 3, no template added (negative control); lane 4, larva from an SMI treatment using a solution lacking pGL (mock-treatment group); lanes 5–14, larvae from an SMI treat-ment using a solution containing 750 ng pGL (experitreat-mental group). A 680-bp fragment amplified from GFP cDNA was indicated.
Fig. 5. Effect of the amount of microinjected DNA on the rates of
fertilization, hatching, and transgenesis of giant freshwater prawns. Plasmid DNA prepared at the concentrations of 28, 250, 750, and 1,000 ng per µl of water were respectively microinjected in a volume of 1 µl into each spermatophore prior to in vivo fertilization. The number of genomes from hatched larvae that were examined to determine the gene transfer rate is indicated above each solid bar.
analysis of genomic DNA extracted from zoea-I-stage embryos in the 10 µg DNase SMI treatment still found a 680-bp band. Moreover, the oligonucleotide primers chosen for PCR amplification of the GFP cDNA (trans-gene) andb-tubulin gene (internal control gene) gener-ated 680 bp and 425 bp DNA fragments, respectively. A 680-bp PCR product was observed when pGL fragments were present in transgenic, zoea-I-stage embryos and hatched larvae. Only nontransgenic, SMI-treated prawns and prawns from the control group contained a 425-bp PCR product. The 680-bp PCR product from the SMI treated samples was positive for Southern blot hybridization using a labeled, GFP cDNA probe. All this evidence indicates that the exogenous plasmid DNA in embryos and larvae were introduced by SMI. Our findings concur with those of Shamila and Mathavan (1998), who demonstrated that a foreign plasmid could be introduced into V instar silkworm larvae by testes microinjection.
Although we have not yet analyzed all the experimen-tal larvae, results from the positive samples of PCR and Southern blot analyses from hatched larvae indicate that SMI achieved a gene transfer rate of about 70%. This success rate is far superior to that reported for microinjection of finfish, which ranged from 16% to 27% for medaka (Ozato et al., 1986; Inoue et al., 1990; Lu et al., 1992) and 0% to 35% for zebrafish, common carp, and channel catfish (Powers et al., 1992). It is also much better than the success rates for electroporation of fertilized finfish eggs, which ranged from 4% to 20% for medaka (Inoue et al., 1990; Lu et al., 1992), and electroporation of finfish sperm, which were only 2.6% to 4.2% for carp, tilapia, and catfish (Muller et al., 1992). The SMI gene transfer rate for giant prawns is comparable to that obtained by electroporation of ze-brafish eggs (35% to 75%, Powers et al., 1992) and the electroporation of loach (50%, Tsai et al., 1995a) and abalone sperm (65%, Tsai et al., 1997). SMI probably achieved a high rate of gene transfer in giant freshwa-ter prawns because the microinjected plasmids are retained in the spermatophore for 2–6 hr before in vivo fertilization begins. The longer the foreign plasmids remain in the spermatophore, the greater the probabil-ity they will enter sperm.
Green fluorescence, which was presumptively ex-pressed by the transgenic GFP cDNA, was not detect-able in hatched larvae. This may be attributed to transient expression of the GFP cDNA and to the functionality of the CMV promoter. It is also possible that the exoskeleton somehow blocks visual detection of GFP gene expression. Gendreau and colleagues (1995) reported that the transient expression of a luciferase gene transferred by ballistics into brine shrimp was detectable only at 12 hr after bombardment. Problems detecting expression of the GFP gene could be overcome in future experiments by using prawn-specific promoter fused with GFP cDNA.
Gendreau and colleagues (1995) reported that ballis-tic shots decreased survival of treated brine shrimp.
Bensheng and Khoo (1997) reported that the early developmental stage of freshwater shrimp embryos is extremely vulnerable to microinjection, and recom-mended that the intermediate stage be used for microin-jection. However, when a gene is transferred to the intermediate stage, the transgene will have a mosaic distribution. In this study, we demonstrated several advantages of SMI gene transfer system, including: (1) a high gene transfer rate; (2) no effect on fertility, survival, and hatching rates; and (3) easy introduc-tion of foreign DNA into oocytes, which definitely helps to minimize transgenic mosaicism. In our opinion, SMI is the most promising technology for prawn trans-genesis.
REFERENCES
Besheng J, Khoo HW. 1997. Transient expression of two luciferase reporter gene constructs in developing embryos of Macrobrachiumm lanchesteri (De Man). Aquaculture Res 28:183–190.
Caceci T, Calson CB, Toth TE, Smith SA. 1996. In vitro embryogenesis of Macrobrachium rosenbergii larvae following in vivo fertilization. Aquaculture 147:169–175.
Chourrout D, Perrot E. 1992. No transgenic rainbow trout produced with sperm incubated with linear DNA. Mol Marine Biol Biotechnol 1:282–285.
Chourrout D, Guyomard R, Houdebine LM. 1986. High-efficiency gene transfer in rainbow trout (Salmo gairdneri) by microinjection into egg cytoplasm. Aquaculture 51:143–150.
Dunham RA, Eash J, Askins J, Townes TM. 1987. Transfer of the metallothionein-human growth hormone fusion gene into channel catfish. Trans Am Fish Soc 116:87–91.
Dunham RA, Ramboux AC, Duncan PL, Hayat M, Chen TT, Lin CM, Knight K, Gonzalez-Villasenor I, Powers DA. 1992. Transfer, expres-sion, and inheritance of salmonid growth hormone genes in channel catfish, Ictalurus punctatusa, and effects on performance traits. Mol Marine Biol Biotechnol 1:380–389.
Fletcher GL, Shears MA, King MJ, Davies PL, Hew CL. 1988. Evidence for antifreeze protein gene transfer in Atlantic salmon (Salmo salar). Can J Fish Aquat Sci 45:352–357.
Gedreau S, Lardans V, Cadoret JP, Mialhe E. 1995. Transient expression of a luciferase reporter gene after ballistic introduction into Artemia franciscana (Crustacea) embryos. Aquaculture 33:199– 205.
Giambernardi TA, Rodeck U, Klebe RJ. 1998. Bovine serum albumin reverses inhibition of RT-PCR by melanin. BioTechniques 25:564-566.
Inoue K, Yamashita S, Hata J, Kabeno S, Asada S, Nagahisa E, Fujita T. 1990. Electroporation as a new technique for producing transgen-ing fish. Cell Differ Dev 29:123–128.
Khoo HW, Ang LH, Lim HB, Wong Y. 1992. Sperm cells as vectors for introducing foreign DNA into zebrafish. Aquaculture 107:1–19. Lu JK, Chen TT, Chrisman CL, Andrisani OM, Dixon JE. 1992.
Integration, expression, and germ-line transmission of foreign growth hormone genes in medaka (Oryzias latipes). Mol Marine Biol Biotechnol 1:366–375.
Muller E, Ivics Z, Erdelyi F, Papp T, Varadi L, Horvath L, MacLean N, Orban L. 1992. Introducing foreign genes into fish eggs with electroporated sperm as a carrier. Mol Marine Biol Biotechnol 1:276–281.
Ozato K, Kondoh H, Inohara H, Iwamatsu T, Wakamatsu Y, Okada TS. 1986. Production of transgenic fish: introduction and expression of chickend-crystallin gene in medaka embryos. Cell Differ 19:234– 237.
Powers DA, Hereford L, Cole T, Chen TT, Lin CM, Knight K, Creech K, Dunham R. 1992. Electroporation: a method for transferring genes into the gametes of zebrafish (Brachydanio rerio) channel catfish
(Ictalurus punctatus), and common carp (Cyprinus carpio). Mol Marine Biol Biotechnol 1:301–308.
Sambrook J, Fritsch EF, Maniatis T. 1989. Large-scale preparations of plasmid DNA. In: Nolan C, editor. Molecular cloning: a laboratory manual. New York: Cold Spring Harbor Laboratory Press. p 1.33–1.46. Shamila Y, Mathavan S. 1998. Sperm-mediated gene transfer in the
silkworm Bombyx mori. Arch Insect Biochem Physiol 37:168–177. Symonds JE, Walker SP, Sin FYT, Sin I. 1994. Development of a mass
gene transfer method in chinook salmon: optimization of gene transfer by electroporated sperm. Mol Marine Biol Biotechnol 3:104–111.
Tsai HJ, Tseng FS. 1994. Electroporation of a foreign gene into black porgy (Acanthopagrus schlegeli) embryos. Fish Sci 60:787–788. Tsai HJ, Tseng FS, Liao IC. 1995a. Electroporation of sperm to
introduce foreign DNA into the genome of loach (Misgurnus anguil-licaudatus). Can J Fish Aquat Sci 52:776–787.
Tsai HJ, Wang SH, Inoue K, Kimura M, Wakamatsu Y, Ozato K. 1995b. Initiation of the transgenic lacZ gene expressed in medaka (Oryzias latipes) embryos. Mol Marine Biol Biotechnol 4:1–9. Tsai HJ, Lai CH, Yang HS. 1997. Sperm as a carrier to introduce an
exogenous DNA fragment into the oocyte of Japanese abalone (Haliotis diversicolor supertexta). Transgenic Res 6:85–95.