國 立 交 通 大 學
應用化學系分子科學碩士班
碩士論文
三原子反應
O(
3P)+HCl OH+Cl(
2P)之反應座標上的
振動與轉動能級
Vibrational and Rotational Levels Along the Reaction
Coordinate for the Triatomic Reaction
O(
3P)+HCl OH+Cl(
2P)
研 究 生:張哲源
指導教授:朱超原 博士
OH+Cl(
2P)之反應座標上的振動與轉動能級
Vibrational and Rotational Energy Levels Along the Reaction Coordinate
for the Triatomic Reaction O(
3P)+HCl
OH+Cl(
2P)
研究生:張哲源 Student:Che-yuan Chang
指導教授:朱超原博士 Advisor:Dr.Chao-yuan Zhu
國立交通大學
應用化學系分子科學碩士班
碩士論文
A Thesis
Submitted to Institute of Molecular Science
College of Science
National Chiao Tung University
In Partial Fulfillment of the Requirements
For the Degree of
Master of Science
In
Applied Chemistry
September 2010
Hsinchu,Taiwan,Republic of China
中華民國九十九年九月
三原子反應
O(
3P)+HCl
三原子反應
O(
3P)+HCl
OH+Cl(
2P)之反應座標上的振動與轉動能級
學生︰張哲源 指導教授︰朱超原博士
國立交通大學應用化學系分子科學碩士班
摘 要
三個從頭算(ab initio)的量子方法:哈特里-福克理論(HF)、達到二級微擾的 Moller-Plesset 微擾理論(MP2)、包含單激發與雙激發的耦合簇理論(Coupled Cluster Theory)(CCSD)被採用於研究 O(3P)+HCl OH+Cl(2P)這個化學反應座標 上的振動與轉動能級。這三個方法都共同搭配同一個基組6-31++G(d,p),以便比 較三個方法的計算準確度。這個反應的座標上,包括反應錯合物、過渡態、生成 錯合物…等眾多錯合物的鍵長、鍵角、振動頻率、轉動常數等都被計算著。沿著 這個反應座標上,電子能量已在波恩-奧本海默近似下被計算出來,並且核的能 量(轉動能與振動能)也在剛體轉子以及諧振子振動模式近似的條件下被計算出 來,於是乎總能的能級(電子能量+核的能量)才能得到。我們發現在 HF 層級的方 法計算中,在總能為2.0eV 之下能決定 404 個能態;在 MP2 層級的方法計算中, 在總能為1.1eV 之下能決定 286 個能態;在 CCSD 層級的方法計算中,在總能為 1.0eV 之下能決定 356 個能態。沿著這個反應座標,造就出振動能的振動量子數, 就看有無虛頻出現(必須被忽略掉),還有該分子是否屬線性分子,可以有兩個到 四個不等的振動量子數(ν1、ν2、ν3、ν4);還有分子的近似對稱性也會造就出 1~2 個轉動量子數(j、k)。這三個方法算出反應錯合物皆為線性分子;至於過渡態的 鍵角,HF 方法算出來的是 119°,MP2 方法算出來的是 135.5°,CCSD 方法算出 來的是137°;而生成錯合物的鍵角,HF 方法算出來的是 179°,MP2 方法算出來 的是56°,CCSD 方法算出來的是 60°。 在計算反應能之前,我們必須先計算出雙原子分子 HCl 與 OH 的相對電子能與振動頻率以及算出O(3P)原子與 Cl(2P)原子的相對電子能。那麼,我們算出 的反應能變化(包含零點校正能)是:HF 方法所得為+8.87kcal/mol,MP2 方法所 得是-0.42kcal/mol,CCSD 方法算出來的是+0.54kcal/mol,三者中以 CCSD 方法 所得最與實驗值+1.19±0.12kcal/mol 接近。
Vibrational and Rotational Levels Along Reaction Coordinate
for the Triatomic Reaction O(
3P)+HCl
OH+Cl(
2P)
Student:Che-yuan Chang Advisor:Dr.Chao-yuan Zhu
Institute of Molecular Science
National Chiao Tung University
Abstract
Three ab initio quantum methods, the Hartree-Fock(HF), Moller-Plesset perturbation theory up to the second order(MP2), and coupled cluster theory with including single and double excitations(CCSD), were employed to study
ro-vibrational energy levels along reaction coordinate for reaction O(3P)+HCl OH+Cl(2P). All three methods utilized the same basis set 6-31++G(d,p) in order to compare accuracy of calculations. The bond lengths, bond angles, vibrational frequencies, and rotational constants along reaction coordinate were all calculated with including structures of reaction complex, transition state, and product complex. Along reaction coordinate, electronic energies were calculated under
Born-Oppenheimer approximation, and nuclear energies (rotational energies and vibrational energies) were computed with rigid rotator and harmonic normal mode approximations. Therefore, potential energy levels (electronic plus nuclear) along reaction coordinate were obtained. We found at transition state geometry that 404 potential energy levels were characterized at HF levels within energy region of 2.0eV, 286 potential energy levels were characterized at MP2 levels within energy region of
1.1eV, and 356 potential energy levels were characterized at CCSD levels within energy region of 1.0eV. Along reaction coordinate, vibrational quantum numbers which contribute to vibrational energies can vary from 2 to 4 modes(ν1、ν2、ν3、ν4) due
to appearance of imaginary vibrational frequencies which are ignored and symmetry top of rotator was approximately adopted with two rotational quantum numbers(j、k). All three methods predicted that reaction complex is linear geometry. Transition state has bond angle 119° predicted from HF method, 135.5° from MP2 and 137° from CCSD. Product complex has bond angle 179° predicted from HF method, 56° from MP2 and 60° from CCSD.
Before we computed the reaction energy, we must compute the relative
electronic energies and vibrational frequencies for diatomic molecules HCl and OH and compute the relative electron energy for atom O(3P) and Cl(2P), from which we obtained reaction potential energy change(including zero-point energy) is
+8.87kcal/mol predicted from HF, -0.42kcal/mol from MP2 and +0.54kcal/mol from CCSD in comparison with the experimental value +1.19±0.12kcal/mol.
致 謝
在碩士生涯中,非常感謝眾多師兄姊:蔡琇雅師姊、李清旭師兄、李軍師 兄、楊玲師姊、宋迪師姐等在實驗上給我的指導與意見以及解答課業上的疑惑。 也很感謝李文凡同學、黃琮偉師弟陪伴我度過了歡笑與類交織的碩士生涯。最後 萬分感謝朱超原教授不厭其煩的指導也非常敬佩老師對學術研究的熱忱。目 錄
頁次 中文摘要... i 英文摘要... iii 致謝... v 目錄... vi 表目錄... viii 圖目錄... ix 附錄目錄... x 第一章、序論... 1 1.1 前言... 1 1.2 振動能級之基本原理... 2 1.2.1 雙原子分子的振動能級... 2 1.2.2 多原子分子的振動能級... 2 1.3 轉動能級之基本原理... 4 1.3.1 球形轉子……… 4 1.3.2 對稱轉子……… 6 1.3.3 線性轉子……… 7 1.3.4 轉動常數之計算……… 7 1.4 振動與轉動聯合能級之基本原理………. 11 1.5 本論文的研究………. 12 第二章、方法... 142.1 劈裂價鍵基組(Pople style basis set)簡介……… 14
2.2 哈特里-福克理論(Hartree-Fock Theory)……… 14
2.2.1 波恩-奧本海默近似(Born-Oppenheimer approximation)…… 15
2.2.2 哈特里-福克方程(Fock equetion)……….. 16
2.2.3 詮釋哈特里-福克近似……… 18
2.3 多體微擾理論(Many-Body perturbation Theory)………. 19
2.3.1 一般微擾理論……….. 19
2.3.2 Moller-Plesset perturbation Theory………. 21
2.4 耦合簇理論(Coupled Cluster Theory)………. 22
2.5 過渡態理論(Transition State Theory)... 23
2.5.1 尋找局部能量最小結構(Local minimum)……….. 25 2.5.2 尋找鞍點與過渡態………... 26 第三章、結果與討論... 28 3.1 雙原子分子 OH, HCl 的鍵長、振動頻率、轉動常數以及全 反應的反應能變化……… 28 3.2 反應障壁(Barrier Height)……… 30 3.3 反應錯合物、中間過渡態、產物錯合物以及 IRC 途徑……. 31 第四章、結論... 39 附錄... A1
表 目 錄
頁次 Table 1:各方法與基組合體所計算出來的雙原子分子 OH 與 HCl 的鍵長、 振動頻率、轉動常數與實驗值之比較。……… 29 Table 2:各方法與基組合體所計算出來的 O(3P)+HCl OH+Cl(2P)反應能 變化。……… 29 Table 3:各方法與基組合體所計算出來的 O(3P)+HCl OH+Cl(2P)反應障壁。 ……… 30 Table 4:各方法與基組合體所計算出來的反應錯合物、中間過渡態、產物 錯合物的鍵長、鍵角。……… 32 Table 5:各方法與基組組合體所計算出來的反應錯合物、中間過渡態、產物 錯合物的振動頻率。……… 33圖 目 錄
頁次 圖 1-1. 量子力學諧振子的振動能級圖。………. 2 圖 1-2. (a)反應錯合物的四個振動模式。 (b)生成錯合物的三個振動模式。………. 3 圖 1-3. 球形轉子與線性轉子的轉動能級圖。………. 5 圖 1-4. 量子數k的意義(a)當 k 接近它的最大值 j 的時候。 (b)當 的時候。……….. 7 圖 1-5. 雙原子分子的振動與轉動聯合能級圖。………. 12 圖 2-1. 化學反應的能量剖面圖。………. 23 圖 2-2. 反應途徑圖解。………. 24 圖 2-3. 已活化的錯合物, ,被定義存在反應障壁頂端的中央位置。…. 24 圖 2-4. 最陡降落最小化法之示意圖。………. 25 圖 2-5. LST 與 QST 法之示意圖。………. 27 圖 3-1. HF/6-31++G(d,p)的 IRC 途徑能級圖(僅虛頻部)。……… 36 圖 3-2. (a) MP2/6-31++G(d,p)的 IRC 途徑能級圖(僅中間過渡態前後各八步)。 36 (b) MP2/6-31++G(d,p)的 IRC 途徑全區能級圖(中間過渡態向左至 Step -151 , 向右至 Step +30.) 。……… 37 圖 3-3. (a)CCSD/6-31++G(d,p)的 IRC 途徑能級圖(僅中間過渡態前後各十步)。 37 (b) CCSD/6-31++G(d,p)的 IRC 途徑全區能級圖(中間過渡態向左至 Step -151 , 向右至 Step +20.)。………. 38 圖 3-4. 反應途徑 1a 與 1b 根據 Ref.22 使用 CCSD(T)/6-311+G(3df,2p)// 0 k= AB + +B3LYP/6-311+G(3df,2p)以及 CCSD(T)/6-311+G(3df,2p)//CCSD(T)/ 6-311+G(d,p)所 繪製的勢能圖。……… 39
附 錄 目 錄
頁次 附錄 A-1 CCSD/6-31++G(d,p)的反應錯合物振動與轉動聯合能級與 總能能級。………. A1 附錄 A-2 CCSD/6-31++G(d,p)的 TS 振動與轉動聯合能級與 總能能級。………. A2 附錄 A-3 CCSD/6-31++G(d,p)的生成錯合物振動與轉動聯合能級與 總能能級。………. A3 附錄 A-4 MP2/6-31++G(d,p)的反應錯合物振動與轉動聯合能級與 總能能級。………. A4 附錄 A-5 MP2/6-31++G(d,p)的 TS 振動與轉動聯合能級與 總能能級。………. A5 附錄 A-6 MP2/6-31++G(d,p)的生成錯合物振動與轉動聯合能級與 總能能級。………. A6第一章 序論
1-1 前言 根據罕德定則,O(3P)是氧原子最基態的狀態以及 Cl(2P)是氯原子最基態的狀 態的原因如下:[1] 氧原子的基態電子組態為2p4:ml +1 0 -1 ↑↓ ↑ ↑→ML=1, MS=1→L=1,S=1→3P 氯原子的基態電子組態為3p5,ml +1 0 -1 ↑↓ ↑↓ ↑→ML=1, MS=1 2→L=1,S= 1 2 →2P 基態的氧原子 O(3P)與氯化氫的反應在大氣化學中扮演了一個重要的角色, 首先臭氧分子被光解後形成O(1D)原子,由於 O(1D)原子化學反應活性極強,因 此很容易跟其他化學物質包括氯化氫反應,造成臭氧層的破壞。 [2] 由上述化學反應式得知,由於形成O(3P)原子的這條途徑放熱最多,因此 O(1D) 與氯化氫的反應會有比較多的機會往形成O(3P)原子的這條途徑進行,而 O(3P) 與氯化氫反應後產生基態氯原子Cl(2P)這個化學反應的反應熱的實驗值為 mol[3] 。本篇會有對於這條反應式有很多項目的量子計算,首 先是在6-31++G(d,p)的相同機組下,用 HF、MP2、CCSD 等三種不同的方法進 行障壁、反應熱、各錯合物的振動能級及轉動能級的計算。 本文是這樣被組織的:第一章先說明振動能級與轉動能級的基礎知識,並解 釋一個分子的轉動常數是如何被計算出來的。第二章討論進行本實驗所用到的方 法。第三章鋪陳進行本實驗所得出來的結果並且討論它們的含意。第四章來為本 實驗下結論。 exp 1.19 / V kcal Δ = +1-2 振動能級之基本原理 1-2-1 雙原子分子的振動能級 一維諧振子的薛丁格方程式表示如下: 2 2 2 ( ) ( ) ( ) 2 d V x x E x dx ψ ψ ψ μ − + = (式 1-1) 其中, ( ) 1 2 2 V x = kx 。而(1-1)式就可以導出以下這個二次微分方程式: 2 2 2 2 2 1 ( ) ( ) 2 d E kx x dx ψ μ ψ + − = (式 1-2) 0 當(式 1-2)解出來的時候即得 ( 1) ( 1) , 0,1, 2, 2 2 vib k E ν ν ω ν μ = + = + = (式 1-3) (式 1-3)就以圖 1-1.來表示之: 圖 1-1. 量子力學諧振子的振動能級圖。 這樣的能級就以 ω 為公差成為等差數列。v=0 對應這個諧振子的零點能 量,其值就是1 2 ω。 1-2-2 多原子分子的振動能級 ㄧ個N 原子的分子其 Normal modes 會有 3N-5(線性)或 3N-6(非線性)個。也 就是說雙原子分子僅ㄧ個Normal mode,但是如果有三個以上原子的分子,它就 不只ㄧ個Normal modes 了,因為所有的鍵長與鍵角都會隨時改變。就拿本論文 所討論的分子為例: [4]
A. 反應錯合物 3 3 5 4× − = 個Normal modes B. 生成錯合物 3 3 6 3× − = 個Normal modes 以上兩個分子的振動模式如下: A. 反應錯合物 (1) 91.22cm−1 (2) 168.09cm−1 (3) 168.09cm−1 (4) 3044.23cm−1 圖 1-2(a). 反應錯合物的四個振動模式。 B. 生成錯合物 (1) 409.64cm−1 (2) 662.08cm−1 (3) 3744.72cm−1 圖 1-2(b). 生成錯合物的三個振動模式。 就如(式 1-3)一樣,多原子分子的振動能級如下: 1 2 3 5 6 1 1 ( , , ) ( ) , 0,1, 2, 2 N or vib i i i i E ν ν ν ω ν − = =
∑
+ = (式 1-4)就拿本論文所討論的分子為例: 反應錯合物為i=1 ~ 4,則ν ν1= 2 =ν3 =ν4 = 的時候,即有零點校正能為0 1 2 3 4 1 ( 2 ω ω ω+ + +ω )。中間過渡態因為三個振動頻率當中有一個為負值(此為虛 頻),而虛頻並不能考慮在振動能的計算當中,因此 只有i =1 ~ 2,則ν ν1= 2 = 的0 時候,即有零點校正能為1 ( 1 2) 2 ω ω+ 。生成錯合物為i=1 ~ 3,則ν ν1= 2 =ν3 =0 的時候,即有零點校正能為1 ( 1 2 3) 2 ω ω ω+ + 。 1-3 轉動能級之基本原理 ㄧ個物體繞著 a 軸轉動的轉動能經典的表達式為: 2 1 2 a a a E = I ω (式 1-5) 為角速度(rad/sec.),Ia 其中ωa 為轉動慣量。而一個物體對著三個軸轉動的轉動能 的表達式為: 1 2 1 2 1 2 2 2 rot a a b b c c 2 E = I ω + I ω + I ω (式 1-6) 又Ja =Iaωa是為角動量,因此(式 1-6)又可以表達成以下這樣的形式: 2 2 2 2 2 a b c rot a b J J J E 2 c I I I = + + (式 1-7) (式 1-7)是一個關鍵的方程式,再此就用它來討論以下的各種形式的轉子。 1-3-1 球形轉子 當Ia =Ib =Ic = I 的時候: 2 2 2 2 2 2 a b c rot J J J J E I I + + = = (式 1-8) 其中, 是角動量的強度(vector),我們可以立即用以下的量子表達式來取代 : 因此,球形轉子的轉動能的值就可以表達為: J J2 2 ( 1) ,2 0,1, 2, J = j j+ j= 2 ( ) ( 1) , 0,1, 2, 2 rot E j j j j I = + = (式 1-9)
(式 1-9)就以圖 1-3.轉動能的能階圖來表示之: 圖 1-3. 這是球形轉子與線性轉子的轉動能級圖,隨著 j 值越大,相鄰兩能階的 差距就越大。 但正常來講,轉動能值的表達方式都是以轉動常數 B 值的形式存在的。其 中, 2 2 hcB I = 所以 4 B cI π = (式 1-10) 然後轉動能級的表達式又可以寫成: (式 1-11) (式 1-10)中轉動常數 ( ) ( 1), 0,1, 2, rot E j =hcBj j+ j= 的單位為cm−1 B ,(式 1-11)中轉動能級的表達式是以焦 耳為單位的,轉動能級的表達式也可以cm−1為單位: (式 1-12) 所以相鄰兩能階的差距為 ( ) ( 1), 0,1, 2, F j =Bj j+ j=
。 (式 1-13) 因為轉動慣量 ( ) ( 1) 2 , 0,1, 2, F j −F j− = Bj j= I 越大時轉動常數 B 就會越小,所以大分子的能階差距通常較小。 1-3-2 對稱轉子 對稱轉子的三個轉動慣量當中有兩個是相等的但卻跟第三個轉動慣量不 同,Ia≠Ib=Ic;我們稱這個比較特別的軸Ia為主軸。對於這個主軸的轉動慣量,定 義為 I ,那麼其他兩個轉動慣量就定義為 I⊥。然後(式 1-7)式就變成了 2 2 2 2 a b rot J J J E 2 c I I⊥ + = + 這個表達式又可以J2 =Ja2+Jb2+Jc2的形式被寫出來: 2 2 2 2 2 1 1 ( ) 2 2 2 2 2 a a rot a J J J J E J I I⊥ I⊥ I I⊥ − = + = + − (式 1-14) 現在可以用 j j( +1) 2來取代 ,又根據角動量的量子理論我們知道任何軸上角 動量的分量都是局限於這個值 2 J k (k= ± ±0, 1, 2, ,± )j 2 2來取代 2 a J ), 。 值是用來標示主軸上那 個分量的量子數 , 最後對稱轉子的轉動能級的表 達式就可以寫成這樣了: (k )因此 也可以用 k 2 ( 1) ( ) , 0,1, 2, ; 0, 1, 2, , , 4 4 rot E hcBj j hc A B k j k A B cI cI π π ⊥ = + + − = = ± ± = = 其中 j ± (式 1-15) (式 1-15)中,當 k=0 的時候,角動量在主軸上就沒有分量了,這個時候轉 動能級就只要依賴 I⊥的值就行了。而當k=±j 的時候,幾乎所有角動量來自於繞 著主軸轉動,此時 I 的大小就對能級有很大地決定性了。k 的正負號不影響能量 的大小,只表示轉動的方向相反。
圖 1-4. 量子數k的意義(a)當 k 接近它的最大值 j 的時候,絕大部分的分子轉動 都是圍繞著主軸。(b)當k=0的時候,這個分子的轉動主軸上沒有角動量。 1-3-3 線性轉子 線性轉子的每個原子的原子核都被視為一個一個的質點,它的轉動行為都 是圍繞著與分子線垂直的軸轉動的,因此線性轉子的主軸上並沒有角動量的分 量,意即(式 1-15)中的k =0。此時(式 1-15)就可簡化為 Erot =hcBj j( +1),j=0,1, 2, (式 1-16) (式 1-16)與(式 1-11)雖然一樣,但線性轉子是因為k=0,球形轉子是 A B= 。[5] 1-3-4 轉動常數之計算 以下就說明慣性矩的觀念以及舉個本文相關例子的分子結構參數來示範如 何計算轉動常數。 任何物體的轉動慣量都是這樣被定義的:
I
=
∫
r dm
2 , 其中 是質量, 是這個點質量(point mass) 等同於所有點質量轉動慣量的總和: m r dm至轉動軸之間的垂直距離,一個體系的總轉動慣量 2 i i 1 N iI
m r
, 同一個物體 ==
∑
,選用不同的轉動軸將會算出不同大小的轉動慣量,除非這個物體是個對所有轉動軸都是對稱 性很好的物體。所以慣性張量矩陣(moment of inertia tensor matrix)是個可以總 結一個物體所有轉動慣量很方便的方法。這個矩陣可以以空間中任何一點當作參 考點,而質心就是最常被用到的參考點。 對於一個有N 個點質量(point mass)mk的物體,慣性矩張量矩陣就如下所示: (式 1-17) 裡面的九個分量都是這樣被定義的: (式 1-18) 其中 ,i j可以是x、y、z, 是點質量rk mk到參考點之間的距離,δij是Kronecker delta。當 i= j時,δij = ;當i1 ≠ 時,j δij = 。 0 2 2 2 2 2 2 2 1 1 2 1 1
(
),
,
1
(
0
(
),
N N.
主慣性矩陣(Principal moments of inertia matrix)
2
)
xx k k xx kx k kx ky kz xx xx k ky kz k k N N xy k k xy kx ky xy xy k kx ky k kI
m r
r
r
r
r
r
I
m r
r
I
m r
r r
I
m r r
δ
δ
δ
δ
= = = ==
−
=
+
+
= ∴
=
=
−
= ∴
= −
∑
∑
∑
∑
∵
∵
Ex
+
主慣性矩陣可以靠把慣性張量矩陣對角化之後得到,簡單的說主慣性矩陣 就是慣性矩張量矩陣的本徵值(eigen value)。得到這樣形式的矩陣: (式 1-19) 所謂矩陣的對角化就是: 1 0 0 det( 0 1 0 ) det( ) 0 0 0 1 xx xy xz xx xy xz yx yy yz yx yy yz zx zy zz zx zy zz I I I I I I I I I I I I I I I I I I λ λ λ λ ⎡ ⎤ ⎡ ⎤ ⎡ − ⎢ ⎥− ⎢ ⎥ = ⎢ − ⎢ ⎥ ⎢ ⎥ ⎢ ⎢ ⎥ ⎢⎣ ⎥⎦ ⎢ − ⎣ ⎦ ⎣ ⎤ ⎥ = ⎥ ⎥⎦ (式 1-20) 即可得到λ1、λ2、λ3為 xx xy xz yx yy yz zx zy zz I I I I I I I I I ⎡ ⎤ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎣ ⎦ 的本徵值(eigen value)。 舉例示範說明: 例一: #CCSD(maxcyc=200)/6-31++G(d,p) opt=(tight,TS) (0.009162019412, 0.026009713299,0)− 首先,可以算出質心座標是 。然後把 所有原子都轉換成質心座標系,也就是說將質心視為新的原點。得到各原子的新 的座標如下:
Atom mass (amu.) X Y Z
H 1.00783 -0.471134748835 0.582463676026 0.000000000000 O 15.99491 0.009316889765 1.691788293986 0.000000000000 Cl 34.96885 0.009316889765 -0.79061867542 0.000000000000 然後就可得到慣性張量矩陣如下:
2 67.97996906 0.282036818 0 0.282036818 0.228129841 0 ( ) 0 0 68.2080989 amu A ⎡ ⎤ ⎢ ⎥ ⋅ ⎢ ⎥ ⎢ ⎥ ⎣ ⎦ 接下來將此慣性張量矩陣對角化, 2 67.97996906 0.282036818 0 det( 0.282036818 0.228129841 0 ( )) 0 0 0 68.2080989 amu A λ λ λ − ⎡ ⎤ ⎢ − ⎥ ⋅ = ⎢ ⎥ ⎢ − ⎥ ⎣ ⎦ 得到 1 2 2 3 0 0 67.9811431 0 0 0 0 0 0.2269558 0 ( ) 0 0 0 0 68.2080989 I I amu A I ⎡ ⎤ ⎡ ⎤ ⎢ ⎥ ⎢= ⎥ ⋅ ⎢ ⎥ ⎢ ⎥ ⎢ ⎥ ⎣ ⎦ ⎣ ⎦ ⎢ ⎥ 2 2 45 2 1 2 4 1 8 2 2 2 45 2 2 3 3 ( ) 8 67.9811431( ) 1.128853209 10 ( ) ( ) 0.2269558( ) 7.434105741 2226.772817 3.768688954 10 ( ) ( ) 68.2080989( ) 1.132621898 10 ( ) 7.4 h B Hz I I amu A kg m G I amu A kg m G B B Hz Hz I amu A kg m B π − − − ≡ ∴ = ⋅ = × ⋅ → = = ⋅ = × ⋅ → = = ⋅ = × ⋅ → = ∵ 09369478(GHz) 對照Gaussian 實際算出的結果: Table 1-1: 轉動常數(GHz) Gaussian03 計算結果 自己手算 B1 7.4341043 7.434105741 B2 2226.7834919 2226.772817 B3 7.4093681 7.409369478 例二: #CCSD(maxcyc=200)/6-31++G(d,p) opt=tight
質心座標是( 。然後把所有原子都轉換成質心座標系,也就 是說將質心視為新的原點。得到各原子的新的座標如下:
0,0,0.024062664745)
Atom mass (amu.) X Y Z
H 1.00783 0.000000000000 0.000000000000 -0.141805899301 O 15.99491 0.000000000000 0.000000000000 -2.468739151669 Cl 34.96885 0.000000000000 -0.000000000000 1.133299401722 然後就可得到慣性張量矩陣如下: 2 142.416868103 0 0 0 142.416868103 0 ( ) 0 0 0 amu A ⎡ ⎤ ⎢ ⎥ ⋅ ⎢ ⎥ ⎢ ⎥ ⎣ ⎦ 而以上這個慣性張量矩陣已經被很完美地對角化了,故同時也是主慣性矩陣。 2 45 2 142.416868103( ) 2.36488725 10 ( ) B 3.548589524( ) I = amu A⋅ = × − kg m⋅ → = GHz 對照Gaussian 實際算出的結果: Table 1-2: 轉動常數(GHz) Gaussian03 計算結果 自己手算 B 3.548589 3.548589524 1-4 振動與轉動聯合能級之基本原理 ㄧ個雙原子分子的振動與轉動聯合能級正如(3)式與(16)式的總和: 1 ( ) ( 1), 0,1, 2, ; 0,1, 2, 2 vib rot E − = ν + ω+hcBj j+ ν = j= (式 1-21) 一般來說ω 會是 B 的幾十倍到幾千倍之間,所以振動能級的能階差距一般來說會
比轉動能級的能階差距大上幾百倍到幾千倍之間。這樣的一個能階圖就整理於圖 1-5.[6] 圖 1-5. 雙原子分子的振動與轉動聯合能級圖。 那麼,ㄧ個多原子分子的對稱轉子,它的振動與轉動聯合能級正如(式 1-4) 與(式 1-15)的總和: 3 5 6 2 1 2 1 1 ( , , , , ) ( ) ( 1) ( ) , 2 0,1, 2, ; 0,1, 2, , 0, 1, 2, N or vib rot i i i i E j k hcBj j hc A j k j ν ν ν ω B k ν − − = = + + + + − = = = ± ± ±
∑
(式 1-22) 當然,νi i N(∈ )= = = 的時候,即有零點校正能為j k 0 3 5 6 1 1 2 N or vib rot i i Eω
− − = =∑
。 1-5 本論文的研究 本論文接下來是這樣被組織的:第二章為方法,第三章為結果與討論,第四 章為結論。本論文主要會針對O(3P)+HCl→OH+Cl(2P)這個化學反應式說明所用 到的計算方法跟基組以及相關的理論基礎,然後第三章的部份會對於這個反應第 一個算出這個反應的反應熱以及雙原子分子的鍵長、振動頻率、轉動常數以及反 應熱並且跟其他文獻的結果和實驗值做比較;第二個算出這個反應的障壁 (Barrier heigh),反應障壁就是中間過渡態的總能減去 O(3P)原子與氯化氫分子的總能;第三個算出反應錯合物、中間過渡態、產物錯合物的鍵長、鍵角、振動頻 率;第四個算出反應的IRC 路徑上幾個錯合物包括反應錯合物、中間過渡態、 產物錯合物的振動能級、轉動能級、振動與轉動聯合能級並且繪成圖形來對其討 論,最後就就在第四章來為本篇論文做總結。 第二章裡面說明到進行本實驗用到了三種方法與機組的組合體,其中基組皆 為同一個6-31++G(d,p);而方法有 HF、MP2、CCSD 三種。而這三種方法就直 接牽扯到三個理論基礎:哈特里-福克理論(Hartree-Fock Theory)、Moller-Plesset 微擾理論(Moller-Plesset perturbation Theory)、耦合簇理論(Coupled Cluster Theory)。 第三章對於O(3P)+HCl→OH+Cl(2P)這個化學反應式利用 HF、MP2、CCSD 等三種方法在6-31++G(d,p)基組下,計算 O(3P)原子、氯化氫分子、Cl(2P)原子、 OH 分子、反應錯合物、中間過渡態、產物錯合物以及其他若干 IRC 路徑上的錯 合物的鍵長、鍵角、振動頻率以及繪圖繪出振動與轉動聯合能級的圖形,並且比 較三種方法與基組的組合體所算出來的數據到底有什麼不同,並且比較三種方法 與基組的組合體跟實驗值的差距又各是如何。
第二章 方法
2-1 劈裂價鍵基組(Pople style basis set)簡介
即 k-nlmG 這類型的基組。k 表示有 k 個初等高斯型軌道(PGTO)被用來代表 核心軌道;n 表示有 n 個初等高斯型軌道被用來代表內層價軌道;l 表示有 l 個初 等高斯型軌道被用來代表中層價軌道;m 表示有 m 個初等高斯型軌道被用來代 表外層價軌道。 以上這些基組都可以加上彌散函數以及極化函數。彌散函數通常以+或者++ 等符號來表示,第一個+號表示有一個p 彌散函數加到了重原子的上面,第二個+ 號表示又有一個s 彌散函數加到了氫原子的上面。加個極化函數到非氫原子上跟 加個彌散函數到非氫原子上是一樣的,差別在於彌散函數都是寫在G 前面,而 極化函數都是寫在G 後面。舉個例子說明:本論文用到的基組是 6-31++G(d,p)。 這個基組表示有6 個初等高斯型軌道被用來代表核心軌道;有 3 個初等高斯型軌 道被用來代表內層價軌道;有1 個初等高斯型軌道被用來代表中層價軌道;有一 套彌散函數在重原子上面,另一套彌散函數在氫原子上面;有一套極化函數 d-function 在重原子上面,另一套極化函數 p-function 在氫原子上面。目前最大的 劈裂價鍵基組是6-311++G(3df,3pd) 。 如果一套極化函數被使用了,另一個替代符號”*”也會被廣泛使用。比方說 6-31G*就等同於 6-31G(d);6-31G**就等同於 6-31G(d,p)。[7] 2-2 哈特里-福克理論(Hartree-Fock Theory) 哈特里-福克方程來自於對多電子體系電子波函數的變分法處理。在Born-Op penheimer approximation,多電子體系的電子運動與能量可以跟原子核的互相分 離,那麼此時就可以用哈密頓算符和多電子波函數還計算種個體系的電子能量。 2-2-1 波恩-奧本海默近似(Born-Oppenheimer approximation)
其哈密頓算符可以寫成以下的形式: tot n e H =T +H (式 2-1) 其中 2 1 2 1 1 1 1 1 2 1 1 2 n e n e e e n n N n R N N N N N N N e i i i j j i e i i j T M Z Z Z H m r R r r R R α α α α β α α α α β α α β = = = = < = < = − ∇ ⎡ ⎤ ⎢ ⎥ = − ∇ − + + − − ⎢ ⎥ ⎣ ⎦
∑
∑
∑∑
∑∑
∑ ∑
− (式 2-2) 整套波函數可以寫成含有原子核跟電子函數的形式: ( , )R r ( ) ( , )Rψ R r Π = Θ (式 2-3) [8] 將(式 2-3)插入薛丁格方程式之後得到(式 2-4)式: ( , ) ( n e) ( , ) tot ( , ) HΠ R r = T +H Π R r =E Π R r (式 2-4) 現在我們將核運動與電子運動的兩波函數分開得到(式 2-5)式: ( , ) ( ) ( , ) ( ( )) ( ) ( ) e n t H R r E R R r T E R R Eot R ψ = ψ + Θ = Θ (式 2-5) 薛丁格方程式中關於電子的部分,假如我們有i 個He的本徵函數,得到 ( , ) ( ) ( , ) e i i i H Ψ R r =E R Ψ R r i=1, 2,3, ,⋅⋅⋅ ∞ (式 2-6) 又∫
Ψi∗( , )R r Ψj( , )R r dr=δij,那麼整套波函數就被展開成: ( , ) j( ) j( , ) j R r R Π =∑
Ψ Ψ R r (式 2-7) 將(式 2-7)帶入(式 2-4)式: 2 1 ( ) ( , ) ( ) ( , ) 2 e j j tot j j j j H R R r E R R M α α α ⎡ ⎤ − ∇ + Ψ Ψ = Ψ Ψ ⎢ ⎥ ⎣ ⎦∑ ∑
∑
r (式 2-8) 其中 2 ( ) ( , ) 2 ( ) ( , ) 2 ( ) ( , ) ( ) 2 ( , ) j R j R r j R j R r j R j R r j R j R r α⎡ ⎤ ⎡ α ⎤ α α ∇ Ψ⎣ Ψ ⎦ ⎣= ∇ Ψ ⎦Ψ + ∇ Ψ ∇ Ψ + Ψ ∇ Ψ (式 2-8)的各項乘上 i ( , ) α R r dr ∗ Ψ∫
這個因子即得: 2 1 ( ) 2 ( ) ( ) ( ) ( ) ( ) ( ) ( ) ( 2 i ij i ij i i i tot j ) i R d R R D R R E R R E R R M α α α α α α ⎡ ⎡ ⎤⎤ − ⎢∇ Ψ + ⎣ ⋅∇ Ψ + Ψ ⎦⎥+ Ψ = Ψ ⎣ ⎦∑
∑
(式 2-9)其中(式 2-9)的dijα( )R = Ψi( , )R r ∇ Ψα j( , )R r ,此為First-order nonadiabatic coupling vector. 而 ( ) ( , ) 2 ( , ) ij i j Dα R = Ψ R r ∇ Ψα R r 就是Second-order nonadiabatic coupling vector. 忽略掉(式 2-9)中的所有非絕熱偶合項即得核的運動方程式: 2 1 ( ) ( ) ( ) ( ) ( ) 2 i R E Ri i R Etot R i R M α α α ⎡ ⎤ −
∑
⎣∇ Ψ ⎦+ Ψ = Ψ i=1, 2,3, ,⋅⋅⋅ ∞ (式 2-10) [9] 2-2-2 哈特里-福克方程(Fock equetion) 為了要推導出 Fock equetion,我們需要一個能量的斯萊特行列(Slater determinant)表達式。根據獨立粒子近似,波函數可寫成以下形式: 1 1 2 2 ( , ) ( ; ) ( ; ) ( e; ) Ne N a a a R r φ r R φ r R φ r Ψ = ⋅⋅⋅ R (式 2-11) 1 1 1 1 ( ) ( ) 2 e e e N N N e n i i j H h i g ij = = = =∑
+∑∑
+V n ,with 1 2 1 ( ) , ( ) , 2 e n e n n N N N N N i nn i i i i j Z Z Z h i g ij V r R r r R R α β α α α α β α> α β = − ∇ − = = − −∑
∑∑
∑ ∑
− (式 2-12) [10] (式 2-12)中的 算符描述i 號電子在所有原子核場的運動, 算符描述電子 與電子間的排斥作用。根據包利不相容原理(Pauli principle),不得有任兩個電子 是所有量子數都相同者。那麼就可以建立一個斯萊特行列式: ( ) h i g ij( ) 1 1 1 2 2 2 (1) (2) ( ) (1) (2) ( ) 1 (1, 2,3, , ) ! (1) (2) ( ) Ne Ne Ne a a a a a a SD e e a a a N N N N N φ φ φ φ φ φ φ φ φ ⋅⋅⋅ ⋅⋅⋅ Φ ⋅⋅⋅ ≡ Φ = ⋅⋅⋅ e e e (式 2-13) 有了斯萊特行列式,能量就可以寫成以下的形式了: 1 1 1 1 ( ) ( ) ( ) ( ) 2 e e e N N N SD e SD nn i i j E R H R h i g ij V = = = = Φ Φ = Φ∑
+∑∑
+ Φ (式 2-14) 於單電子算符,只有波函數與算符為同一個號次才能夠不等於零,比方說: 1 2 1 2 1 1 2 2 1 1 1 1 1 1 1 (1) (2) ( ) (1) (2) ( ) (1) (1) (2) (2) ( ) ( ) (1) (1) Ne Ne Ne Ne a a a e a a a e a a a a a e a e a a h N h N h N N φ φ φ φ φ φ φ φ φ φ φ φ φ φ Φ Φ = = = h =h於雙電子算符: 1 2 1 2 1 2 1 2 1 2 1 2 12 12 12 12 12 (1) (2) ( ) (1) (2) ( ) (1) (2) (1) (2) ( ) ( ) (1) (2) (1) (2) Ne Ne Ne Ne a a a e a a a e a a a a a e a e a a a a g N g g N N g J φ φ φ φ φ φ φ φ φ φ φ φ φ φ φ φ Φ Φ = = = = N (式 2-15) 又 1 2 2 1 1 2 2 1 1 2 2 1 12 12 12 12 (1) (2) ( ) (1) (2) ( ) (1) (2) (1) (2) ( ) ( ) (1) (2) (1) (2) Ne Ne Ne Ne a a a e a a a e a a a a a e a e a a a a N g N g N g K φ φ φ φ φ φ φ φ φ φ φ φ φ φ φ φ = = = N (式 2-16) (式 2-15)中的 就是庫倫積分項(Coulomb integral);(式 2-16)中的 就是交換 積分項(Exchange integral)。然後能量因此就能夠表達成為: 12 J K12 1 12 1 1 1 1 1 1 1 (1) (1) ( (1) (2) (1) (2) (1) (2) (1) (2) ) 2 1 ( ) 2 e e e i i i j i j i j j i e e e N N N a a a a a a a a a a i i j N N N i ij ij i i j E h g g h J K φ φ φ φ φ φ φ φ φ φ = = = = = = = + − = + −
∑
∑∑
∑
∑∑
12 (式 2-17) 現在的目標是找到ㄧ組可以讓能量極小化的分子軌道,然而變分法只能夠在所有 分子軌道的波函數都是歸一化的以及互為正交的情況下使用,所以這個時候就只 能使用拉格朗日乘因子法(Lagrange multipliers method)來解決了:( ) ( ) e e N ij i j ij ij N ij i j i j ij L E L E λ φ φ δ δ δ λ δφ φ φ δφ = − − = − − =
∑
∑
0 (式 2-18) 其中 1 ( ) ( 2 ) e e N N i i i i i i i j j i i ij i j j i j i i j j i i j E h h J K J K J K J K δ δφ φ φ δφ δφ φ φ δφ δφ φ φ δφ = + + − − + − + −∑
∑
+ (式 2-19) (式 2-19)可以簡化為:( ) ( ( ) e e e N N i i i i i i i j j i i j j i i ij N i i i i i i i E h h J K J K F F ) δ δφ φ φ δφ δφ φ φ δφ δφ φ φ δφ = + + − + − = +
∑
∑
∑
(式 2-20) 其中Fock operator e ( ) N i i j j j F = +h∑
J −K Fock operator 是個很有效的單電子能量算符,它描述著一個電子的動能與電 子與其他原子核的吸引作用( ),也描述著電子跟跟其他電子之間的交互作用 ( & )。然後(式 2-18)的拉格朗日變分函數就變成(式 2-21)了: i h J K ( ) ( e e N N i i i i i i ij i j i j i ij L F F δ =∑
δφ φ + φ δφ −∑
λ δφ φ − φ δφ ) 0= (式 2-21) 由於複數的共軛性質, φ δφ = δφ φ ∗, φ F δφ = δφ F φ ∗,(式 2-21)又可以 被表達成(式 2-22): 0 e e e e N N N N i i i ij i j i i i ij j i i ij i ij L F F δ δφ φ λ δφ φ δφ φ ∗ λ δφ φ ∗ =∑
−∑
+∑
−∑
= (式 2-22) 不論是 δφ 還是 δφ ∗都應該使得δL=0,又再利用複數的共軛性質: ( ) e N ij ji i j ij λ λ− ∗ δφ φ =0∑
(式 2-23) (式 2-23)的意思是說拉格朗日乘因子就是 Hermitian matrix(λij =λji∗)裡面的元素, 然後得到Fock equation: e N i i ij j j Fφ =∑
λ φ (式 2-24) (式 2-24)中i≠ j時λij = ;0 i= j時λij=εi。故最後Fock equation 應為 ' ' i i i i Fφ ε φ= [11] (式 2-25) 2-2-3 詮釋哈特里-福克近似[12] 1. 每個電子感觸到所有其他的電子是一個整體,換言之就是每個電子都在其他 電子所建構出來的平均場中運動。 2. 一個福克算符(Fock operator)是被第 個分子軌道中一個既定的電子所引進的: i ( ) ( ) i ( ) F i ϕ i =ε ϕ i 。2-3 多體微擾理論(Many-Body perturbation Theory) 2-3-1 一般微擾理論 從哈密頓算符開始: 0 1 0 , , 1, 2, i i i H H H H E i λ = + Φ = Φ = (式 2-26) (式 2-26)中 H0是參考項,也就是非微擾項(unperturbrd term);H1是微擾項;λ 就 是決定微擾項影響力有多大的參數(假定為小量)。現在我們就只直接考慮最低的 能態,那麼含微擾的薛丁格方程式就如下: HΨ = ΨW (式 2-27) (式 2-26)中當 λ=0 時,H= H0,Ψ=Φ0,W= E0。當微擾加入時,能量跟波函數也 勢必要持續地改變,於是能量跟波函數的表達式也可以含有微擾參數λ(假定為小 量)的泰勒展開式表示: 2 3 4 0 1 2 3 4 2 3 4 0 1 2 3 4 W W W W W W λ λ λ λ λ λ λ λ Ψ = Ψ + Ψ + Ψ + Ψ + Ψ + = + + + + + (式 2-28) 直接使用中間歸一化的波函數: Ψ Φ =0 1; Ψi≠0 Φ =0 0 有了(式 2-28),(式 2-27)的薛丁格方程式就可以寫成: k 1 0 0 0 1 1 0 0 [ ] n k n n m n n k n m H λ + H + H W λ = = + ⎛ ⎞ Ψ + Ψ + Ψ = ⎜ Ψ ⎟ ⎝ ⎠
∑
∑ ∑
= (式 2-29) 現在將(式 2-29)中等號兩邊相同 λ 冪次的項集合起來: (式 2-30) 根據(式 2-30),現在來算一級微擾的部份: 0 0 0 0 0 1 0 1 1 0 0 1 1 0 2 0 2 1 1 0 2 1 1 2 0 3 0 3 1 2 0 3 1 2 2 1 3 0 : : : : H W H H W W H H W W W H H W W W W λ λ λ λ Ψ = Ψ Ψ + Ψ = Ψ + Ψ Ψ + Ψ = Ψ + Ψ + Ψ Ψ + Ψ = Ψ + Ψ + Ψ + Ψ0 0 1 1 1 0 1 0 0 0 1 1 0 0 ( ) ( ) 0 with ( ) ( ) i i i i i i H W H W c H W c H W ∞ = ∞ = − Ψ + − Φ = Ψ = Φ → − ⋅ Φ + − Φ =
∑
∑
0 (式 2-31) (式 2-31)的等號兩邊各乘以 jd r ∗ Φ∫
得到: 0 1 0 1 0 0 1 0 1 0 ( ) ( ) 0 i i ji j j i j j j j c E E H W c E E H W δ δ δ = − + Φ Φ − = → − + Φ Φ − =∑
0 0 (式 2-32) j=0 時,W1= Φ0 H1 Φ0 j≠0 時, 1 0 1 1 0 0 0 0 j j j j j j j H H c E E ≠ E E Φ Φ Φ Φ = − → Ψ = − Φ −∑
− (式 2-33) 根據(式 2-30),現在來算二級微擾的部份: 0 0 2 1 1 1 2 0 2 1 0 0 0 0 1 1 2 0 0 0 ( ) ( ) 0 with ( ) ( ) i i i i i i i i i i i i H E H W W d c H E d H W c W ∞ ∞ = = ∞ ∞ = = − Ψ + − Ψ − Φ = Ψ = Φ Ψ = Φ → − ⋅ Φ + − Φ − Φ =∑
∑
∑
∑
0 (式 2-34) (式 2-34)的等號兩邊各乘以 jd r ∗ Φ∫
得到: 0 1 1 0 0 0 1 1 2 0 ( ) ( ) 0 i i ji i j i j i i j j i j i j i d E E c H W W d E E c H W W δ δ δ = = = − + Φ − Φ − = → − + Φ − Φ − = 2 0 0 0∑
∑
∑
(式 2-35) j=0 時, 0( 0 0) d E −E 0 1 1 0 0 0 i i i i i i c H W c = = +∑
Φ Φ −∑
Φ Φ 2 0 0 0 1 1 0 2 0 1 0 0 0 0 i i i i i i i W H H W c H E E = ≠ − Φ Φ = Φ Φ Φ Φ → = Φ Φ = − −∑
∑
……… j≠0 時, 0 1 1 2 0 0 ( ) j j i j i j i d E E c H W Wδ = − +∑
Φ − Φ − 0 1 1 1 0 0 1 1 0 1 0 0 1 0 2 0 0 0 0 0 ( ) ( )( ) ( ) j j i j i i j i j i i j i i j j i j i j d E E c H W c H W c H H H H d E E E E E E = ≠ ≠ = → − = − Φ − Φ = − Φ Φ + Φ Φ Φ Φ Φ Φ Φ Φ → = − − − −∑
∑
∑
( 1 式2-36)2-3-2 Moller-Plesset perturbation Theory[13] 從電子的哈密頓算符開始: 1 1 1 ( ) ( ) 2 e e e N N N e i i j i H h i g = = ≠ =
∑
+∑∑
ij ,with ( ) 1 2 , ( ) 1 2 e n e N N N i i i i i Z h i g ij r R r r α α α = − ∇ − = j − −∑
∑∑
上式中He =H0+(He−H0),又設 0 1 2 1 1 ( , , , ) e ( ) e e N N N i i i H r r r F r F i ∧ ∧ = = =∑
≡∑
( ) (式 2-37) 又根據(式 2-20), ( ) ( ) j( ) j( ) j F i h i Jα i Kα i α ∧ ∧ ⎡∧ ∧ ⎤ = + ⎢ − ⎥ ⎣ ⎦∑
(式 2-38) 再根據(式 2-25), ( ) ( ) ( ) (式 2-39) j j j F iφα i ε φα α i ∧ = 又設H1 =He−H0,則 1 1 1 1 ( ) ( ) ( ) 2 e e e j j j N N N i j i i H g ij Jα i Kα α ∧ ∧ = ≠ = i ⎡ ⎤ = − ⎢ − ⎥ ⎣ ⎦∑∑
∑∑
(式 2-40) 又設H1 Vee , (式 2-41) ∧ ∧ = − Ω 其中 1 1 ( ) 2 e e N N ee i j i V g ij 0 1 1 ( ) ( ) ( ( ) ( )) ( ) e e j j j N N i i Jα i Kα i F i h i H h i α ∧ ∧ ∧ ∧ ∧ ∧ = = ⎡ ⎤ Ω = ⎢ − ⎥= − = − ⎣ ⎦∑∑
∑
∑
∧ = ≠ =∑∑
, 1 e N i ∧ = 。 為了要讓微擾理論能夠適用於相關能的計算,非微擾項部分的哈密頓算符 必須被選擇。最常用的選擇就是將所有福克算符總加起來然後才引入MP 微擾理 論。但是這種把所有福克算符總加起來的作法會把平均電子與電子的排斥作用重 複多算了一次,所以這個微擾項就變成了真正的Vee算符減掉兩倍的 Vee 。 得到: 1 ee 2 ee H V V ∧ ∧ = − (式 2-42) 根據(式 2-33),W1= Φ0 H1 Φ0 。 那麼就W1 0 H1 0 0 Vee 0 2 Vee Vee ∧ ∧ = Φ Φ = Φ Φ − = − ∧ (式 2-43)所以 1 0 1 ( 0) ( 1) e j N ee ( ) J MP W W E MP E MP α V E HF α ε = + = + =
∑
− = (式 2-44) (式 2-44)的意思就是先把所有分子軌道的能量加起來再扣掉重複多算一次的電 子與電子排斥勢能的平均值,這樣才是HF 這方法算出來的電子能量值。 得 ( ) 1 [ 2 ] j i i j j i j E HF hα α Jα α Kα α α α α =∑
+∑∑
− i j (式 2-45) 根據(式 2-36), 0 1 1 0 2 0 0 A A A A H H W E E ≠ Φ Φ Φ Φ = − −∑
。 那麼就 0 1 1 0 0 0 2 0 0 0 0 A A ee A A ee A A A A H H V V W E E E E ∧ ∧ ≠ ≠ Φ Φ Φ Φ Φ Φ Φ Φ = − = − − −∑
∑
(式 2-46)2-4 耦合簇理論(Coupled Cluster Theory)
微擾的方法是把對零級波函數Φ 所有型式的校正能都相加到一定的級別0 (二級、三級、四級…等等),而耦合簇的方法則是把所有型式,從一級到無限級 的校正能通通考慮進來。現在,我們就開始定義激發態算符 T 於(式 2-47): 1 2 3 N T = + + +T T T T e (式 2-47) Ti算符作用在HF 的零級波函數Φ 上面,造就了 th 的激發態斯萊特行列式: 0 在耦合簇理論中,很常用到t 當做展開係數。 (式 2-48) 耦合簇波函數是: (式 2-49) i 1 0 2 0 3 0 occ vir a a i i i a occ vir ab ab ij ij i j a b occ vir abc abc ijk ijk i j k a b c T t T t T t < < < < < < Φ = Φ Φ = Φ Φ = Φ
∑∑
∑∑
∑ ∑
0 T e Ψ = Φ 2 0 1 1 with 1 2 ! T n n e T T n ∞ = = + + + =∑
T 再根據(式 2-47),則: 2 3 1 2 1 3 2 1 1 1 1 1 ( ) ( ) 2 6 T e = + +T T + T + T +T T + T + (式 2-50)有了(式 2-49),則薛丁格方程式是 T 0 T e H e Φ =Ee Φ (式 2-51) 0 (式 2-51)的等號兩邊各乘以
∫
Φ0∗dτ得到: 2 0 0 0 0 1 2 1 0 1 1 ( ) 2 T T e H e E e T T T E Φ Φ = Φ Φ =0 E Φ + + + + Φ = (式 2-52) 2 2 0 1 2 1 0 0 0 0 1 0 0 2 0 0 1 1 1 (1 ) 2 2 e e e e E= Φ H + + +T T T Φ = Φ H Φ + Φ H TΦ + Φ H TΦ + Φ H Te Φ0 0 ( ) 0occ vir occ vir
a a ab a b b a ab HF i e i ij i j i j e ij i a i j a b E t H t t t t t H < < = +
∑∑
Φ Φ +∑∑
+ − Φ Φ (式 2-53) 而根據 Brillouin’s theorem, 0 a 0 0 0 e i e H H T Φ Φ = ⇒ Φ 1Φ =0 [14] 所以 ( )[ occ vir ab a b b a HF ij i j i j i j a b i j b a i j a b E E t t t t t φ φ g φ φ φ φ g φ φ ] < < = +∑∑
+ − − (式 2-54) [15] (式 2-47)中,CCSD 就是T = + ,那麼T1 T2 eT ≈eT T1+2。2-5 過渡態理論(Transition State Theory)
圖 2-1. 化學反應的能量剖面圖。要變成生成物之前,反應物必須獲得足夠的能 量以便越過反應障壁。反應座標(Reaction coordinate)代表著這個反應從反應物走 到生成物所發生的結構與能量的變化。[16]
如果說活化能是個使得反應物能夠起反應的能量,我們可以在圖 2-1.描述一 個化學反應。我們可以說這個化學反應從反應物沿著反應座標來到生成物。
圖 2-2.反應途徑圖解。 [17]
圖 2-2 關係到一個典型熱反應的單一能量表面(Single energy surface)。反應
座標帶領整個系統沿著最小能量途徑(MEP)從反應物走到生成物,而過渡態理論 將過渡態擺在最小能量途徑的最高點位置。 圖 2-3. 這個已活化的錯合物, AB + +,被定義存在於δ這個小區域裡,反應障壁 頂端的中央位置。 [18]
2-5-1 尋找局部能量最小結構(Local minimum) 計算化學的優化問題充滿了變數,但至少 General function 的一階導數(梯度 向量)g 可被用來分析以及計算,有些方法則是會用到 General function 的二階導 數 H 來做計算。 該注意的是這個函數及其各階導數們精準度是有限的,因此一個駐點並沒 有辦法被很精確地定位出來,梯度也只能夠被減小到某個程度而已。所以實務 上,如果我們發現梯度已經減小到某個規定的數量級以下了或是說這個函數在某 兩次迭代之間的變化已經足夠小了,我們就可以視為已收斂了。那麼有以下兩種 用來找到極小點的優化技巧,分別各用到一階及二階導數: 最陡降落法(Steepest descent) 在這個法裡面,一連串函數值都往梯度的負值那個方向移動,定義Fi = −gi。 如果這個線最小化(Line minimization)被實行淂足夠精準的話,它往往都能夠降低 函數值,這樣才能夠因此保證說趨近一個最小值。圖 2-4 解說著最陡降落途徑是 沿著這個最小化路徑振盪的,最陡降落法並不能夠到達真正的極小點,它只能夠 以越來越慢的降落速度來接近真正的極小點。 圖 2-4. 最陡降落最小化法之示意圖。 [19] 一個精準的線搜尋(Line search)是需要幾個函數沿著每個搜尋方向做評估 的。在執行優化的過程中,步長(Step Size)往往會不斷地小幅變動;如果前一步 把函數值給減少了,那麼後一步的步長會小幅拉長,反之如果前一步把函數值給
增加了,那麼後一步的步長會變短。若是沒有了精準的線搜尋,就無法再把這個 函數值降低了,並且優化過程就會在無法收歛的狀態中被迫中斷。 牛頓逼近法(Newton-Raphson methods) 牛頓逼近法(NR 法)將函數展開到二次微分項: f(x)≒f(x0)+g(x- x0)+1 2H(x- x0) 2 (式 2-55) 又因為g=f’(x),則 g=0 時,(x- x0)=-H-1g (式 2-56) 當接近極小點時,所有的 Hessian 本徵值都是正的,並且步行方向與梯度 相反。但是如果萬一有出現一個Hessian 本徵值是負的時候,步行方向便會與梯 度相同並導致函數值的增加,這個時候優化過程可能就會結束在一個叫做第ㄧ鞍 點的地方。一般來講NR 法會嘗試著在最近處的駐點收斂起來,不管那是極小 點、鞍點、還是極大點。 2-5-2 尋找鞍點與過渡態 定位出一個函數的極小點是較為容易的。但是要找到第一鞍點,也就是過 渡態結構是較困難的,並沒有任何的方法是保證有效的!許多不同的方法都被用 上了,但這眾多方法可以被歸類為兩類,有內插法與局部資訊法。內插法就是假 設反應物與生成物都是已知的情況下,認為過渡態結構肯定是會座落在這兩個端 點之間的,該注意的是內插法還是不能找到最確切的過渡態結構,只能說找到與 此最確切的結構相近的結構而已。局部資訊法則是事前根本不知道反應物與生成 物的結構,只能靠初始猜足夠良好以便於優化過程到最後能夠收歛。一旦過渡態 結構找到了以後,整個反應的途徑就可以用IRC 勾勒出來了。 最直觀最簡單的方法就是利用反應物與生成物的結構去尋找過渡態結構 了。那麼反應物與生成物這兩個已知的結構就是兩個固定值了,那麼這個優化過 程中一個個被優化出來的“擬"結構都是變動的,當走到最後一個“擬"結構
時,其梯度就已經足夠小到可以收斂了,這個最後一個“擬"結構就是本次計算 所找到的過渡態結構了。 LST 法是假設沿著這條反應途徑的所有變動都是等速的,而過渡態結構結構 就只是這條內插線上能量最高者,但LST 法並不是一個很好的近似法,它只對 簡單的小的系統管用而已。如果LST 法是直線型的近似法的話,那 QST 法就是 拋物線型的近似法。圖 2-5.就說明了 LST 與 QST 有何不同,其中 IRC 代表真實 的反應座標。 圖 2-5. LST 與 QST 法之示意圖,能量極大與極小點以 * 與 ‧分別表示之。 [20]
第三章 結果與討論
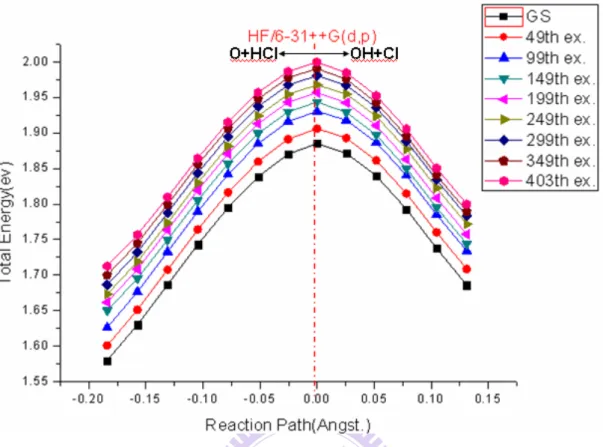
3-1 雙原子分子 OH, HCl 的鍵長、振動頻率、轉動常數以及全反應的反應能變 化 這裡,我們使用了從頭算(Ab initio),也就是先從低精度的方法 HF 開始對於 OH, HCl 的鍵長、振動頻率、轉動常數以及全反應的反應熱的計算,而其中反應 熱就是: (OH 的電子能加零點校正能+ Cl(2P)原子的電子能)- (HCl 的電子能加零點校正能 + O(3P))原子的電子能) 接著擴展到使用中精度的方法MP2,進而使用高精度的方法 CCSD 進行本項計 算。這些數據也會與實驗值做比較皆列於表一與表二中。 那麼來分析表一與表二:除了MP2/6-31++G(d,p)在 OH 的鍵長與轉動常數的 計算結果較其他二者精準以外,其他大抵上CCSD/6-31++G(d,p)是三種方法與基 組合體當中最精準的。HF/6-31++G(d,p)對於振動頻率、轉動常數、反應熱的計 算都有明顯高估的現象,OH 的振動頻率更是算到 4000 1 cm− 出頭,跟實驗值差距 有了300 1 cm− ,其他二者跟實驗值的差距僅幾十個cm−1而已;OH 的轉動常數更 是算到接近19.5 1,跟實驗值差距有了0.58 cm− cm−1,其他二者跟實驗值的差距 僅0.15~0.25 HF/6-31++G(d,p) 大約 的結果,但是實驗值僅吸熱約 MP2/6-31++G(d,p) 0.42 的結果, 1.6 , 差距並不若 算出來的是吸熱 0.54 , 1 此, 跟實際上的差距是最大的,CCSD/6-31++G(d,p)是三者當中比較精準的方法與基 組合體。 1 − 而已; l HF/6-31++G(d,p) 的結果 cm / al m 雖然算出來的是放熱 / l mol 對於反應能變化的計算,更是算到吸熱 mol, 但跟實驗值的差距僅 l。因 9Kc a o 1 CCSD/6-31++G(d,p) Kcal / Kcal / mo / Kcal mol 這麼大, 與實驗值的差距不足 / Kcal mol HF/6-31++G(d,p) KcTable 1:各方法與基組合體所計算出來的雙原子分子 OH 與 HCl 的鍵長、振動頻 率、轉動常數與實驗值之比較。 鍵長(A) 振動頻率(cm )−1 轉動常數(cm−1) HCl HF/6-31++G(d,p) 1.26628 3177.3 10.7323 MP2/6-31++G(d,p) 1.26968 3115.1 10.6747 CCSD/6-31++G(d,p) 1.27309 3063.6 10.6177 Experimenta 1.27458 2990.9 10.5934 OH HF/6-31++G(d,p) 0.95509 4054.8 19.4923 MP2/6-31++G(d,p) 0.97360 3823.0 18.7581 CCSD/6-31++G(d,p) 0.97593 3770.3 18.6687 Experimenta 0.96966 3737.8 18.9108 aSee reference 3. Table 2:各方法與基組合體所計算出來的 O(3P)+HCl OH+Cl(2P)反應能變化。 V Kcal mol)( / (不含零點校正) V Kcal mol( / )(含零點校正) HF/6-31++G(d,p) +7.62 +8.87
MP2/6-31++G(d,p) -1.44 -0.42 CCSD/6-31++G(d,p) -0.47 +0.54
Experimenta -0.57±0.12 +1.19±0.12
3-2 反應障壁(Barrier Height) Table 3:各方法與基組合體所計算出來的 O(3P)+HCl OH+Cl(2P)反應障壁。 V (eV (不含零點校正) (含零點校正) + + ) ( ) zp V eV + + HF/6-31++G(d,p) 1.704 1.633 MP2/6-31++G(d,p) 0.815 0.728 CCSD/6-31++G(d,p) 0.716 0.626 UMP2/6-31G(d,p)a 0.817 0.730 UMP2/cc-pVTZa 0.695 0.610 Experimentb 0.377( 8.7Kcal/mol)≒ a See reference 21. [21] b See reference 22. [22] 為了要畫出準確的能級圖,本實驗的一個關鍵的計算工程就是計算出 O(3P)+ HCl OH+Cl(2P)這個反應的反應障壁,所謂反應障壁就是中間過渡態的能量減 去反應物O(3P)+ HCl 的能量,零點校正能就是所有振動與轉動量子數都是 0 的 時候振動加轉動能量,當考慮到零點校正能的時候,就必須要把HCl 跟中間過 渡態的零點校正能加進去,O(3P)是單原子所以是沒有振動能跟轉動能的。表三 摘錄了各種方法與基組合體所計算出來的反應障壁,含與不含零點校正能所得的 結果都有總結在表三當中。而同樣是6-31++G(d,p)這個基組下,反應障壁的計算 結果其大小隨著方法的精度增加而降低,從HF 的 1.633eV 到 CCSD 的 0.626eV; 同樣是MP2 這個方法下,反應障壁的數值亦隨著基組的大小的增加而下降,從 6-31G(d,p)的 0.730eV 到 cc-pVTZ 的 0.610eV,UMP2/cc-pVTZ 與 CCSD/6-31++G(d,p)所計算出來的結果差距極小。而 Ref. 22 中閱讀到反應障壁實 驗值約為8.7Kcal/mol(也就是大約 0.377ev),所以從表三當中可以發現 HF/6-31++ G(d,p)對於本反應的反應障壁有明顯高估的現象,高出實驗值約 29kcal/mol, MP2/6-31++G(d,p)僅高出約 8kcal/mol,CCSD/6-31++G(d,p)則是僅高出不到
6kcal/mol。 3-3 反應錯合物、中間過渡態、產物錯合物以及 IRC 途徑 反應錯合物即O(3P)原子與 HCl 分子之間藉著凡得瓦力結合起來的錯合物, 它也是O(3P)原子與 HCl 開始發生反應的那一瞬間所形成的第一個錯合物,然後 經過各鍵長與鍵角隨著時間的變化,HCl 分子之間的共價鍵逐漸弱化終致消失, 然後形成中間過渡態,中間過渡態以及其IRC 路徑上附近的幾個錯合物不論是 氧原子與氫原子之間或是氫原子與氯原子之間都是僅有凡得瓦力維繫著彼此,一 旦各原子之間僅有凡得瓦力維繫著彼此振動頻率就會有虛頻的現象發生。形成中 間過渡態之後,氧原子與氫原子之間的共價鍵逐漸增強,鍵長逐漸縮短但氫原子 與氯原子之間的距離逐漸疏離,然後形成本反應的最後一個錯合物,產物錯合 物。產物錯合物的Cl(2P)原子與 OH 分子之間也是藉著凡得瓦力結合起來的錯合 物,其接下來的發展就是形成Cl(2P)原子與 OH 分子兩個生成物,此時反應才告 終。 表四與表五總結了反應錯合物、中間過渡態、產物錯合物的鍵長、鍵角、振 動頻率的計算結果。反應錯合物在此三個方法與基組組合體中都是呈現出線性分 子的結果,除HCl 鍵長三者差異不大之外,OH 鍵長 HF/6-31++G(d,p)就長達 2.4 左右,MP2/6-31++G(d,p)與 CCSD/6-31++G(d,p)二者所計算出來的 OH 鍵長就都 有在2.3 A ° A ° 者彼此差異不大 ~ r A ° 左右。中間過渡態的計算結果大抵上不論是OH 鍵長還是 HCl 鍵長三 (Ref 3. 所用的各方法與基組合體算出的接果都是 之間, 之間;Ref 22[22].算出的是 1.20 1.30 OH r A ° = 1.21 OH = , HCl 1.40 ~ 1.50 HCl r A ° = r A ° ),但是 1.46 = ∠ OHCl 119°的結果, 二者所計算出來的 的鍵角計算結果則呈現極大的差異, MP2/6-31++G(d,p)與 HF/6-31++G(d,p) CCSD/6-31++G(d,p) 就算出僅大約 ∠OHCl 鍵角的結果都有在 135°~140°之 間,而事實上Ref 3.與 Ref 22.(Ref 22.算出的是 139.5°的)都是算出 130°~140°之
間的結果。產物錯合物在此三個方法與基組合體中,HF/6-31++G(d,p)與其它二 者的差異就更大了,不但OH 鍵長比其他二者偏短僅大約 0.95 左右, MP2/6-31++G(d,p)與 CCSD/6-31++ G(d,p)二者所計算出來的 OH 鍵長皆是大約 0.98 左右;HCl 鍵長比其他二者偏長,接近 3 了,但 MP2/6-31++G(d,p)與 CCSD/6-31++ G(d,p)二者所計算出來的 HCl 鍵長之結果卻僅有 2.4 ~2.5 之 間。(Ref 3. 所用的各方法與基組合體算出的接果都是 之間, A ° ~ 2.40 A ° = le 4 A ° 0.98 A ° 0.99 r A ° ) 、 A ° 產物錯合物 OHCl 0.97 OH r A ° = , HCl = 。 、中間過渡態 ( ) HCl r A ° 2.40 ~ 2.60 HCl r A ° Tab :各 的鍵長、鍵角。 之間;Ref 22.算出的是 方法與基組合體所計算出來的反應錯合物 OH r A ° = ( ) OH r A ° min( . .) E a u θ O CCSD/6-31++G(d,p) T MP2/6-31++G(d,p) -535.0631 CCSD/6-31++G(d,p) -HC HF/6-31++G(d,p) -534.8562531 2.42128 1.26813 180 MP2/6-31++G(d,p) -535.0960166 2.29021 1.27278 180 -535.1286388 2.32693 1.2751 S HF/6-31++G(d,p) -534.7915991 1.16109 1.51830 1 1 OH-Cl HF/6-31++G(d,p) -534.843071 0.95534 2.99163 179.93561 MP2/6-31++G(d,p) -535.0934602 0.97682 2.44602 56.32998 CCSD/6-31++G(d,p) -535.1209637 0.97823 2.47131 59.60701 l 1 180 18.94999 172 1.20680 1.43368 135.50019 -535.0995479 1.20890 1.45471 137.2971


![圖 2-1. 化學反應的能量剖面圖。要變成生成物之前,反應物必須獲得足夠的能 量以便越過反應障壁。反應座標(Reaction coordinate)代表著這個反應從反應物走 到生成物所發生的結構與能量的變化。 [16]](https://thumb-ap.123doks.com/thumbv2/9libinfo/8033954.161549/36.892.136.706.185.881/過反應障壁反應座Reaction代表著這個反應從反應物走所發生結構與能量的.webp)