Synthesis of Dinuclear and Trinuclear Ruthenium
Cyclopropenyl Complexes
Chiung-Cheng Huang, Ying-Chih Lin,* Shou-Ling Huang, Yi-Hong Liu, and
Yu Wang
Department of Chemistry, National Taiwan University, Taipei, Taiwan, 106 Republic of China
Received November 4, 2002
Dinuclear ruthenium cyclopropenyl complexes
{
[Ru]CdC(CHR)
}
2C6H4([Ru] ) (η
5-C5H5)(PPh3)2Ru, R ) CN, 3a; R ) CH2
dCH
2, 3b; R ) Ph, 3c) are prepared by deprotonationof corresponding vinylidene complexes
{
[Ru]dCdC(CH
2R)}
2C6H42+(2). For the vinylidene
complex 2d (R ) CO2Me) with an ester group, the deprotonation reaction leads to formation
of the dinuclear bis-furyl complex
{
[Ru]CdC(CHdC(O)OMe)
}
2C6H4(5d). Electrophilic
addition of TCNQ to both three-membered rings of 3a yields the zwitterionic bis-vinylidene
complex
{
[Ru]dCdC[CH(TCNQ)CN]
}
2C6H4(4a), which, in the presence of
MeOH/n-Bu4-NOH, gives the methoxy-substituted bis-cyclopropenyl complex
{
[Ru]CdC(C(OMe)CN)
}
2C6H4(6a). The proton-induced demethoxylation of 6a generates
{
[Ru]CC(C(CN))
}
2C6H42+(7a).
The reaction of TMSN3
with 3a gives the bis-tetrazolate complex
{
[Ru](N4C)CH(CH2-CN)
}
2C6H4(8a). Trinuclear tris-cyclopropenyl complexes
{
[Ru]CdC(CHR)C6H4CtC
}
3C6H3(R ) CN, 11a; R ) CH2
dCH
2, 11b; R ) Ph, 11c) are obtained from deprotonation of{
1,3,5-{
[Ru]dCdC(CH2R)C6H4CtC
}
3C6H3}
3+(10). Complex 2b is characterized by X-ray diffraction
analysis, and other complexes are characterized by spectroscopic methods.
Introduction
Cyclopropene is believed to be the most highly strained
cycloalkene, with an estimated strain energy of more
than 50 kcal/mol.
1This molecule has hence been under
intense investigation
2and has played a crucial role in
the development of the concept of aromaticity.
3Chemi-cal reactivity of this molecule has also been addressed.
4-7However, transition metal cyclopropenyl complexes are
rare,
8even though participation of d orbitals in these
complexes is expected to significantly stabilize the
molecule. Previously we reported the facile synthesis of
several mononuclear ruthenium cyclopropenyl
com-plexes
9by deprotonation of (η
5-C5H5)(PPh3)2RudCd
C(Ph)CH
2R
+in which CR
of the vinylidene ligand is
known to be electron deficient. Thus deprotonation at
C
γcauses intramolecular nucleophilic addition at CR,
leading to the formation of cyclopropenyl complexes. As
applications of dendrimers are currently being
investi-gated for use as biomimetic catalysts,
10building blocks
for fabrication of designed materials,
11molecular
car-riers for chemical catalysts,
12and potential vehicles for
delivery of drugs and immunogens,
13we extend our
synthesis to a few small preliminary dendrimeric
sys-tems. Herein we report the preparation of dinuclear and
trinuculear ruthenium vinylidene and cyclopropenyl
complexes using 1,4-diethynylbenzene and 1,3,5-(HCt
CC6H4CtC)3C6H3
14as core backbones, respectively.
(1) (a) Special issue on strained organic compounds: Chem. Rev.1989, 89. (b) Liebman, J. F.; Greenberg, A. Strained Organic Molecules; Wiley: New York, 1978; p 91.
(2) (a) Marier, G.; Periss, T.; Reisenauer, H. P.; Hess, B. A., Jr.; Schand, L. J. J. Am. Chem. Soc. 1994, 116, 2014. (b) Hopf, H.; Plagens, A.; Walsh, R. J. Chem. Soc., Chem. Commun. 1994, 1467.
(3) (a) Liebman, J. F.; Greenberg, A. Chem. Rev. 1976, 76, 311. (b) Halton, B.; Banwell, M. G. In The Chemistry of the Cyclopropnyl Group; Patai, S., Rappoport, Z., Eds.; Wiley: Chichester, 1987; Part 2, Chapter 21, p 1223.
(4) (a) Lahti, P. M.; Berson, J. A. J. Am. Chem. Soc. 1981, 103, 7011. (b) Rigby, J. H.; Kierkus, P. C. J. Am. Chem. Soc. 1989, 111, 4125. (c) Deem, M. L. Synthesis 1972, 675. (d) Galloway, N.; Deut, B. R.; Halton, B. Aust. J. Chem. 1983, 36, 593. (e) Gompper, R.; Choenafinder, K. Chem. Ber. 1979, 112, 1529. (f) Mueller, P.; Bernardinelli, G.; Pfyffer, J.; Schaller, J. P. Helv. Chim. Acta 1991, 74, 993.
(5) Bailey, I. M.; Walsh, R. J. Chem. Soc., Faraday Trans. 1 1978, 74, 1146.
(6) (a) Padwa, A.; Blacklock, T. J.; Getman, D.; Hatanaka, N.; Loza, R. J. Org. Chem. 1978, 43, 1481. (b) Padwa, A. Acc. Chem. Res. 1979, 12, 310. (c) Arnold, D. R.; Humphreys, R. W.; Leigh, W. J.; Palmer, G. E. J. Am. Chem. Soc. 1976, 98, 6625. (d) Zimmerman, H. E.; Aasen, S. M. J. Am. Chem. Soc. 1977, 99, 2342.
(7) (a) Franck-Neumann, M.; Miesch, M.; Kempf, H. Tetrahedron 1988, 44, 2933. (b) Dombrovskii, V. S.; Yakushikina N. I.; Bolesov, I. G. Zh. Org. Khim. 1979, 15, 1184.
(8) Gompper, R.; Bartmann, E. Angew. Chem., Int. Ed. Engl. 1985, 24, 3.
(9) (a) Ting, P. C.; Lin, Y. C.; Cheng, M. C.; Wang, Y. Organome-tallics 1994, 13, 2150. (b) Ting, P. C.; Lin, Y. C.; Lee, G. H.; Cheng, M. C.; Wang, Y. J. Am. Chem. Soc. 1996, 112, 6433. (c) Lo, Y. H.; Lin, Y. C.; Lee, G. H.; Wang, Y. Organometallics 1999, 18, 982. (d) Chang, C. W.; Lin, Y. C.; Lee, G. H.; Wang, Y. Organometallics 2000, 19, 3211. (10) Huck, W. T. S.; Prins, L. J.; Fokkens, R. H.; Nibbering, N. M. M.; van Veggel, F. C. J. M.; Reinhoudt, D. N. J. Am. Chem. Soc. 1998, 120, 6240.
(11) Mongin, O.; Gossauer, A. Tetrahedron Lett. 1996, 37, 3825. (12) Knapen, J. W. J.; van der Made, A. W.; de Wilde, J. C.; van Leeuwen, P. W. W. N. M.; Wijkens, P.; Grove, D. M.; van Koten, G. Nature 1994, 372, 659.
(13) (a) Duncan, R.; Kopecek, J. Adv. Polym. Sci. 1984, 57, 51. (b) Peppas, N. A.; Nagai, T.; Miyajima, M. Pharm. Technol. Jpn. 1994, 10, 611. (c) Bieniarz, C. Dendrimers: Applications to Pharmaceutical and Medicinal Chemistry. In Encyclopedia of Pharmaceutical Technol-ogy; Marcel Dekker: New York, 1999; p 55.
10.1021/om020913x CCC: $25.00 © 2003 American Chemical Society Publication on Web 02/22/2003
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
Other dinuclear ruthenium complexes obtained from the
bis-vinylidene complex are also reported.
Results and Discussion
Preparation of Dinuclear Vinylidene Complexes.
Treatment of [Ru]Cl with 1,4-diethynylbenzene in the
presence of NaPF6
afforded a deep red solution
contain-ing a vinylidene intermediate, which underwent
depro-tonation in the presence of sodium methoxide to give
the dinuclear bis-acetylide complex 1 in 84% yield.
15,16With half an equivalent of bisalkyne the reaction yields
no mononuclear complex. The singlet resonance at δ
50.98 in the
31P NMR spectrum of 1 is in the region of
a regular ruthenium acetylide complex.
17The mass
spectrum of 1 gives the parent peaks at m/z ) 1506 as
well as fragmentations due to loss of phosphines. Bruce
and co-workers carried out the reaction of
1,4-bis-(trimethylsilylethynyl)benzene with 1 equiv of [Ru]Cl
in the presence of KF to give first [Ru](CtCC6H4Ct
CSiMe3). Then addition of another equivalent of
[Ru]-Cl and KF cleaved the remaining C-Si bond with
concomitant formation of the other Ru-C bond to afford
the same acetylide complex 1. Direct use of 2 equiv of
[Ru]Cl and KF also afforded 1.
18Reactions of 1 with various alkyl halides RCH
2X
generate dinuclear bis-vinylidene complexes
{
[Ru]dCd
C(CH
2R)
}
2C
6H
42+(2) in high yield (Scheme 1). For
example, the reaction of 1 with ICH2CN at 40 °C yields
the dicationic bis-vinylidene complex
{
[Ru]dCdC(CH2-CN)
}
2C6H42+(2a). Several analogous vinylidene
com-plexes 2 (R ) CH2
dCH
2, 2b; R ) Ph, 2c; R ) CO2CH3,2d; R ) CO
2Et, 2e) are similarly prepared. All thesevinylidene complexes, 2a-e, display a characteristic
deep red color and deshielded
13C resonances at δ 345
( 5 assignable to CR
of the vinylidene ligand.
19 31P NMR
resonances of 2 appear at around δ 42 ( 1 in CDCl3
as
singlets due to the fluxional behavior of the vinylidene
ligand at room temperature.
20Complexes 1 and 2 are
less soluble than their corresponding mononuclear
complexes. Previously we reported
9bthe transformation
of a mononuclear ruthenium cyclopropenyl complex to
the dimeric dication vinylidene complex
{
[Ru]dCd
C(Ph)CH(CN)-
}
22+, which, upon deprotonation, yielded
the bis-cyclopropenyl complex
{
[Ru]CdC(Ph)C(CN)-
}
2.The two cyclopropenyl groups are bound together
di-rectly by the sp
3carbon of the three-membered ring. The
formation of this complex probably involves the cationic
ruthenium vinylidene radical
21formed from the reaction
of the mononuclear ruthenium cyclopropenyl complex
with allyl iodide.
Single crystals of 2b suitable for X-ray diffraction
analysis are obtained by recrystallization from CDCl3.
Complex 2b crystallized with only one independent
molecule in the unit cell and cocrystallized with
coun-terion and chloroform molecules. The solid-state
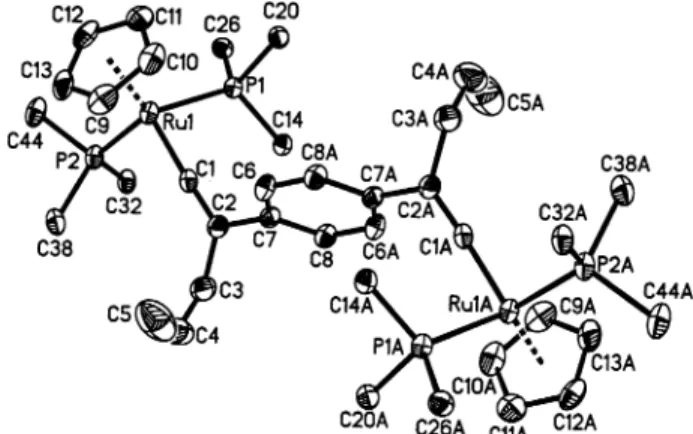
struc-ture of 2b is shown in Figure 1, and representative bond
lengths and bond angles are reported in Table 1. The
molecule possesses an inversion center at the center of
the core phenyl group. The RudC bond length of
1.853-(4) Å is in the range of a regular RudC bond of other
crystallographically characterized ruthenium vinylidene
complexes.
16bThe disorder of an allyl group usually
observed for metal complexes containing such a ligand
is not observed in 2b, possibly due to bulky phosphine
ligands that restrict the number of accessible
conforma-(14) (a) Uno, M.; Dixneuf, P. H. Angew. Chem., Int. Ed. 1998, 37,1714. (b) McDonagh, A. M.; Humphrey, M. G.; Samoc, M.; Davies, B. L.; Hiubrechts, S.; Wada, T.; Sasabe, H.; Persoon, A. J. Am. Chem. Soc. 1999, 121, 1405.
(15) Bruce, M. I.; Wallis, R. C. Aust. J. Chem. 1979, 32, 1471. (16) For general reviews, see: (a) Bruce, M. I.; Swincer, A. G. Adv. Organomet. Chem. 1987, 52, 3940. (b) Bruce, M. I. Chem. Rev. 1991, 91, 197.
(17) (a) Whittall, I. R.; Humphrey, M. G.; Persoons, A.; Houbrechts, S. Organometallics 1996, 15, 1935. (b) Wu, I. Y.; Lin, J. T.; Luo, J.; Li, C. S.; Tsai, C.; Wen, Y. S.; Hsu, C. C.; Yeh, F. F.; Liou, S. Organome-tallics 1998, 17, 2188.
(18) (a) Bruce, M. I.; Hall, B. C.; Kelly, B. D.; Low, P. J.; Skelton B. W.; White, A. H. J. Chem. Soc., Dalton Trans. 1999, 3719. (b) Bruce, M. I.; Hall, B. C.; Low, P. J.; Skelton B. W.; White, A. H. J. Organomet. Chem. 1999, 592, 74.
(19) Werner, H.; Bachmann, P.; Martin, M. Can. J. Chem. 2001, 79, 519.
(20) (a) Allen, D. L.; Gibson, V. C.; Green, M. L.; Skinner, T. F.; Bashikin, J.; Grebenik, P. D. J. Chem. Soc., Chem. Commun. 1985, 895. (b) Consiglio, G.; Morandini, F. Chem. Rev. 1987, 87, 761.
(21) (a) Rabier, A.; Lugan, N.; Mathieu, R.; Geoffroy, G. L. Orga-nometallics 1994, 13, 4676. (b) Antinolo, A.; Otero, A.; Fajardo, M.; Garcia-Yebra, C.; Gil-Sanz, R.; Lopez-Mardomingo, C.; Martin, A.; Gomez-Sal, P. Organometallics 1994, 13, 4679.
Scheme 1
Figure 1. ORTEP drawing of complex 2b (30% probability
ellipsoids).
Table 1. Selected Bond Distances (Å) and Angles (deg) of 1,4-{[Ru]CdC(CH2CHdCH2)}2C6H42+(2b) Ru(1)-C(1) 1.853(4) Ru(1)-C(9) 2.235(4) Ru(1)-C(10) 2.240(4) Ru(1)-C(13) 2.243(4) Ru(1)-C(11) 2.285(4) Ru(1)-C(12) 2.295(4) Ru(1)-P(1) 2.3429(11) Ru(1)-P(2) 2.3708(11) C(1)-C(2) 1.311(6) C(2)-C(7) 1.497(6) C(2)-C(3) 1.525(7) C(3)-C(4) 1.465(10) C(4)-C(5) 1.237(12) C(2)-C(1)-Ru(1) 174.0(3) C(1)-C(2)-C(7) 119.7(4) C(1)-C(2)-C(3) 120.8(4) C(7)-C(2)-C(3) 119.3(4) C(4)-C(3)-C(2) 116.2(6) C(5)-C(4)-C(3) 127.5(4)
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
tions. The torsion angle C(3)-C(2)-C(7)-C(6) is
54.9-(5)° and C(1)-C(2)-C(7)-C(8) is 60.754.9-(5)°, indicating
that the core phenyl group is not coplanar with the
vinylidene plane.
Dinuclear Cyclopropenyl and Furyl Complexes.
Deprotonation of the vinylidene complex 2a by
n-Bu4-NOH in acetone is accompanied with a cyclization
reaction affording the bis-cyclopropenyl complex
{
[Ru]-CdC(CHCN)
}
2C6H4(3a). To prevent attack of halide
anion to the metal, NH4PF6
was added. With two
stereogenic carbon centers in 3a, it is not surprising to
see two sets of coupled doublets at δ 51.9, 49.3 (JP-P
)
36.4 Hz) and 51.8, 49.2 (JP-P
) 35.2 Hz) in the
31P NMR
spectrum of 3a. The intensity ratio of 1:1 attributed to
stereoisomers indicates no diastereoselectivity. For the
1H NMR spectrum of 3a, only in C6D6,
1H resonances
of two diastereomers are distinguishable. Complex 3a
is more stable than the neutral 2,2
′
-bicyclopropenyl
complex
{
[Ru]CdC(Ph)CCN
}
2previously reported by
us.
9bUsing the same method two other dinuclear
cyclopropenyl complexes
{
[Ru]CdC(CHCHdCH2)
}
2C6H4(3b) and
{
[Ru]CdC(CHPh)
}
2C
6H
4(3c) are prepared.
Characteristic spectroscopic data of 3b and 3c are
similar to those of 3a. The
31P NMR data of 3b and
3c reveal the presence of diastereomers both in 1:1
ra-tio. Protonation of 3 readily regenerates 2.
Prepara-tion of the organic phenyl bridged biscyclopropene
1,4-[PhCdC(C(Ph)(t-BuO))]C6
H
4by the addition of
1,4-bis-(phenylethynyl)benzene to 2 equiv of chlorocarbene
PhClC: generated from Ph-CHCl
2/t-BuOK has been
reported.
22aAdditionally,
1,4-bis[3,3-dimethyl-2-(tri-methylsilyl)-1-cyclopropen-1-yl]benzene was obtained
from the reaction of cyclopropenylzinc chloride and
p-diiodobenzene.
22bThe unsubstituted 2,2
′
-bicyclopro-pene has been prepared,
23and its structure has been
determined by X-ray diffraction analysis at 103 K.
24However, deprotonation of the dinuclear
bis-vi-nylidene complexes 2d and 2e, each containing an ester
substituent at C
γof the vinylidene ligand, yields the
bis-furyl complexes
{
[Ru]CdC(CHdC(O)OR
′
)
}
2C6H4(R
′
)
Me, 5d; R
′
) Et, 5e) (Scheme 2). The
31P NMR spectrum
of 5d displays a singlet resonance at δ 51.2, indicating
no stereogenic carbon center.
9b 31P NMR data at the
initial stage of the reaction indicate formation of a
mixture of 5d and the bis-cyclopropenyl complex
{
[Ru]-CdC(CHCOOMe)
}
2C6H4(3d); the latter readily converts
to 5d in solution. The less-strained five-membered ring
relative to the cyclopropenyl ligand and better oxygen
Lewis basicity are driving forces for the formation of 5.
A few organic bis-furans linked by a phenyl group have
been reported.
25Preparation of Dinuclear Cyclopropenylium
Complex. Electrophilic addition of TCNQ
(tetracyano-quinodimethane) to two C
γof the bis-cyclopropenyl
ligand of 3a leads to the zwitterionic bis-vinylidene
complex
{
[Ru]dCdC(CH(TCNQ)CN)
}
2C6H4(4a) (Scheme
3). The TCNQ-containing complex 4a displays a typical
deep purple-red color and is only moderately soluble in
DMSO. The
31P NMR spectrum displays one set of
two-doublet resonances at δ 47.2, 37.9 with JH-H
) 26.5 Hz.
Deprotonation of 4a in acetone with n-Bu4NOH in
MeOH yields
{
[Ru]CdC(C(OMe)CN)
}
2C
6H
4(6a),
pos-sibly via formation of an unobserved
TCNQ-substi-tuted cyclopropenyl complex (A) (Scheme 3).
Electro-philic attack of a methoxide at CR
of the
three-mem-bered ring accompanied with removal of TCNQ is
followed by a migration of the methoxide to C
γto give
6a. The chemical reactivity of 6a, containing a
meth-oxy group in the three-membered ring, differs from
that of cyclopropenyl complexes with no methoxy group,
which, in the presence of acid, readily undergo
ring-opening to give vinylidene. Protonation of 6a with
HPF
6, however, results in demethoxylation, yielding
{
[Ru]CC(CCN)
}
2C6H42+(7a) without opening of the
three-membered ring (Scheme 3). This is similar to the
reactivity of organic cyclopropene containing a methoxy
(22) (a) Eicher T.; Berneth H. Tetrahedron Lett. 1973, 2039. (b)Untiedt S.; de Meijere, A. Chem. Ber. 1994, 127, 1511.
(23) Billups, W. E.; Haley, M. M. Angew. Chem., Int. Ed. Engl. 1989, 28, 1711.
(24) Bordalla, D.; Mootz, D.; Roese, R.; Oswald, W. J. Appl. Crytal-logr. 1985, 18, 316.
(25) (a) Pelter, A.; Rowlands, M.; Jenkins, I. H. Tetrahedron Lett. 1987, 28, 5213. (b) Teng, X.; Wada, T.; Okamoto, S.; Sato, F. Tetrahedron Lett. 2001, 42, 5501. (c) Kang, S. K.; Baik, T. G.; Song, S. Y. Synth. Lett. 1999, 3, 327. (d) Lee, C. F.; Yang, L.-M.; Hwu, T. Y.; Feng, A. S.; Tseng, J. C.; Luh, T. Y. J. Am. Chem. Soc. 2000, 122, 4992.
Scheme 2 Scheme 3
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
substituent.
26The symmetrical planar structure of the
three-membered ring in 7a is revealed by its
31P NMR
spectrum, which shows only a singlet resonance at δ
46.8. The reaction of
1,4-bis[3-tert-butoxylphenyl-2-phenyl-1-cyclopropen-1-yl]benzene with HClO4
resulted
in elimination of t-BuOH, leading to a
1,4-bis(diphenyl-cyclopropenylium)benzene dication.
22aReaction of Me
3SiN
3with 3a. Treatment of 3a with
more than 10-fold excess of Me
3SiN
3afforded 1,4-
{
[Ru]-(N4C)CH(CH2CN)
}
2C6H4(8a) (Scheme 3). The reaction
yields diastereoisomers in a 1:1 ratio, as indicated by
two sets of two doublet resonances at δ 43.3, 41.9 and
43.1, 41.6 in the
31P NMR spectrum of the product. The
reaction may proceed via an electrophilic attack of TMS
at C
γof the three-membered ring followed by
nucleo-philic addition of an azide at CR
with subsequent loss
of N2
to first yield an unobserved nitrile complex
27(B).
Then a [2+3] cycloaddition of the coordinated nitrile
ligand with a second azide satisfactorily accounts for
formation of the product. Organic tetrazole compounds
are usually synthesized via a [3+2] cycloaddition
reac-tion of a nitrile group with azide.
28Metal-coordinated
azide ligands undergo 1,3-dipolar cycloaddition reactions
with carbon-carbon and carbon-heteroatom multiple
bonds. The metals involved are mostly Pd(II),
29Pt(II),
30or Co(III),
31although a whole range of other transition
metals
32-35have been used. However, formation of a
tetrazolate ring in our ruthenium complex should not
proceed via such a pathway since the reaction of organic
nitrile with [Ru]N3
does not yield the ruthenium
tetra-zolate complex.
27Trinuclear cyclopropenyl Complexes.
Tris-(alkynylmetal) derivatives with identical Pt(II),
36Fe-(II),
37or Ru(II)
38moieties have been synthesized from
the reaction of 1,3,5-triethynylbenzene with appropriate
metal precursors. We use the tripodal arylalkynyl ligand
1,3,5-(HCtCC6H4CtC)3C6H3,
14awhich is an extended
version of 1,3,5-triethynylbenzene. The trinuclear
acetyl-ide complex 1,3,5-
{
[Ru]CtCC6H4CtC
}
3C6H3(9) is
pre-pared in 86% yield from the reaction of [Ru]Cl in excess
with 1,3,5-(HCtCC6H4CtC)3C6H3. In the
1H NMR
spectrum of 9 no signal for alkynyl proton is detected;
i.e., a complex with only one or two metals is not
observed. The
31P NMR spectrum of 9 displays a singlet
resonance for six equivalent phosphines at δ 50.88,
showing high symmetry of this complex. A similar
complex containing different auxiliary ligands on the
ruthenium metal center has been reported.
14Electro-philic additions of alkyl halide RCH2X to three C
βatoms
of bridging acetylide ligands give the tricationic
tris-vinylidene complexes 1,3,5-
{
[Ru]dCdC(CH2R)-C6H4Ct
C
}
3C6H33+(R ) CN, 10a; R ) CHdCH2, 10b; R ) Ph,
10c) (Scheme 4). Excess organic halide was used to give
the single tris-vinylidene product. The downfield
13C NMR resonances at δ 345 ( 5 and
31P NMR
resonances at δ 40 ( 2 of these complexes clearly
indicate the presence of the tris-vinylidene ligand. The
tris-vinylidene complexes 10 are readily deprotonated
by n-Bu
4NOH, leading to the formation of 1,3,5-
{
[Ru]-CdC(CHR)C6H4CtC
}
3C6H3(R ) CN, 11a; R ) CHd
CH2, 11b; R ) Ph, 11c) (Scheme 4). Again only a single
product is obtained; namely, no mixed
vinylidene-cyclopropenyl complex is observed. There is only one set
of AX patterns at δ 51.5 and 49.5 (d, JP-P
) 35.0 Hz) in
the
31P NMR spectrum possibly due to distal
cyclopro-penyl moieties. Tris-cycloprocyclopro-penyl complexes 11
gradu-ally decompose in air or in CDCl3, producing the
tris-acetylide complex 9 and some unidentified compounds.
Furthermore, tris-cyclopropenyl complexes are less
stable than the corresponding mono- and dinuclear
cyclopropenyl complexes. The stability of cyclopropenyl
complexes follows the trend for trinuclear < dinuclear
< mononuclear system.
Concluding Remarks. We report the preparation
of dinuclear ruthenium cyclopropenyl complexes 3a-c
by deprotonation of vinylidene complexes 2a-c.
Dia-stereomeric pairs in a 1:1 ratio are obtained. However,
the deprotonation reaction of complexes 2d,e each
containing an ester substituent at C
γgives the dinuclear
bis-furyl complexes 5d,e. Additionally, the
bis-methoxy-substituted cyclopropenyl complex 6a is synthesized
(26) Breslow, R.; Chang, H. W. J. Am. Chem. Soc. 1961, 83, 2367.(b) Krebs, A. W. Angew. Chem., Int. Ed. Engl. 1965, 4, 10. (c) Closs, G. L.; Boll, W. A.; Heyn, H.; Dev, V. J. Am. Chem. Soc. 1968, 90, 173. (27) (a) Chang, K. H.; Lin, Y. C. Chem. Commun. 1998, 1441. (b) Chang, K. H.; Lin, Y. C.; Liu, Y. H.; Wang, Y. J. Chem. Soc., Dalton Trans. 2001, 3154.
(28) (a) Abbe`, G. L. Chem. Rev. 1969, 69, 345. (b) Butler, R. N. Comprehensive Heterocyclic Chemistry; Katritzky, A. R., Rees, C. W., Eds.; Pergamon: Oxford, 1984; Vol. 5, Part 4A, p 791.
(29) (a) Fehlhammer, W. P.; Beck, W. Z. Naturforsch. Teil B 1983, 38, 546. (b) Geisenberger, J.; Erbe, J.; Heidrich, J.; Nagel, U.; Beck, W. Z. Naturforsch. Teil B 1987, 42, 55.
(30) Beck, W.; Schorpp, K. Chem. Ber. 1975, 108, 3317.
(31) (a) Hsieh, B. T.; Nelson, J. H.; Milosavljevic, E. B.; Beck, W.; Kemmerich, T. Inorg. Chim. Acta 1987, 133, 267. (b) Kemmerich, T.; Nelson, J. H.; Takach, N. E.; Bohme, H. Jablonski B.; Beck, W. Inorg. Chem. 1982, 21, 1226.
(32) Blunden, S. J.; Mahon, M. F.; Molloy, K. C.; Waterfield, P. C. J. Chem. Soc., Dalton Trans. 1994, 2135.
(33) Guilard, R.; Perrot, I.; Tabard, A.; Richard, P.; Lecomte, C. Inorg. Chem. 1991, 30, 19. (b). Guilard, R.; Perrot, I.; Tabard, A.; Richard, P.; Lecomte, C. Inorg. Chem. 1991, 30, 27.
(34) Erbe, J.; Beck, W. Chem. Ber. 1983, 116, 3867.
(35) Nomiya, K.; Noguchi R.; Oda, M. Inorg. Chim. Acta 2000, 298, 24.
(36) Ohshiro, N.; Takei, F.; Onitsuka, K.; Takahashi, S. Chem. Lett. 1996, 871. (b) Khan, M. S.; Schwartz, D. J.; Pasha, N. A.; Kakkar, A. K.; Lin, B.; Raithby, R.; Lewis, J. Z. Anorg. Allg. Chem. 1992, 616, 121.
(37) Weyland, T.; Lapinte, C.; Frapper, G.; Calhorda, M. J.; Halet, J.-F.; Toupet, L. Organometallics 1997, 16, 2024. (b) Fink, H.; Long N.; J.; Martin, A. J.; Opromolla, G.; White, A. J. P.; Williams, D. J.; Zanello, P. Organometallics 1997, 16, 2646.
(38) Long, N. J.; Martin, A. J.; Biani, F. F. de; Zanello, P. J. Chem. Soc., Dalton Trans. 1998, 2017.
Scheme 4
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
from the zwitterionic TCNQ-containing bis-vinylidene
complex 4a prepared from 3a. The proton-induced
demethoxylation of 6a generates 7a. The bis-tetrazolate
complex 8a is obtained from the reaction of TMSN3
with
3a. Trinuclear tris-cyclopropenyl complexes 11 are
obtained from deprotonation of trinuclear tris-vinylidene
complexes 10, which are readily prepared from 9.
Experimental Section
General Procedures. All manipulations were performed
under nitrogen using vacuum-line, drybox, and standard Schlenk techniques. CH2Cl2 was distilled from CaH2, and
diethyl ether and THF were distilled from Na/diphenylketyl. All other solvents and reagents were of reagent grade and were used as received. NMR spectra were recorded on Bruker AC-300 and DMX-500 FT-NMR spectrometers at room tempera-ture (unless stated otherwise) and are reported in units of δ with residual protons in the solvents as a standard (CDCl3, δ
7.24; C6D6, δ 7.16). FAB mass spectra were recorded on a JEOL
SX-102A spectrometer. Complex [Ru]Cl ([Ru] ) (η5-C
5H5)(PPh3)2
-Ru) was prepared according to the literature method,39as were
1,4-diethynylbenzene40 and 1,3,5-(HCtCC
6H4CtC)3C6H3.14a
Elemental analyses and X-ray diffraction studies were carried out at the Regional Center of Analytical Instrument located at National Taiwan University.
Synthesis of 1,4-{[Ru]CtC}2C6H4(1). A solution of
[Ru]-Cl (230 mg, 0.32 mmol) and NaPF6(260 mg, 1.58 mmol) in
methanol (25 mL) was heated to reflux for 40 min to give an orange-red suspension, to which 1,4-diethynylbenzene (20 mg, 0.16 mmol) was added. The mixture was heated to reflux for 40 min and then cooled to room temperature. Addition of 5 equiv of sodium methoxide (86 mg) resulted in rapid precipita-tion of a yellow powder. The mixture was filtered, and the yellow solid was washed with cold methanol and dried under vacuum to give 1 (200 mg, 0.27 mmol, 84%). Spectroscopic data for 1 are as follows. 31P NMR (CDCl
3): δ 50.98. 1H NMR
(CDCl3): δ 7.45-6.93 (m, 64H, Ph, C6H4), 4.29 (s, 10H, C5H5).
MS (FAB) m/z: 1506 (M+), 1244 (M+- PPh3). Anal. Calcd for
C92H74P4Ru2: C, 73.39; H, 4.95. Found: C, 73.60; H, 4.86. Synthesis of{1,4-{[Ru]dCdC(CH2CN)}2C6H4}I2(2a). To
a Schlenk flask charged with 1 (150 mg, 0.10 mmol) in CH2
-Cl2 (15 mL) was added ICH2CN (145 µL, 20 mmol). The
resulting solution was stirred at 40 °C for 24 h, then cooled to room temperature, and the solvent was reduced to about 2.5 mL. The mixture was slowly added to 25 mL of vigorously stirred diethyl ether. The red precipitate thus formed was filtered off and washed with diethyl ether and dried under vacuum to give 2a (169 mg, 0.92 mmol, 92% yield). Spectro-scopic data for 2a are as follows.31P NMR (CDCl
3): δ 40.9.1H
NMR (CDCl3): δ 7.43-6.89 (m, 64H, Ph), 5.42 (s, 10H, C5H5),
3.42 (s, 4H, CH2CN).13C NMR (CD3SOCD3): δ 349.2 (t, CR, JP-C) 15.3 Hz), 134.1-129.5 (m, Ph), 124.0 (Cβ), 119.7 (CN), 96.4 (Cp), 13.4 (CH2). MS (FAB) m/z: 1713 (M+ - I). Anal.
Calcd for C96H78N2P4Ru2I2: C, 62.68; H, 4.27; N, 1.52. Found:
C, 62.44; H, 4.37; N, 1.49.
Synthesis of {1,4-{[Ru]dCdC(CH2R)}2C6H4}X2 (R ) CHdCH2, 2b; R ) Ph, 2c; R ) CO2CH3, 2d; R ) CO2C2H5, 2e). Synthesis of 2b-e followed the same procedure as that
used for the preparation of 2a from complex 1 (150 mg, 0.10 mmol). Spectroscopic data for 2b (166 mg, 0.90 mmol, 90% yield) are as follows. 31P NMR (CDCl
3): δ 42.5. 1H NMR
(CDCl3): δ 7.44-6.85 (m, 64H, Ph), 5.31 (s, 10H, C5H5),
5.48-5.19 (m, 2H, dCH), 4.78 (d, 2H, J ) 5.9 Hz, dCH), 4.68 (s, 2H, dCH), 2.79 (d, 4H, CH2).13C NMR (CDCl3): δ 349.2 (t,
CR, JP-C) 15 Hz), 134.5-127.5 (m, Ph, CH2dCH), 117.3 (Cβ),
94.6 (Cp), 30.2 (CH2). MS (FAB) m/z: 1715 (M+ - I). Anal.
Calcd for C98H84P2Ru2I2: C, 62.71; H, 4.94. Found: C, 62.34;
H, 4.89. Red single crystals of 2b are obtained from the CDCl3
solution used for NMR data. Spectroscopic data for 2c (177 mg, 0.96 mmol, 96% yield) are as follows.31P NMR (CDCl
3): δ 42.2.1H NMR (CDCl
3): δ 7.33-6.83 (m, 74H, Ph), 5.34 (s,
10H, C5H5), 3.38 (s, 4H, CH2).13C NMR (CDCl3): δ 350.3 (t,
CR, JP-C) 15.3 Hz), 139.8-127.0 (m, Ph), 122.2 (Cβ), 95.3 (Cp), 31.8 (CH2). MS (FAB) m/z: 1767 (M+- Br). Anal. Calcd for
C106H88P4Ru2Br2: C, 68.90; H, 4.80. Found: C, 69.74; H, 4.95.
Spectroscopic data for 2d (161 mg, 0.89 mmol, 89% yield) are as follows.31P NMR (CDCl
3): δ 41.9.1H NMR (CDCl3): δ
7.48-6.89 (m, 74H, Ph), 5.42 (s, 10H, C5H5), 3.23 (s, 6H, CH3), 2.92
(s, 4H, CH2).13C NMR (CDCl3): δ 349.6 (t, CR, JP-C) 15.0
Hz), 172.3 (CO2), 135.0-129.3 (Ph), 125.8 (Cβ), 95.8 (Cp), 52.6 (CH3), 31.9 (CH2). MS (FAB) m/z: 1731 (M+- Br). Anal. Calcd
for C98H84O4P4Ru2Br2: C, 64.97; H, 4.67. Found: C, 65.35; H,
4.48 Spectroscopic data of 2e (172 mg, 0.91 mmol, 91% yield) are as follows.31P NMR (CDCl 3): δ 41.9.1H NMR (CDCl3): δ 7.51-6.90 (m, 74H, Ph), 5.41 (s, 10H, C5H5), 3.77 (q, 4H, JH-H ) 7.1 Hz, OCH2), 2.91 (s, 4H, CH2COO), 0.97 (t, 6H, JH-H) 7.1 Hz, CH3).13C NMR (CDCl3): δ 349.3 (t, CR, JP-C) 15.0 Hz), 171.8 (CO2), 135.0-129.4 (m, Ph), 126.0 (Cβ), 95.8 (Cp), 61.6 (CH2CO2), 32.3 (OCH2), 14.6 (CH3). MS (FAB) m/z: 1767
(M+- I). Anal. Calcd for C100H88O4P4Ru2I2: C, 62.11; H, 4.59.
Found: C, 62.37; H, 4.81.
Synthesis of 1,4-{[Ru]CdC(CHCN)}2C6H4 (3a). To a
solution of 2a (203 mg, 0.11 mmol) in 10 mL of CH2Cl2was
added NH4PF6(41 mg, 0.25 mmol). After stirring at room
temperature for 6 h, the mixture was filtered through Celite to remove NH4I, and the solvent of the filtrate was removed
under vacuum. Then 5 mL of acetone and a solution of n-Bu4
-NOH (2 mL, 1 M in MeOH) were added. The mixture was stirred for 6 h, yielding yellow microcrystalline precipitates, which were filtered off and washed with 2× 5 mL of acetone, then dried under vacuum. The product contains two diaster-eomers and is identified as 3a (148 mg, 0.94 mmol, 85% yield). Spectroscopic data for 3a are as follows.31P NMR (CDCl
3): δ 51.9 (d, JP-P) 36.4 Hz), 49.3 (d, JP-P) 36.4 Hz), 51.8 (d, JP-P ) 35.2 Hz), 49.2 (d, JP-P) 35.2 Hz) (1:1).1H NMR (CDCl3): δ 7.62-6.41 (m, 64H, Ph), 4.28 (s, 10H, Cp), 1.32 (s, 2H, CH). 1H NMR (C 6D6): δ 7.34-6.86 (m, 64H, Ph), 4.70, 4.69 (s, 10H, Cp), 1.72, 1.71 (s, 2H, CH).13C NMR (CDCl 3): δ 140.4-128.1 (m, Ph, CR) 120.0 (CN), 86.3 (Cp), 8.8 (CH). MS (FAB) m/z: 1585 (M++ 1), 1324 (M+- PPh3), 1061 (M+- 2PPh3). Anal.
Calcd for C96H76N2P4Ru2: C, 72.81; H, 4.84; N, 1.77. Found:
C, 72.69; H, 4.91; N, 1.81.
Synthesis of 1,4-{[Ru]CdC(CHCHdCH2)}2C6H4 (3b).
Complex 3b (155 mg, 0.098 mmol, 65% yield) was prepared from 2b (276 mg, 0.15 mmol) in analogy with the synthesis of
3a. Spectroscopic data for 3b are as follows.31P NMR (C 6D6): δ 53.2 (d, JP-P) 37.3 Hz), 49.7 (d, JP-P) 37.3 Hz), 53.1 (d, JP-P) 36.9 Hz), 49.5 (d, JP-P) 36.9 Hz), (1:1).1H NMR (C6D6): δ 7.46-6.84 (m, 74H, Ph), 6.30-6.16 (m, 2H, dCH), 5.63, 5.62 (dd, JH-H) 17.0, 2.5 Hz, 2H, dCH), 5.13, 5.12 (dd, JH-H) 10.0, 2.5 Hz, 2H, dCH), 4.67 (s, 10H, Cp), 2.46, 2.45 (d, JH-H ) 8.6 Hz, 2H, CH2).13C NMR (C6D6): δ 154.6 (dCH), 141.2-123.6 (m, Ph, CR), 106.4 (dCH2), 86.2 (Cp), 33.5 (CH). MS (FAB) m/z: 1587 (M++ 1), 1547 (M++ 1 - CHCHdCH2), 1326
(M++ 1 - PPh3). Anal. Calcd for C98H82P4Ru2: C, 74.23; H,
5.21. Found: C, 74.01; H, 5.33.
Synthesis of 1,4-{[Ru]CdC(CHPh)}2C6H4(3c). Complex 3c (121 mg, 0.072 mmol, 55% yield) was prepared from 2c (240
mg, 0.13 mmol) in analogy with the synthesis of 3a. Spectro-scopic data for 3c are as follows.31P NMR (C
6D6): δ 54.8 (d, JP-P) 36.8 Hz), 48.2 (d, JP-P) 36.8 Hz), 54.8 (d, JP-P) 37.0 Hz), 48.1 (d, JP-P) 37.0 Hz) (1:1).1H NMR (C6D6): δ 7.70-6.80 (m, 74H, Ph), 4.43, 4.40 (s, 10H, Cp), 2.87, 2.86 (s, 2H, CH).13C NMR (CDCl 3): δ 141.2-123.6 (m, Ph, CR), 86.1 (Cp),
(39) Bruce, M. I.; Hameister, C. A. Swincer G.; Wallis, R. C. Inorg. Synth. 1990, 28, 270.
(40) Pelter, A.; Jones, D. E. J. Chem. Soc., Perkin Trans. 1 2000, 2289.
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
33.9 (CH). Anal. Calcd for C106H86P4Ru2: C, 75.51; H, 5.14.
Found: C, 75.82; H, 5.06.
Synthesis of{[Ru]CdC(CHdC(O)OR)}2C6H4(R ) Me, 5d; R ) Et, 5e). The synthesis and workup were similar to
those used in the preparation of complex 3a. Complex 5d (119 mg, 0.072 mmol, 80% yield) was prepared from 2d (163 mg, 0.09 mmol). Spectroscopic data for 5d are as follows.31P NMR
(CDCl3): δ 51.2.1H NMR (CDCl3): δ 7.25-6.99 (m, 64H, Ph),
5.06 (s, 2H, CH), 4.10 (s, 10H, Cp), 3.04 (s, 6H, OCH3).13C
NMR (CDCl3): δ 163.9 (CO2), 155.0 (CR), 140.6-127.1 (Ph),
87.1 (Cγ), 84.00 (Cp), 58.1 (CH3). Anal. Calcd for C98H82O4P4
-Ru2: C, 71.35; H, 5.01. Found: C, 71.50; H, 4.89. Complex 5e
(110 mg, 0.066 mmol, 82% yield) was prepared from 2e (151 mg, 0.08 mmol). Spectroscopic data for 5e are as follows.31P
NMR (CDCl3): δ 51.7.1H NMR (CDCl3): δ 7.35-6.96 (m, 64H,
Ph), 5.11 (s, 2H, CH), 4.11 (s, 10H, Cp), 3.10 (q, JH-H) 7.07
Hz, 4H, OCH2), 0.93 (t, JH-H) 7.07 Hz, 6H, CH3).13C NMR
(CDCl3): δ 163.2 (CO2), 155.3(CR), 141.6-127.7 (Ph), 89.6 (Cγ), 84.5 (Cp), 67.3 (CH2), 15.4 (CH3). Anal. Calcd for C100H86O4P4
-Ru2: C, 71.59; H, 5.17. Found: C, 71.40; H, 5.30.
Reaction of 3a with TCNQ. To a mixture of 3a (40 mg,
0.025 mmol) in CH2Cl2(5 mL) was added TCNQ (10 mg, 0.05
mmol). The solution was stirred at room temperature for 40 min, and the solvent was removed under vacuum. The residue was washed with 3× 5 mL of methanol to produce the purple-red powder 4a (46 mg, 0.023 mmol, 92% yield). Spectroscopic data for 4a are as follows.31P NMR (d
6-DMSO): δ 47.2, 37.9
(2d, JP-P) 26.5 Hz).1H NMR (d6-DMSO): δ 7.62-6.9 (m, Ph),
5.50 (s, Cp).
Synthesis of 1,4-{[Ru]CdC(C(OMe)CN)}2C6H4(6a). To
a solution of 4a (120 mg, 0.06 mmol) in 7 mL of acetone was added 0.7 mL of CH3OH/n-Bu4NOH (1 M in MeOH). The color
of the solution immediately changed to dark green. The solution was further stirred at room temperature for 1.5 h, and then the solvent was removed under vacuum. The residue was washed with 3× 5 mL of methanol to produce yellow-green microcrystals of complex 6a (76 mg, 0.046 mmol, 77% yield). Spectroscopic data for 6a are as follows. 31P NMR
(CDCl3): δ 51.9 (d, JP-P) 36.4 Hz), 49.9 (d, JP-P) 36.4 Hz),
51.2 (d, JP-P) 36.4 Hz), 49.3 (d, JP-P) 36.4 Hz) (1:1).1H NMR
(CDCl3): δ 7.16-6.39 (m, 64H, Ph), 4.65 (s, 10H, Cp), 3.42, 3.38
(s, 6H, OMe).1H NMR (C
6D6): δ 7.44-6.68 (m, 64H, Ph), 4.89
(s, 10H, Cp), 3.61 (s, 3H, OMe), 3.59 (s, 3H, OMe). MS (FAB)
m/z: 1644 (M+), 1618 (M+- CN).13C NMR (CDCl
3): δ
139.5-126.7 (Ph, CR), 86.3 (Cp), 59.3, 59.1 (C(CN)(OMe)), 55.7, 55.5
(OMe). Anal. Calcd for C98H80O2N2P4Ru2: C, 71.61; H, 4.91;
N, 1.70. Found: C, 71.42; H, 4.99; N, 1.73.
Reaction of 6a with HPF6. To a solution of 6a (30 mg,
0.018 mmol) in 2 mL of CH2Cl2at 0 °C was added 2.5 µL of
HPF6(60 wt % in H2O). The color of the solution immediately
changed from yellow to amber-red. The solution was stirred at 0 °C for 10 min and then was added to 10 mL of an ether solution in an ice-bath. The orange precipitate thus formed was filtered and washed with diethyl ether to give the product {[Ru]CC(C(CN))}2C6H4(PF6)2(7a). Spectroscopic data for 7a
are as follows.31P NMR (C
6D6): δ 46.79.1H NMR (C6D6): δ
7.67-6.89 (m, 74H, Ph), 5.20 (s, 10H, Cp).
Synthesis of 1,4-{[Ru]N4CCH(CH2CN)}2C6H4(8a). To a
solution of complex 3a (30 mg, 0.019 mmol) in THF (3 mL) was added (CH3)3SiN3(30 µL, 0.23 mmol). After stirring at
room temperature for 7 h, the mixture was concentrated to ca. 1 mL and slowly added to vigorously stirred hexane (8 mL). The yellow precipitate thus formed was filtered off and washed with 2× 5 mL of hexane. The product was analytically pure and was identified as complex 8a (24 mg, 0.014 mmol, 75% yield). Spectroscopic data for 8a are as follows. 31P NMR
(C6D6): δ 43.3, 41.9 (d, JP-P) 38.4 Hz) 43.1, 41.6 (d, JP-P) 38.2 Hz) (1:1).1H NMR (C 6D6): δ 7.41-6.74 (m, 64H, Ph), 4.49, 4.43 (dd, 2H,3J H-H) 7.75, 3JH-H) 7.84 Hz), 4.29 (s, 10H, Cp), 2.77-2.63, 2.47-2.36 (m, 4H, CH2).13C NMR (CDCl3): δ 163.9 (NCN), 138.3-123.6 (Ph), 118.7 (CN), 83.1 (Cp), 39.6, 39.5 (CH), 23.7, 23.5 (CH2). MS (FAB) m/z: 1700 (M+) 1437 (M+- PPh
3) 1176 (M+- 2PPh3). Anal. Calcd for C96H78N10P4
-Ru2: C, 67.91; H, 4.63; N, 8.25. Found: C, 68.02; H, 4.54; N,
8.20.
Synthesis of 1,3,5-{[Ru]CtCC6H4CtC}3C6H3(9).
Com-plex [Ru]Cl (290 mg, 0.04 mmol) in methanol (25 mL) was heated to reflux for 40 min to give an orange-red solution, to which 1,3,5-(HCtCC6H4CtC)3C6H3(60 mg, 0.13 mmol) was
then added. The mixture was stirred and heated to reflux for 1 h and then cooled to room temperature. Addition of 10 equiv of triethylamine resulted in rapid precipitation of a yellow powder. The mixture was stirred for 1 h and filtered, and the yellow solid washed with cold methanol to give 9 (289 mg, 0.034 mmol, 86% yield). Spectroscopic data for 9 are as follows.
31P NMR (CDCl
3): δ 50.88.1H NMR (CDCl3): δ 7.56-7.03 (m,
105H, Ph), 4.32 (s, 15H, Cp).13C NMR (CDCl
3): δ 138.7 (t,
CR, JP-C) 20.9 Hz), 133.8-127.2 (Ph), 85.3 (Cp), 115.3 (tC),
91.5, 88.2 (tC). MS (FAB) m/z: 2521(M++ 1). Anal. Calcd for C159H120P6Ru3: C, 75.79; H, 4.80. Found: C, 75.92; H, 4.64.
Preparation of{1,3,5-{[Ru]dCdC(CH2CN)C6H4CtC}3C6 -H3}I3(10a). A Schlenk flask was charged with 9 (330 mg,
0.131 mmol) in 7 mL of CH2Cl2, and ICH2CN (282 µL 3.9
mmol) was added under nitrogen. The resulting solution was stirred at 40 °C for 24 h, then cooled to room temperature, and the solvent was reduced to about 2.5 mL. The mixture was slowly added to 25 mL of vigorously stirred diethyl ether. The pale red precipitate thus formed was filtered off and washed with diethyl ether, then dried under vacuum to give
10a (364 mg, 0.120 mmol, 92% yield). Spectroscopic data for 10a are as follows. 31P NMR (CDCl
3): δ 40.94. 1H NMR
(CDCl3): δ 7.62-6.92 (m, 105H, Ph), 5.38 (s, 15H, Cp), 3.55 (s,
6H, CH2).13C NMR (CD3SOCD3): δ 345.7 (t, CR, JP-C) 15.0
Hz), 134.4-129.5 (m, Ph), 124.0 (Cβ), 119.7 (CN), 96.2 (Cp), 91.1, 88.8 (tC), 13.4 (CH2). Anal. Calcd for C165H126N3P6
-Ru3I3: C, 65.61; H, 4.20; N, 1.39. Found: C, 65.35; H, 4.31; N,
1.31.
Preparation of{1,3,5-{[Ru]dCdC(CH2CHdCH2)C6H4Ct C}3C6H3}I3(10b). Complex 10b (376 mg, 0.120 mmol, 92%
yield) was prepared from 9 (330 mg, 0.131 mmol) and ICH2
-CHdCH2in analogy with the synthesis of 10a. Spectroscopic
data for 10b are as follows. 31P NMR (CDCl
3): δ 42.35. 1H NMR (CDCl3): δ 7.76-6.87 (m, 105H, Ph), 5.67-5.53 (m, 3H, dCH), 5.17 (s, 15H, C5H5), 5.01 (d, 3H, J ) 9.9 Hz, dCH2), 4.93 (d, 3H, J ) 17.1 Hz, dCH2), 2.79 (d, 4H, J ) 8.6 Hz, CH2). 13C NMR (CDCl 3): δ 349.0 (t, CR, JP-C) 15.5 Hz), 140.6-122.9 (m, Ph), 117.9 (Cβ), 94.8 (Cp), 90.6, 88.9 (tC), 30.7 (CH2). Anal.
Calcd for C168H135P6Ru3I3: C, 66.73; H, 4.50. Found: C, 66.91;
H, 4.68.
Preparation of{1,3,5-{[Ru]dCdC(CH2Ph)C6H4CtC}3C6 -H3}Br3(10c). Complex 10c (354 mg, 0.117 mmol, 89% yield)
was prepared from 9 (330 mg, 0.131 mmol) and BrCH2Ph in
analogy with the synthesis of 10a. Spectroscopic data for 10c are as follows.31P NMR (CDCl 3): δ 42.10.1H NMR (CDCl3): δ 7.57-6.90 (m, 120H, Ph), 5.21 (s, 15H, Cp), 3.59 (s, 6H, CH2). 13C NMR (CDCl 3): δ 348.6 (t, CR, JP-C) 15.8 Hz), 137.7-126.7 (m, Ph), 122.0 (Cβ), 94.7 (Cp), 90.9, 88.6 (tC), 31.8 (CH2). Anal.
Calcd for C180H141P6Ru3Br3: C, 71.28; H, 4.69. Found: C, 71.62;
H, 4.55.
Preparation of{1,3,5-{[Ru]CdC(CHR)C6H4CtC}3C6H3} (11a, R ) CN). To a solution of 10a (302 mg, 0.10 mmol) in
10 mL of CH2Cl2was added NH4PF6(82 mg, 0.5 mmol). After
stirring at room temperature for 6 h, the mixture was filtered through Celite and the solvent was removed by vacuum. Then 4 mL of acetone and a solution of n-Bu4NOH (2 mL, 1 M in
MeOH) were added. After stirring for 8 h, the solvent was reduced to about 1.5 mL. The mixture was slowly added to 8 mL of vigorously stirred CH3CN. The yellow precipitate thus
formed was filtered off, washed with CH3CN, and dried under
vacuum to give 11a (217 mg, 0.082 mmol, 82%). Spectroscopic
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
data for 11a are as follows.31P NMR (CDCl 3): δ 51.5, 49.5 (d, JP-P) 35.0 Hz).1H NMR (CDCl3): δ 7.56-6.54 (m, 71H, Ph), 4.58 (s, 15H, Cp), 1.48 (s, 3H, CH).13C NMR (CDCl 3): δ 138.6-127.4 (Ph), 122.3 (CN), 116.4 (tC), 90.7, 85.2 (tC), 86.5 (Cp), 8.13 (CH). Anal. Calcd for C165H123N3P6Ru3: C, 75.16; H, 4.07;
N, 1.59. Found: C, 75.43; H, 4.21; N, 1.44.
Complex 11b (R ) CHdCH2) (198 mg, 0.075 mmol, 75% yield) was prepared from 10b (302 mg, 0.10 mmol) in analogy with the synthesis of 11a. Spectroscopic data for 11b are as follows.31P NMR (C 6D6): δ 53.0, 49.5 (d, JP-P) 36.5 Hz).1H NMR (C6D6): δ 7.61-6.84 (m, 71H, Ph), 6.34-6.22 (m, 3H, d CH), 5.73 (dd, JH-H) 17.0, 2.5 Hz, 2H, dCH2) 5.22 (dd, JH-H ) 10.0, 2.5 Hz, 2H, dCH2) 4.69 (s, 15H, Cp), 2.57 (d, JH-H) 8.6 Hz, 3H, CH).13C NMR (CDCl 3): δ 153.1 (dCH), 143.1 (t, CR, JP-C) 20.7 Hz), 140.2-123.6 (Ph), 117.2 (Cβ), 106.4 (d CH2), 91.8, 87.4 (tC), 85.8 (Cp), 32.9 (CH). Anal. Calcd for
C168H132P6Ru3: C, 76.43; H, 5.04. Found: C, 76.98; H, 4.82. Complex 11c (R ) Ph) (173 mg, 0.062 mmol, 62% yield)
was prepared from 10c (303 mg, 0.10 mmol) in analogy with the synthesis of 11a. Spectroscopic data for 11c are as follows.
31P NMR (C 6D6): δ 54.4, 47.6 (d, Jp-p) 36.7 Hz). 1H NMR (C6D6): δ 7.68-6.85 (m, 86H, Ph), 4.43 (s, 15H, Cp), 3.0 (s, 3H, CH).13C NMR (CDCl 3): δ 143.1(t, CR, JP-C) 20.1 Hz), 140.6-127.4 (Ph), 117.2(Cβ), 91.8, 87.5 (tC), 85.3 (Cp), 33.0 (CH). Anal. Calcd for C180H138P6Ru3: C, 77.49; H, 4.99. Found: C,
77.26; H, 4.84.
Single-Crystal X-ray Diffraction Analysis of 2b. Single
crystals of 2b suitable for an X-ray diffraction study were grown as mentioned above. A single crystal of dimensions 0.40 × 0.20 × 0.15 mm3was glued to a glass fiber and mounted on
an SMART CCD diffractometer. The diffraction data were collected using 3 kW sealed-tube molybdenum KR radiation (T ) 295 K). Exposure time was 5 s per frame. SADABS (Siemens area detector absorption) absorption correction was applied, and decay was negligible. Data were processed, and the structures were solved and refined by the SHELXTL program. The structure was solved using direct methods and confirmed by Patterson methods refining on intensities of all data (67 315 reflections) to give R1 ) 0.0531 and wR2 ) 0.1325 for 12 547 unique observed reflections (I > 2σ(I)). Hydrogen atoms were placed geometrically using the riding model with thermal parameters set to 1.2 times that for the atoms to
which the hydrogen is attached and 1.5 times that for the methyl hydrogens. (Data collection parameters are listed in Table 2.)
Acknowledgment. We thank the National Science
Council, Taiwan, Republic of China, for support of this
work.
Supporting Information Available: Tables of atomic
coordinates, bond lengths and angles, anisotropic thermal parameters, and hydrogen atom positions for 2b. This material is available free of charge via the Internet at http://pubs.acs.org. OM020913X
Table 2. Crystal and Intensity Collection Data for 1,4-{[Ru]CdC(CH2CHdCH2)}2C6H42+(2b) mol formula C104H84D6Cl18I2P4Ru2
mol wt 2563.68
cryst syst triclinic
space group P1h a, Å 9.5660(1) b, Å 15.2030(1) c, Å 20.8060(2) R, deg 101.413(1) β, deg 103.079(1) γ, deg 103.824(1) V, Å3 2758.58(4) Z 1 cryst dimens, mm3 0.40× 0.20 × 0.15 Mo KR radiation: γ, Å 0.71073 θ range, deg 1.04-27.47 limiting indices -12 e h e 12 -19 e k e 19 -26 e l e 26 no. of reflns collected 67 315 no. of ind reflns (Rint) 12 612 (0.0760)
max. and min. transmn 0.874 and 0.653
refinement method full-matrix least-squares on F2
no. of data/restraints/params 12547/0/587
GOF 1.020
final R indices [I > 2σ(Ι)] R1 ) 0.0531, wR2) 0.1325 R indices (all data) R1 ) 0.0936, wR2) 0.1630
∆F (in final map), e/Å-3 -0.888 and +0.967
Downloaded by NATIONAL TAIWAN UNIV on August 10, 2009
![Table 2. Crystal and Intensity Collection Data for 1,4- {[Ru]CdC(CH 2 CHdCH 2 ) } 2 C 6 H 4 2+ (2b) mol formula C 104 H 84 D 6 Cl 18 I 2 P 4 Ru 2](https://thumb-ap.123doks.com/thumbv2/9libinfo/8868836.247190/7.918.475.833.93.474/table-crystal-intensity-collection-data-cdc-chdch-formula.webp)
