A Pharmacophore-Based Evolutionary Approach for
Screening Selective Estrogen Receptor Modulators
Jinn-Moon Yang* and Tsai-Wei ShenDepartment of Biological Science and Technology, and Institute of Bioinformatics, National Chiao Tung University, Hsinchu, Taiwan
ABSTRACT We developed a pharmacophore-based evolutionary approach for virtual screening. This tool, termed the Generic Evolutionary Method for molecular DOCKing (GEMDOCK), combines an evolutionary approach with a new pharmacophore-based scoring function. The former integrates dis-crete and continuous global search strategies with local search strategies to expedite convergence. The latter, integrating an empirical-based energy func-tion and pharmacological preferences (binding-site pharmacological interactions and ligand prefer-ences), simultaneously serves as the scoring func-tion for both molecular docking and postdocking analyses to improve screening accuracy. We apply pharmacological interaction preferences to select the ligands that form pharmacological interactions with target proteins, and use the ligand preferences to eliminate the ligands that violate the electro-static or hydrophilic constraints. We assessed the accuracy of our approach using human estrogen receptor (ER) and a ligand database from the com-parative studies of Bissantz et al. (J Med Chem 2000;43:4759 – 4767). Using GEMDOCK, the average goodness-of-hit (GH) score was 0.83 and the average false-positive rate was 0.13% for ER antagonists, and the average GH score was 0.48 and the average false-positive rate was 0.75% for ER agonists. The performance of GEMDOCK was superior to compet-ing methods such as GOLD and DOCK. We found that our pharmacophore-based scoring function in-deed was able to reduce the number of false posi-tives; moreover, the resulting pharmacological inter-actions at the binding site, as well as ligand preferences, were important to the screening accu-racy of our experiments. These results suggest that GEMDOCK constitutes a robust tool for virtual data-base screening. Proteins 2005;59:205–220.
©2005 Wiley-Liss, Inc.
Key words: estrogen receptor; evolutionary ap-proach; hot spots; pharmacophore-based scoring function; SERMs; virtual screening
INTRODUCTION
Virtual screening (VS) of molecular compound libraries has emerged as a powerful and inexpensive method for the discovery of novel lead compounds for drug
develop-ment.1,2Given the structure of a target protein active site
and a potential small ligand database, VS predicts the binding mode and the binding affinity for each ligand and ranks a series of candidate ligands. There are 4 main reasons for the rapid acceptance and success of VS: (1) the availability of the growing number of protein crystal structures; (2) the advent of structural proteomics technolo-gies; (3) the enrichment and speed of VS1,3; and (4) the
contribution of VS to the reduction in the cost of drug discovery. VS generally encompasses 4 phases based on both high-throughput molecular docking methods and the crystal structures of the target protein. These include target protein preparation, compound database prepara-tion, molecular docking, and postdocking analysis.1 The
molecular docking method screens the compound library to find lead compounds for the target protein, whereas postdocking analysis enriches the hit rate and optimizes the confirmed lead molecules through structure–activity relationship.4
The VS computational method involves 2 basic critical elements: efficient molecular docking and a reliable scor-ing method. A molecular dockscor-ing method for VS should be able to screen a large number of potential ligands with reasonable accuracy and speed. The many molecular dock-ing approaches that have been developed can be roughly categorized as rigid docking,5 flexible ligand docking,6,7
and protein flexible docking. Most current VS methods employ flexible docking tools, such as incremental and fragment-based approaches (DOCK8and FlexX7) and
evo-lutionary algorithms (GOLD,6 AutoDock,9 and
GEM-DOCK10).
Scoring methods for VS should effectively discriminate between correct binding states and non-native docked conformations during the molecular docking phase and distinguish a small number of active compounds from hundreds of thousands of nonactive compounds during the postdocking analysis. The scoring functions that calculate
Grant sponsor: National Science Council of Taiwan; Grant numbers: NSC-92-2113-M-009-024 and NSC-93-2113-M-009-010. Grant spon-sor: Veterans General Hospitals, University System of Taiwan; Grant number: VGHUST93-G5-05-3.
*Correspondence to: Jinn-Moon Yang, Department of Biological Science and Technology, and Institute of Bioinformatics, National
Chiao Tung University, Hsinchu, 30050, Taiwan. E-mail:
Received 9 June 2004; Accepted 18 October 2004
Published online 22 February 2005 in Wiley InterScience (www.interscience.wiley.com). DOI: 10.1002/prot.20387
the binding free energy mainly include knowledge-based,11 physics-based,12 and empirical-based13 scoring
functions. The performance of these scoring functions is often inconsistent across different systems from a data-base search.14,15 It has been proposed that combining
multiple scoring functions (consensus scoring) improves the enrichment of true positives.14,15
While the field of VS may be maturing,1–3 and many
good VS methods have been proposed, the promise of the virtual compound library16to rapidly increase the number
of candidate ligands demands further improvement in terms of the computational efficiency of flexible docking algorithms.6,7,9In addition, some VS methods are capable
of identifying so-called “pharmacological preferences” that are often the important interactions or binding-site hot spots typically evolved from known active ligands and the target protein.17,18 These preferences might improve
screening accuracy and guide the design and selection of lead compounds for subsequent investigation and refine-ment during lead discovery and lead optimization pro-cesses. Finally, the screening quality of docking methods using energy-based scoring functions alone is often influ-enced by the molecular weight and the structure of the ligand being screened (e.g., the numbers of charged and polar atoms). These methods are often biased toward both the selection of high molecular weight compounds (due to the contribution of the compound size19,20) and charged
polar compounds (due to the pair-atom potentials of the electrostatic energy and hydrogen-bonding energy).
To address the above issues, we developed a new VS method, termed GEMDOCK (Generic Evolutionary Method for molecular DOCKing), modified from our previous stud-ies.10,21 GEMDOCK is an evolutionary-based approach
that was applied in some fast VS algorithms.6,9 Our
approach uses multiple operators (e.g., discrete and con-tinuous genetic operators) that cooperate using family competition (similar to a local search procedure) to balance exploration and exploitation. Like some VS methods,18,22,23
GEMDOCK evolves the pharmacological preferences from a number of known active ligands to take advantage of the similarity of a putative ligand to those that are known to bind to a protein’s active site, thereby guiding the docking of the putative ligand. However, unlike existing pharma-cophore-based docking methods, we developed and incorpo-rated a new scoring function that evolves a pharmacologi-cal consensus (e.g., hot spots) and ligand preferences using the target protein and known active ligands. This scoring function not only serves as the basis for molecular docking but also ranks the screened ligands prior to postdocking analysis by reducing the deleterious effect of certain structural features within some of the ligands.
While GEMDOCK is generally applicable, in particular, it has been validated by its application to the docking of a number of selective estrogen receptor modulators (SERMs) that are of great interest in cancer chemotherapy, as well as estrogen replacement therapy in postmenopausal women.24 –26To evaluate the strengths and limitations of
GEMDOCK, and to compare it with several widely used methods (DOCK, GOLD, and FlexX), we evaluated the
screening utility of GEMDOCK by testing human estrogen receptor (ER) with the ligand data set, as proposed by Bissantz et al.14We also assessed whether our new scoring
function was applicable to both the molecular docking and ligand scoring during VS. The screening performance of GEMDOCK on this ligand data set is superior to that of the best available methods, and the docking accuracy is also comparable. Thus, GEMDOCK constitutes a rapid method that reduces the number of false positives during the screening of large databases when both pharmacologi-cal interactions and ligand preferences are mined from known active compounds. When known active ligands are not available, the screening accuracy of GEMDOCK is somewhat influenced and is comparable to that of compara-tive methods on this ligand data set.
MATERIALS AND METHODS
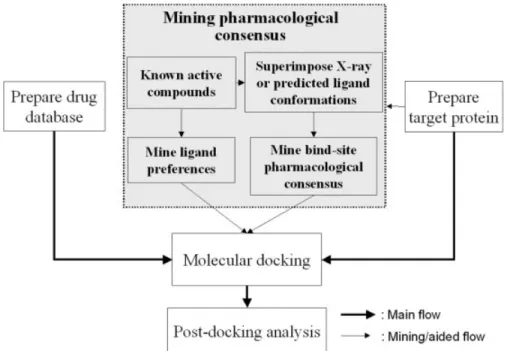
GEMDOCK was modified and enhanced from our previ-ous tool10for VS (Fig. 1). GEMDOCK can be sequentially
applied to prepare target proteins and ligand databases, predict docked conformations and binding affinity using flexible ligand docking, and rank a series of candidates for postdocking analysis. Several programs were developed separately for each phase, and Linux shell script was used to integrate these programs and automate the process. In this section, we give details of the ligand database and target protein preparations, outline the scoring function used in this study, describe details of mining binding-site pharmacological interactions (e.g., hot spots) and ligand preferences, and briefly describe the docking method. Preparations of Ligand Databases and Target Proteins
SERMs exert their physiological effects by binding to the 2 currently known estrogen receptors (ER␣ or ER), which are members of the nuclear receptor superfamily of ligand-dependent transcription factors; moreover, SERMs dis-play tissue-selective estrogen agonistic or antagonistic profiles.24 –26SERMs often beneficially affect the
cardiovas-cular and central nervous systems, and exert significant estrogenlike effects on some estrogen targets such as bone, lipid, breast, and uterine cells. Despite the benefits of SERMs, long-term treatment with SERMs is often limited by intolerable side effects, such as benign and malignant uterine lesions. Therefore, the design of new SERMs has become a challenging task.
We used the ligand data set and initial ligand conforma-tion from the comparative studies of Bissantz et al.14(e.g.,
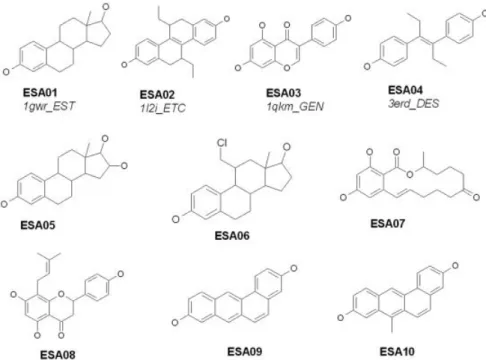
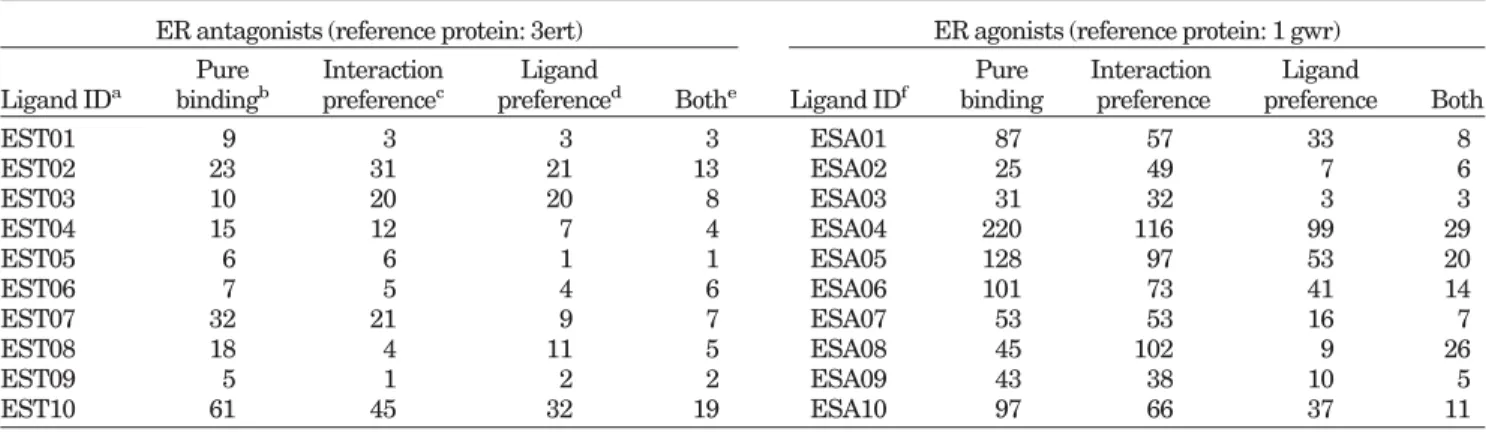
DOCK, FlexX, and GOLD) to evaluate the screening accuracy of GEMDOCK using the ER antagonists. The ligand data set included the 10 known active compounds (EST01–EST10) listed in Figure 2 and 990 randomly chosen compounds from the Available Chemical Directory (ACD). The data set is available on the Web at http:// gemdock.life.nctu.edu.tw/dock/download.php. For screen-ing ER agonists, a set of 10 known ER agonists (Fig. 3, ESA01–ESA10) used in this study was identical to that reported earlier.27In total, the database used for
Data Bank (PDB) code: 3ert26] and ER-agonist complex
(PDB code: 1gwr28) contained 1000 molecules; that is, 990
random compounds were the same for the 2 screens. In addition, 3 ER-antagonist complexes (PDB codes: 1err, 3ert, and 1hj1) and 4 ER-agonist complexes (PDB codes: 1gwr, 1l2i, 1qkm, and 3erd) with experimentally
deter-mined X-ray structures from the PDB were selected to evaluate not only the docking accuracy but also the pharmacological consensuses evolved from known active ligands (i.e., Figs. 2 and 3) and reference proteins (Fig. 4). Each ligand from the PDB was represented systematically by 4 characters followed by 3 characters. For example, in Fig. 1. The main steps of GEMDOCK for virtual database screening, including the target protein and
compound database preparation, flexible docking, and postdocking analysis. GEMDOCK mines a pharmaco-logical consensus from the target protein and known active ligands when available.
Fig. 2. Ten known ER antagonists are studied with respect to evolving the pharmacological consensus and docking against the ER-antagonist complex. Three ligands, EST01–EST03, are obtained from the PDB and each ligand is denoted by 4 characters followed by 3 characters, as in the PDB (e.g., 3ert.OHT, “3ert” denotes the PDB code and “OHT” is the ligand name in the PDB).
the ligand “3ert.OHT,” “3ert” denotes the PDB code and “OHT” is the ligand code in the PDB. These ligand structures are shown in Figure 2 (e.g., EST01, EST02, and EST03) and Figure 3 (e.g., ESA01, ESA02, ESA03, and ESA04).
The antagonist complex (PDB code: 3ert) and ER-agonist complex (PDB code: 1gwr) were selected as reference proteins for virtual screening. These complexes were reason-able choices, because their ligand-binding cavities are wide enough to accommodate a broad variety of ligands and therefore did not require binding-site modifications. As shown in Figure 4, the structures of these 2 reference proteins complexed with tamoxifen (3ert) or estradiol (1gwr) show that both ligands bind at the same site within the core of the ligand-binding domain and that each ligand induces a differ-ent conformation of helix 12 (H12). Comparison of the structures of these 2 complexes reveals that the H12 (blue) sits above the ligand-binding cavity in the ER-agonist com-plex (1gwr), thereby forming a lid. In contrast, the side-chains of antagonists (e.g., tamoxifen and raloxifene) in the ER-antagonist complexes prevent the agonistlike induced conformational change of H12 (green), projecting out of the ligand-binding pocket. When preparing the size and location of the ligand-binding site, we considered the protein atoms located less than 10 Å from each ligand atom. The metal atoms were retained, and all structured water molecules were removed from the active site. GEMDOCK then as-signed a formal charge and atom type for each protein atom based on our previous study.10
Scoring Function
We developed a new scoring function that simulta-neously serves as the scoring function for both molecular
docking and the ranking of screened compounds for post-docking analysis. This function consists of a simple empiri-cal binding score and a pharmacophore-based score to reduce the number of false positives. The energy function can be dissected into the following terms:
Etot⫽ Ebind⫹ Epharma⫹ Eligpre, (1)
where Ebindis the empirical binding energy, Epharmais the energy of binding site pharmacophores (hot spots), and
Eligpre is a penalty value if a ligand does not satisfy the ligand preferences. Epharmaand Eligpre(see subsection on mining pharmacological consensuses) are especially useful in selecting active compounds from hundreds of thousands of nonactive compounds by excluding ligands that violate the characteristics of known active ligands, thereby improv-ing the number of true positives. The values of Epharmaand
Eligpre are determined according to the pharmacological
consensus derived from known active compounds and the target protein. In contrast, the values of Epharma and
Eligpreare set to zero if active compounds are not available. The empirical-binding energy (Ebind) is given as
Ebind⫽ Einter⫹ Eintra⫹ Epenal, (2)
where Einterand Eintraare the intermolecular and intramo-lecular energies, respectively, and Epenalis a large penalty value if the ligand is out of the range of the search box. For our present work, Epenalwas set to 10,000. The intermolecu-lar energy is defined as
Einter⫽
冘
i⫽1 lig冘
j⫽1 pro冋
F共rij Bij兲 ⫹ 332.0qiqj 4rij 2册
, (3)Fig. 3. Ten known ER agonists are docked against the ER-agonist complex (PDB code: 1gwr), and the pharmacological consensus is evolved. Four ligands, ESA01–ESA04, are obtained from the PDB, and each ligand is represented by 4 characters followed by 3 characters in the PDB.
where rijis the distance between the atoms i and j, qiand qj are the formal charges, and 332.0 is a factor that converts the electrostatic energy into kilocalories per mole. The lig and pro denote the numbers of the heavy atoms in the ligand and receptor, respectively. F(rij
Bij) is a simple atomic pairwise potential function (Fig. 5), as defined in our previous study,10where r
ij
Bijis the distance between atoms
i and j with interaction type Bijformed by pairwise heavy atoms between ligands and proteins, and Bij is either a hydrogen bond or a steric state. In this atomic pairwise model, these 2 potentials are calculated by the same function form but different parameters, V1,…, V6, given in
Figure 5. The energy value of a hydrogen bonding should be larger than that for steric potential. In this model, atoms are divided into 4 different atom types10: donor,
acceptor, both, and nonpolar. A hydrogen bond can be formed by the following pair-atom types: donor–acceptor (or acceptor– donor), donor– both (or both– donor), acceptor– both (or both–acceptor), and both– both. Other pair-atom combinations are used to form the steric state. We used the atom formal charge to calculate the electrostatic energy,10
which is set to 5 or⫺5, respectively, if the electrostatic energy is more than 5 or less than⫺5. These parameters,
V1 to V6, and the maximum electrostatic energy were
refined according to the docking accuracies of our previous work10on a highly diverse data set of 100 protein–ligand
complexes proposed by Jones et al.6
The intramolecular energy of a ligand is
Eintra⫽
冘
i⫽1 lig冘
j⫽i⫹2 lig冋
F共rij Bij兲 ⫹ 332.0qiqj 4rij 2册
⫹冘
k⫽1 dihed A关1 ⫺ cos共mk⫺ 0兲兴, (4)where F(rijBij) is defined as for Eq. (3) except the value is set to 1000 when rijBij ⬍ 2.0 Å, and dihed is the number of rotatable bonds in a ligand. We followed the work of Gehlhaar et al.13to set the values of A, m, and
0. For the
sp3O sp3bond, A⫽ 3.0, m ⫽ 3, and
0⫽ ; for the sp 3O sp2
bond, A⫽ 1.5, m ⫽ 6, and 0⫽ 0.
Fig. 4. Comparing the binding sites of the ER reference proteins by superimposing the complexes of the ER agonists (yellow; PDB code: 1gwr) and ER antagonists (blue; PDB code: 3ert). The bound ligands (estradiol and tamoxifen) are shown in red. In the ER-agonist complex, helix 12 (H12) (blue) sits above the ligand-binding cavity, forming a lid. H12 in the ER-antagonist complex protrudes from the pocket.
Fig. 8. Docking energy is influenced by ligand structures generated by CORINA. (a) The fraction of polar atoms in ESA01-C is the smallest among these 3 ligands, whereas that of ESA01-COO is the largest. (b) The docked positions are similar, but the docking energies differ: ⫺91.32 for ESA01, ⫺76.86 for ESA01-C, and ⫺99.64 for ESA01-COO.
Fig. 9. The influence of molecular weight on docking energy. (a) ESA01 (blue) and EST03 (yellow) have a common group A, and EST03 has an additional substructure group B. (b) The docked conformations (into reference protein 3ert) are similar, and the docking energies are ⫺82.82 for ESA01 and ⫺127.27 for EST03.
Mining Pharmacological Consensuses
GEMDOCK evolves the binding-site pharmacological consensus and ligand preferences from both known active ligands and the target protein to improve screening accu-racy. We used the premise that previously acquired inter-actions (hot spots) between ligands and the target protein can be used to guide the selection of lead compounds for subsequent investigation and refinement. When known active ligands were available, GEMDOCK used a pharma-cophore-based scoring function [Eq. (1)]. On the other
hand, LPelec and LPhbwere set to zero, and GEMDOCK used a purely empirical-based scoring function [Eq. (2)] if known active compounds were not available.
For each known active ligand, GEMDOCK first yielded 5 docked ligand conformations by docking the ligand into the target protein, and only the docked ligand conformation with the lowest energy was retained for pharmacological consensus analysis. The protein–ligand interactions were extracted by overlapping these lowest energy docked con-formations, and the interactions were classified into 2 Fig. 5. The linear energy function of pairwise atoms for steric interactions (light line), hydrogen bonds (bold
line), and electrostatic potential in GEMDOCK.
Fig. 6. The binding-site pharmacological consensuses are identified by overlapping the docked conforma-tions of (a) 10 known ER antagonists and (b) 10 known ER agonists against the reference proteins 3ert and 1gwr, respectively. (a) Four pharmacological interactions were identified and circled as A (phenolic hydroxyl group), B (phenolic hydroxyl group), and C (piperidine nitrogen). (b) Three pharmacological interactions were identified and circled as A⬘ (phenolic hydroxyl group) and B⬘ (phenolic hydroxyl group). The dashed lines indicate the hydrogen bonds formed between the ligand and the target protein. These pharmacological interactions are consistent with those evolved from X-ray structures.
different types, including bonding and hydrogen-charged interactions. After all of the protein–ligand inter-actions were calculated, and the atom interaction-profile weight of the target protein representing the pharmacolog-ical consensus of a particular interaction was given as
Qjk⫽
fjk
N, (5)
where N is the number of known active compounds and fj k is the total interaction number of an atom j (in a protein) interacting with an atom of known active ligands with the interaction type k (e.g., bonding or hydrogen-charged interactions). In this work, an atom j (in a protein) was considered to interact with an atom i (in a ligand) if the distance between the atoms j and i ranges from (V1⫹
V2)/2 to (V3⫹ V4)/2, where V1,…, V4are given in Figure 5.
An atom j in the reference protein was considered a hot-spot atom when Qjkwas more than 0.5.
The pharmacophore-based interaction energy (Epharma) between the ligand and the protein is calculated by summing the binding energies of all hot spot atoms:
Epharma⫽
冘
i⫽1 lig冘
j⫽1 hs CW共Bij兲F共rij Bij兲, (6)where CW(Bij) is a pharmacological weight function of a hot spot atom j with interaction type Bij, F(rijBij) is defined as in Eq. (3), lig is the number of heavy atoms in a screened ligand, and hs is the number of hot spot atoms in the protein. The CW(Bij) is given as
CW共Bij兲 ⫽
再
1.0 if Qjkⱕ 0.5 or Bij⫽ k 1.5⫹ 5共Qj k⫺ 0.5兲 if Q j k⬎ 0.5 and B ij⫽ k . (7) Qjkis the atomic pharmacological profile weight [Eq. (5)] and k is the interaction type of the hot spot atom j.
We evolved the ligand preferences (Eligpre) from known ligands to reduce the deleterious effects of screening ligand structures that are rich in charged or polar atoms. Docking methods using energy-based scoring functions are often biased toward such compounds, which abound with charged and polar atoms (i.e., hydrogen donor or acceptor atoms) because the pair-atom potential of the electrostatic energy and hydrogen bonding energy is always larger than the steric energy. For example, the atomic pairwise potential energies of the electrostatic, hydrogen bond, and steric potential were set to⫺5, ⫺2.5, and ⫺0.4 in this work. The ligand preference (Eligpre) is a penalty value for those screened ligands that violate the electrostatic or hydro-philic constraints. The Eligpreis given as
Eligpre⫽ LPelec⫹ LPhb, (8)
where LPelec and LPhb are the penalties for the electro-static (i.e., the number of charged atoms of a screened ligand) and hydrophilic (i.e., the fraction of polar atoms in a screened ligand) constraints, respectively. LPelec is de-fined as
LPelec⫽
再
10NAelec if NAelec⬎ UBelec
0 if NAelecⱕ UBelec , (9) where UBelec⫽ elec⫹ elec.
NAelecis the number of charged atoms of a screened ligand and UBelecis the upper bound number of charged atoms derived from known active compounds.elec is the maxi-mum number of charged atoms among known active compounds, and elec is the standard deviation of the charged atoms of known active compounds. LPhbis defined as LPhb⫽
再
5NAhb if rhb⬎ Urhb 0 if rhbⱕ Urhb , (10) where rhb⫽ NAhb NAt and Urhb⫽ hb⫹ hb.rhbis the fraction of polar atoms (i.e., the atom type is both, donor, or acceptor) in a screened ligand, and Urhb is the upper bound of the fraction of polar atoms calculated from known active ligands. NAhb and NAtare the number of polar atoms and the total number of the heavy atoms of a screened ligand, respectively.hb andhb are the maxi-mum ratio and the standard deviation of the ratios of polar atoms evolved from known ligands, respectively.
In order to reduce the deleterious effects of biasing toward the selection of high molecular weight compounds, we formulate a normalization strategy defined as
EbindMW ⫽ Ebind 共NAt兲K , where K ⫽
冦
0.5 ifmwⱕ 15 0.5⫺0.45共mw⫺ 15兲 25 if 15⬍ mwⱕ 40 0.05 ifmw⬎ 40 , (11)where Ebindis the empirical binding energy [Eq. (2)], NAt is the total number of the heavy atoms in a screened ligand, andmwis the mean of the number of heavy atoms in known active compounds. When the normalization strategy is applied, the energy function [Eq. (1)] is given as
Etot⫽ Ebind
MW⫹ E
pharma⫹ Eligpre. (12)
Flexible Docking Algorithm
Here, we present the outline of our molecular docking method that is a generic evolutionary method enhanced from our original technique.10The core idea of our
evolu-tionary approach was to design multiple operators that cooperate using the family competition model, which is similar to a local search procedure. The rotamer-based mutation operator, a discrete operator, is used to reduce the search space of ligand structure conformations. The Gaussian and Cauchy mutations, continuous genetic opera-tors, search the orientation and conformation of the ligand relating to the center of the target protein.
After the ligand database and the target protein were prepared and the pharmacological preferences were evolved, we first specified the crystal coordinates of the protein atoms from the PDB and assigned a formal charge SELECTIVE ESTROGEN RECEPTOR MODULATORS
and atom type for each protein atom. GEMDOCK then automatically decides the search cube of a binding site based on the maximum and minimum values of coordi-nates among these selected protein atoms. For each ligand in the database, GEMDOCK takes the atomic coordinates from the ligand database and assigns a formal charge and atom type for each atom. It then sequentially predicts the binding conformation and estimates the binding affinity for each ligand. Finally, GEMDOCK ranks these docked ligand conformations for use in the postdocking analysis.
Our docking method works as follows: It randomly generates a starting population with N docked structures by initializing the orientation and conformation of the ligand relating to the center of the target protein. Each solution is represented as a set of 3 n-dimensional vectors (xi,i,i), where n is the number of adjustable variables of a docking system and i⫽ 1,…,N, where N is the population size. The vector x is the adjustable variables representing a particular orientation and conformation space of a ligand to be optimized, in which x1, x2, and x3 are the
three-dimensional (3D) location of the ligand relating to the center of the target protein; x4, x5, and x6are the rotational
angles of the ligand relating to axes; and x7to xnare the twisting angles of the rotatable bonds inside the ligand. and are the step-size vectors of decreasing-based Gauss-ian mutation and self-adaptive Cauchy mutation. In other words, each solution x is associated with some parameters for step-size control. The initial values of x1, x2, and x3are
randomly chosen from the feasible box, and the others, from x4to xn, are randomly chosen from 0 to 2 in radians. The initial step size is 0.8 and is 0.2. After GEMDOCK initializes the solutions, it enters the main evolutionary loop, which consists of 2 stages in every iteration: decreas-ing-based Gaussian mutation and self-adaptive Cauchy mutation. Each stage is realized by generating a new quasi-population (with N solutions) as the parent of the next stage. These stages apply a general procedure “FC-_adaptive” with only different working population and the mutation operator.
The FC_adaptive procedure employs 2 parameters, namely, the working population (P, with N solutions) and mutation operator (M), to generate a new quasi-popula-tion. The main work of FC_adaptive is to produce offspring and then conduct the family competition. Each individual in the population sequentially becomes the “family father.” With a probability pc, this family father and another solution that is randomly chosen from the rest of the parent population are used as parents for a recombination operation. Then the new offspring or the family father (if the recombination is not conducted) is operated by the rotamer mutation or by differential evolution to generate a quasi-offspring. Finally, the working mutation is operates on the quasi-offspring to generate a new offspring. For each family father, such a procedure is repeated L times, called the family competition length. Among these L offspring and the family father, only the one with the lowest scoring function value survives. Since we create L children from one “family father” and perform a selection, this is a family competition strategy. This method avoids
the population prematureness but also keeps the spirit of local searches. Finally, the FC_adaptive procedure gener-ates N solutions because it forces each solution of the working population to have one final offspring. In the following, genetic operators are briefly described. We use
a⫽ (xa,a,a) to represent the “family father” and b⫽ (xb, b,b) as another parent. The offspring of each operation is represented as c⫽ (xc,c,c). The symbol x
j
sis used to denote the jth adjustable optimization variable of a solu-tion s,@j 僆 {1,…, n}.
Recombination operators
GEMDOCK implemented modified discrete recombina-tion and intermediate recombinarecombina-tion. A recombinarecombina-tion operator selected the “family father (a)” and another solution (b) randomly selected from the working popula-tion. The former generates a child as follows:
xjc⫽
再
xj
awith probability 0.8
xjbwith probability 0.2 .
The generated child inherits genes from the “family father” with a higher probability 0.8. Intermediate recom-bination works as
wjc⫽ wja⫹ 共wjb⫺ wja兲/2,
where w is or based on the mutation operator applied in the FC_adaptive procedure. The intermediate recombi-nation only operated on step-size vectors and the modified discrete recombination was used for adjustable vectors (x).
Mutation operators
After the recombination, a mutation operator, the main operator of GEMDOCK, is applied to mutate adjustable variables (x). Gaussian and Cauchy Mutations are accom-plished by first mutating the step size (w) and then mutating the adjustable variable x:
w⬘j⫽ w⬘jA
x⬘j⫽ xj⫹ w⬘jD,
where wj and xj are the ith component of w and x, respectively, and wj is the respective step size of the xj, where w is or . A( 䡠 ) is evaluated as exp[⬘N(0, 1) ⫹ Nj(0, 1)] if the mutation is a self-adaptive mutation, where N(0, 1) is the standard normal distribution, Nj(0, 1) is a new value with distribution N(0, 1) that must be regenerated for each index j. When the mutation is a decreasing-based mutation A(䡠 ) is defined as a fixed decreasing rate ␥ ⫽ 0.95. D(䡠 ) is evaluated as N(0, 1) or C(1) if the mutation is, respectively, Gaussian or Cauchy. For example, the self-adaptive Cauchy mutation is defined as
jc⫽ jaexp关⬘N共0, 1兲 ⫹ Nj共0, 1兲兴,
xjc⫽ xja⫹ jcCj共t兲.
We set and ⬘ to (公2n)⫺1and (公2公2n)⫺1, respectively, according to the suggestion of evolution strategies. A random variable is said to have the Cauchy distribution [C(t)] if it has the density function: f( y; t)⫽ (t/)/(t2⫹ y2),
decreasing-based Gaussian mutation uses the step-size vector with a fixed decreasing rate␥ ⫽ 0.95 and works as c⫽ ␥aand
xj c⫽ x
j a⫹ cN
j(0, 1).
Our rotamer mutation is only used for x7to xnto find the conformations of the rotatable bonds inside the ligand. For each ligand, this operator mutates all of the rotatable angles according to the rotamer distribution and works as
xj⫽ ␥kiwith probability pki, where␥kiand pkiare the angle value and the probability, respectively, of ith rotamer of
kth bond type including sp3Osp3and sp3Osp2bond. The
values of␥kiand pkiare based on the energy distributions of these 2 bond types.
RESULTS AND DISCUSSION Parameters of GEMDOCK
In our studies, GEMDOCK parameters in the flexible search phase included the initial step sizes ( ⫽ 0.8 and ⫽ 0.2), family competition length (L ⫽ 2), population size (N⫽ 200), and recombination probability (pc⫽ 0.3). For each ligand screened, GEMDOCK optimization stopped either when the convergence was below a certain threshold value or the iterations exceeded the maximal preset value of 60. Therefore, GEMDOCK generated 800 solutions in one generation and terminated after it exhausted 48,000 solutions for each docked ligand. The average GEMDOCK docking run took 135 s using a Pentium 1.4-GHz personal computer with a single processor.
Mining the Pharmacological Consensus
Figure 6 and Table I show the pharmacological interac-tion preferences (hot-spot atoms), and Table II shows the ligand preferences. We evolved these pharmacological consensuses and steric binding interactions by overlap-ping the docked ligand conformations, yielded by GEM-DOCK, of all known active compounds. Figure 6(a and b)
shows the overlap of 10 docked poses of 10 known active ligands in the vicinity of the ER-antagonist target protein and ER-agonist target protein, respectively. The dashed lines indicate the hydrogen bonds formed between the ligand and the reference proteins. For the ER-antagonist target protein, 4 binding-site pharmacological interactions were identified and circled as A (hydroxyl group26,29 –32), B
(hydroxyl group26,29 –31), and C (dimethylamino group26,31
or piperidine nitrogen29,30). These interactions, evolved
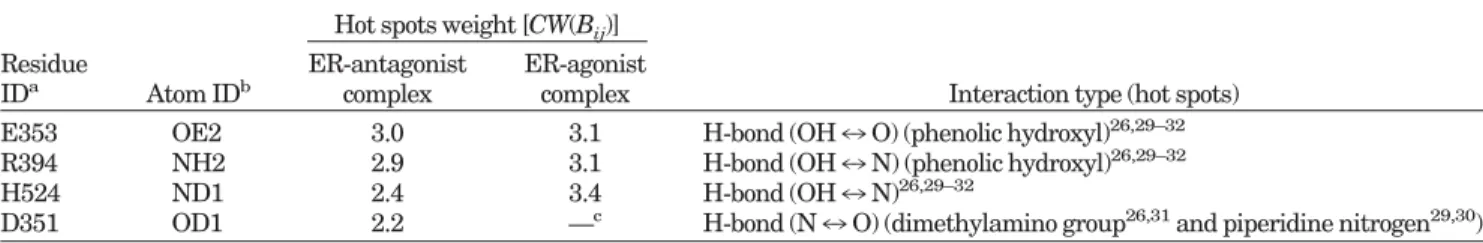
from docked conformations, are consistent with the inter-actions evolved from superimposing 3 X-ray structures with that from related studies.26,29 –31As shown in Table I,
the pharmacological weights [CW(Bij) defined in Eq. (7)] and the interaction type for the ER-antagonist complex included E353-OE2 (3.0), R394-NH2 (2.9), H524-ND1 (2.4), and D351-OD1 (2.4). For the ER-agonist target protein, 2 binding-site pharmacological interactions were identified (e.g., A⬘ hydroxyl group and B⬘ hydroxyl group). The pharmacological weights and the interaction type for the ER-agonist complex included E353-OE2 (3.1), R394-NH2 (3.1), and H524-ND1 (3.4). These interactions are also consistent with those evolved by superimposing 4 X-ray structures [Fig. 6(b)].
For screening ER antagonists and agonists, Table II shows the parameter values of ligand preferences evolved from known ER antagonists (Fig. 2) and agonists (Fig. 3). These ligand preferences improve the screening accuracy by reducing the deleterious effects of ligand molecular weights and ligand structures that are rich in charged or polar atoms. The electrostatic parameter values [see Eq. (9)] for ER antagonists included the maximum number of charged atoms (elec ⫽ 2.0), standard deviation of the charged atoms (elec⫽ 0.63), and upper bound number of charged atoms (UBelec⫽ 2.63). For the hydrophilic prefer-ences [see Eq. (10)], the maximum ratio (hb) was 0.15, the
TABLE I. Pharmacological Weights of Hot Spot Atoms of the ER-Antagonist and ER-Agonist Complexes Evolved by Overlapping Docked Conformations of Known Active Ligands
Residue
IDa Atom IDb
Hot spots weight [CW(Bij)]
Interaction type (hot spots) ER-antagonist
complex
ER-agonist complex
E353 OE2 3.0 3.1 H-bond (OH7 O) (phenolic hydroxyl)26,29–32 R394 NH2 2.9 3.1 H-bond (OH7 N) (phenolic hydroxyl)26,29–32
H524 ND1 2.4 3.4 H-bond (OH7 N)26,29–32
D351 OD1 2.2 —c H-bond (N7 O) (dimethylamino group26,31and piperidine nitrogen29,30)
a
One-letter amino acid code, with the residue sequence numbered as in the PDB.
bAtom name in the PDB. c
D351-OD1 is not a hot spot atom in the ER-agonist reference complex.
TABLE II. Ligand Preferences Evolved from Known Active Ligands Screen Lead Compounds for the ER-Antagonist and ER-Agonist Complexes
Ligand name Electrostatic preferences [Eq. (9)] Hydrophilic preferences [Eq. (10)] Molecular weight [Eq. (11)] elec elec UBelec hb hb Urhb mw K
ER antagonist 2.0 0.63 2.63 0.15 0.02 0.17 34.0 0.16
ER agonist 0 0 0 0.25 0.06 0.31 21.4 0.38
standard deviation (hb) of the ratios was 0.02, and the upper bound ratio (Urhb) of polar atoms was 0.17. For molecular weight [see Eq. (11)], the mean of heavy atoms (mw) was 21.6 and linear normalization parameter K was 0.16. In contrast, for ER agonists the values of UBelecand
Urhbwere 0 and 0.31, respectively, and K was 0.38. Evaluation of Virtual Screening Accuracy
Some common factors were used to evaluate the screen-ing quality, includscreen-ing coverage (the percentage of active ligands retrieved from the database), yield (the percentage of active ligands in the hit list), false-positive (FP) rate, enrichment, and goodness-of-hit (GH). The coverage (true positive rate) is defined as Ah/A (%),Ah/Th(%) is the yield (hit rate), and the FP rate is defined as (Th⫺ Ah)/(T⫺ A) (%). The enrichment is defined as (Ah/Th)/(A/T). Ahis the number of active ligands among the Th highest ranking compounds, which is called the hit list, A is the total number of active ligands in the database, and T is the total number of compounds in the database. The GH score is defined as33
GH⫽
冉
Ah共3A ⫹ Th兲4ThA
冊冉
1⫺Th⫺ Ah
T⫺ A
冊
. (13)The GH score contains a coefficient to penalize excessive hit list size and, when evaluating hit lists, is calibrated by weighting the score with respect to the yield and coverage. The GH score ranges from 0.0 to 1.0, where 1.0 represents a perfect hit list (i.e., containing all of, and only, the active ligands). In the data sets for screening the ER agonists or ER antagonists, A and T are 10 and 1000, respectively. Here, we also took the averages of hit rates, enrichments, GH scores, and FP rates. For example, the averages of the hit rates and enrichments are defined as (¥iA⫽1i/Th
i)/A and {¥iA⫽1(i/Th
i)/( A/T)}/A, respectively, where T h
i is the number of compounds in a hit list containing i active compounds.
Molecular Recognition of Antagonist and ER-Agonist Complexes
We tested GEMDOCK10on a highly diverse data set of
100 protein–ligand complexes proposed by Jones et al.6
and on 2 cross-docking ensembles of protein structures. Upon consideration of the solutions at the first rank, in 79% of these complexes, the docked lowest energy ligand structures had root-mean-square deviations (RMSDs) be-low 2.0 Å with respect to the corresponding crystal struc-tures. The success rate increased to 85% if the structured water molecules were retained. In contrast, GOLD6yielded
a 71% success rate in identifying the experimental binding model based on the GOLD assessment categories, and the rate was 66% if based on the top-ranked solutions with RMSD values of less than 2 Å. FlexX7achieved 70% and
46.5% success rates for solutions at any rank and the first rank, respectively.
The main objective of this study was to evaluate whether the new scoring function was applicable to both molecular docking and ligand scoring during VS. First, GEMDOCK was evaluated by docking each ligand of 7 ER complexes in the PDB into its respective complex and into its reference protein. Table III shows the overall predicted accuracies of GEMDOCK and GOLD. Ten independent docking runs were performed for each active compound, and the docked ligand conformation with the lowest energy was used to calculate RMSD values for ligand heavy atoms between the docked conformation and the crystal structure. The RMSD values of 7 docked conformations (docking each ligand back into its respective complex) were less than 2.0 Å. When these ligands were docked into the reference protein using GEMDOCK, all docked conformations had an RMSD of less than 2.0 Å except for EST03 and ESA03 (genistein). EST03 docked well in the binding site, with the exception of the long acyclic side-chain. The agonist ESA03 could not be docked into its corresponding pose in the reference protein (1gwr) due to a fundamental
differ-TABLE III. Comparing GEMDOCK and GOLD With Respect to Docking 7 Ligands Back Into Respective Complexes and Reference Proteins
Ligand ID GEMDOCK GOLD Native proteinb Reference proteinc
Native proteinb Reference proteinc Etotd E bind d E tot Ebind EST01 (1err.RALa) 0.66 0.65 1.37 1.36 1.02 1.68 EST02 (3ert.OHT) 0.60 0.75 0.60 0.75 1.15 1.15 EST03 (1hj1.AOE) 1.41 1.05 3.27 3.35 5.07 3.92 ESA01 (1gwr.EST) 0.66 0.64 0.66 0.64 0.54 0.54 ESA02 (1l2i_ETC) 0.61 0.48 0.62 0.69 0.55 0.76 ESA03 (1qkm.GEN) 0.69 1.53 3.32 4.83 0.24 7.16 ESA04 (3erd.DES) 0.67 0.51 1.44 1.43 1.10 1.76 a
Four characters followed by 3 characters (separated by a period) denote the PDB code and the ligand name in the PDB, respectively.
b
The RMSD value for docking each ligand back into its respective complex.
cThe RMSD value for docking each ligand into its reference complex, 3ert for ER antagonists (e.g., EST01⬃ EST03) and 1gwr
for ER agonists (e.g., ESA01⬃ ESA04).
dE
ence between the binding site of ER␣ (1gwr) and ER (1qkm). As shown in Table III, GEMDOCK and GOLD yielded results of equal quality, and GEMDOCK yielded similar results regardless of whether the pharmacological preferences (i.e., Epharmaand Eligpre) were considered. Virtual Screening of ER Antagonists and ER Agonists
We compared the overall accuracy of GEMDOCK using 4 variations of energy terms to screen ER antagonists and agonists from a data set of 1000 compounds proposed by Bissantz et al.14 (Fig. 7 and Table IV). Each variation
combined 3 scoring terms applied in GEMDOCK: binding energy (Ebind), pharmacological interaction preferences (Epharma), and ligand preferences (Eligpre). For example, the approach “Pure binding” used only the binding energy (Ebind) as the scoring function; the approach “Interaction preference” integrated Ebind and Epharma for the scoring
function; “Ligand preference” integrated Ebindand Eligpre for the scoring function; and “Both” integrated Ebind,
Eligpre, and Epharmafor the scoring function. The parame-ter values for inparame-teraction preferences (Epharma) and ligand preferences (Eligpre) are shown in Tables I and II, respec-tively. The various ranks of 10 known active ligands in the ligand screening database are shown in Table V, and the comparison of results obtained with other methods is shown in Table VI.
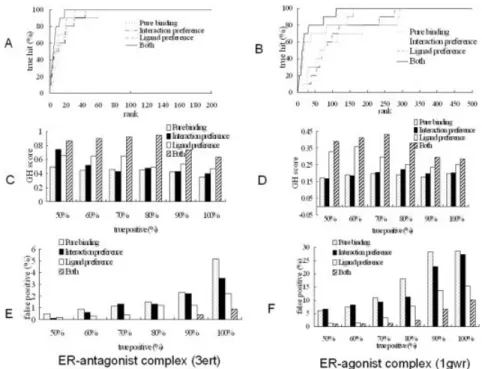
As shown in Table IV and Figure 7, GEMDOCK gener-ally improves the screening quality when both interaction preferences and ligand preferences are considered. The latter was more important than the former for this data set. For the ER antagonists that were screened, average hit rates were 92.19% (Both), 71.58% (Ligand preference), 57.93% (Interaction preference), and 34.8% (Ebind). The average GH scores were 0.83 (Both), 0.67 (Ligand prefer-ence), 0.57 (Interaction preferprefer-ence), and 0.39 (Ebind). Fig-Fig. 7. GEMDOCK screening accuracies of ER antagonists and ER agonists assessed by (A and B) true
hits, (C and D) GH scores, and (E and F) the false-positive rates against different true-positive rates ranging from 50% to 100%. The performance of GEMDOCK was consistently superior when using both ligand preferences and pharmacological-interaction preferences.
TABLE IV. GEMDOCK Screening Accuracies Using Different Combinations of Pharmacological Preferences on the Data Set Proposed by Bissantz et al.14
Measure factor
ER antagonists (reference protein: 3ert) ER agonists (reference protein: 1gwr) Pure bindinga Interaction preferenceb Ligand preferencec Bothd Pure bindinga Interaction preferenceb Ligand preferencec Bothd Average hit rate (%) 34.88 57.93 71.58 92.19 6.94 7.52 25.02 45.66
Average enrichment 34.88 57.93 71.58 92.19 6.94 7.52 25.02 45.66 Average false-positive rate (%) 1.32 0.94 0.56 0.13 7.83 6.34 2.56 0.75 Average GH score 0.39 0.57 0.67 0.83 0.17 0.18 0.32 0.48 a,b,c,d
Using Ebind, Ebind⫹ Epharma, Ebind⫹ Eligpre, and Etot, respectively, for the scoring function. These energy terms are defined in Eq. (1). SELECTIVE ESTROGEN RECEPTOR MODULATORS
ure 7(C and E) shows that the GH scores and FP rates of the true positive rates ranged from 50% to 100%. For the ER agonists that were screened, average hit rates were 45.66% (Both), 25.02% (Ligand preference), 7.52% (Interac-tion preference), and 6.94% (Ebind). The average GH scores were 0.48 (Both), 0.32 (Ligand preference), 0.18 (Interac-tion preference), and 0.17 (Ebind). Figure 7(D and F) shows the GH scores and FP rates with different true positive rates ranging from 50% to 100%.
The screening accuracy of GEMDOCK for ER antago-nists was better than that of ER agoantago-nists on this data set. These results might be caused by using the same 990 random compounds proposed by Bissantz et al.14for these
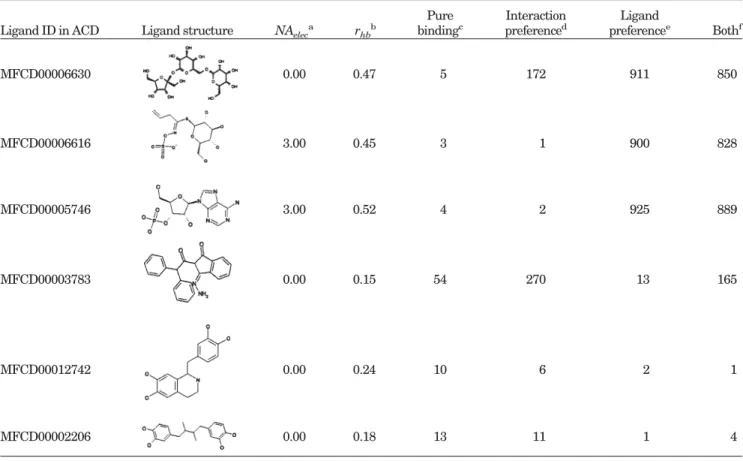
2 screens. When they prepared the random ligand set, only the chemical reagents of the ER-antagonist complex were eliminated and therefore the ER-agonist-like compounds might be selected. For example, GEMDOCK screened two ligands, MFCD00012742 and MFCD00002206 (Table VII), which are similar in structures to ESA03 and ESA04 (Fig. 4), respectively. At the same time, the numbers of the ligands that violate the ligand preferences (e.g., LPelecand
LPhbshown in Table II) of ER antagonists and ER agonists are 400 and 289 compounds, respectively. The MFCD compounds were the random ligands in the data set.
GEMDOCK was superior to other approaches (Surflex, DOCK, FlexX, and GOLD) for screening the ER antago-nists (Table VI). All of these methods were tested using the same reference protein and screening database with true-positive rates ranging from 80% to 100%. When the true
positive rate was 90%, the FP rates were 2.3% DOCK without pharmacological preferences), 0.4% (GEM-DOCK with pharmacological preferences), 1.6% (Surflex), 17.4% (DOCK), 70.9% (FlexX), and 8.3% (GOLD).
The Influences of Pharmacological Preferences When using interaction energy scoring alone for choos-ing ligands, dockchoos-ing methods (e.g., GEMDOCK and GOLD) favor the selection of not only highly charged polar com-pounds but also high molecular weight comcom-pounds. Fig-ures 8 and 9 show the influences of the ligand structFig-ures and molecular weight, respectively, when the binding scoring (Ebind) alone was used in GEMDOCK. The docking energy of a ligand with charged or polar atoms is often lower than the energy of a noncharged ligand when the docked conformations are similar. For example, the dock-ing energies are⫺76.86 for ESA01-C (rhbis the smallest), ⫺91.32 for ESA01, and ⫺99.64 for ESA01-COO (with charged atoms, and rhb is the largest) when the docked positions of these ligands are similar (Fig. 8). At the same time, ESA01 and ESA01-COO form the pharmacological interactions shown in Figure 6(B) (e.g., A⬘ phenolic hy-droxyl group and B⬘ phenolic hydroxyl group). In contrast, ESA01-C has no polar atoms to form these pharmacologi-cal interactions. We obtained these ligand structures (EAS01-C and ESA01-COO) using the 3D structure genera-tor CORINA.34
Tables VII and VIII show the effect of pharmacological preferences of some typical ligand structures on screened
TABLE V. Ranks of 10 Known ER Antagonists and 10 Known ER Agonists Using GEMDOCK With Different Combinations of Pharmacological Preferences on the Data Set Proposed by Bissantz et al.14
ER antagonists (reference protein: 3ert) ER agonists (reference protein: 1 gwr) Ligand IDa Pure bindingb Interaction preferencec Ligand
preferenced Bothe Ligand IDf
Pure binding Interaction preference Ligand preference Both EST01 9 3 3 3 ESA01 87 57 33 8 EST02 23 31 21 13 ESA02 25 49 7 6 EST03 10 20 20 8 ESA03 31 32 3 3 EST04 15 12 7 4 ESA04 220 116 99 29 EST05 6 6 1 1 ESA05 128 97 53 20 EST06 7 5 4 6 ESA06 101 73 41 14 EST07 32 21 9 7 ESA07 53 53 16 7 EST08 18 4 11 5 ESA08 45 102 9 26 EST09 5 1 2 2 ESA09 43 38 10 5 EST10 61 45 32 19 ESA10 97 66 37 11 a
Defined in Figures 2 and 3, respectively.
b,c,d,eUsing E
bind, Ebind⫹ Epharma, Ebind⫹ Eligpre, and Etot, respectively, for the scoring function. These energy terms are defined in Eq. (1).
TABLE VI. Comparing GEMDOCK With Other Methods on Screening the ER Antagonists by False-Positive Rates (%) on the Data Set Proposed by Bissantz et al.14
True positive (%) GEMDOCKa GEMDOCKb Surflexc DOCKc FlexXc GOLDc
80 1.5 (15/990)d 0.0 (0/990) 1.3 13.3 57.8 5.3
90 2.3 (23/990) 0.4 (4/990) 1.6 17.4 70.9 8.3
100 5.2 (51/990) 0.9 (9/990) 2.9 18.9 —e 23.4
aGEMDOCK without pharmacological interactions and ligand preferences (e.g., E
bindfor the scoring function). bGEMDOCK with pharmacological interactions and ligand preferences (e.g., E
totfor the scoring function). cDirectly summarized from the references.1,35
dThe false-positive rate from 990 random ligands (percentage). eFlexX could not calculate the docked solution for EST09.
ER agonists and antagonists, respectively. When the bind-ing energy (Ebind) alone was used to screen ER agonists, GEMDOCK selected 2 ligands, MFCD00012742 (first) and MFCD00002206 (fourth), which are similar in structure to ESA03 and ESA04, respectively, and satisfy the ligand preferences. Due to higher numbers of polar atoms at critical sites, these ligands formed greater numbers of pharmacological interactions compared with known active ligands. At the same time, GEMDOCK was able to exclude
ligands such as MFCD00006630 (rhb ⫽ 0.47),
MFCD00006616 (rhb ⫽ 0.45 and NAelec ⫽ 3), and MFCD00005746 (rhb⫽ 0.52 and NAelec ⫽ 3) that violate the ligand preferences of known ER agonists (Table II). For example, their rhbvalues were larger than the upper bound ratio (Urhb ⫽ 0.31) of polar atoms or the upper bound number (UBelec ⫽ 0) of charged atoms. When the penalty for the ligand preferences (Eligpre) was considered, the ranks of MFCD00006630 (911th), MFCD00006616 (900th), and MFCD00005746 (928th) lagged substantially. Ligands such as MFCD00003783 lagged (244th), since it is unable to interact with 3 important residues [Glu353, Arg394, and His524; Fig. 6(B) in the reference protein].
GEMDOCK yielded similar results when the ER antago-nists were screened (Table VIII). When the binding energy (Ebind) alone was used, the ranks of ligands
MFCD00016941 (rhb⫽ 0.35), MFCD00016787 (rhb⫽ 0.32), and MFCD00001218 (rhb⫽ 0.34) were 8th, 51st, and 13th, respectively. When both Ebind and ligand preferences (Eligpre) were considered for the scoring function, the ranks of these ligands were 661st (MFCD00016941), 747th (MFCD00016787), and 954th (MFCD00001218) since their
rhbvalues were larger than the upper bound ratio (e.g.,
Urhb⫽ 0.17 in Table II) derived from known ER antago-nists. These total scoring values were penalized by hydro-philic preferences [i.e., LPhb in Eq. (10)]. Ligand MFCD00001218 was also penalized by the electrostatic preferences [i.e., LPelecin Eq. (9)], because the number of charged atoms (NAelec ⫽ 6) was larger than the upper bound (Urelec⫽ 2.63 in Table II). The screening of ligand MFCD00010009, which has no polar atoms to form phar-macological interactions [Fig. 6(A)], often fell behind when GEMDOCK used both Ebind and Epharma for the scoring function. In contrast, ligands MFCD00002371 and MFCD00002206 yielded good ranks for various combina-tions of energy terms, since they are able to form binding-site pharmacological interactions and satisfy the ligand preferences.
Figures 9 and 10 show the effect of molecular weight on screening accuracy. A docking method using energy-based scoring alone is often biased toward large molecular
TABLE VII. GEMDOCK Ranks Using Different Combinations of Pharmacological Preferences for Some Typical Ligands on Screening ER Agonists on the Data Set Proposed by Bissantz et al.14
Ligand ID in ACD Ligand structure NAeleca rhbb
Pure bindingc Interaction preferenced Ligand preferencee Bothf MFCD00006630 0.00 0.47 5 172 911 850 MFCD00006616 3.00 0.45 3 1 900 828 MFCD00005746 3.00 0.52 4 2 925 889 MFCD00003783 0.00 0.15 54 270 13 165 MFCD00012742 0.00 0.24 10 6 2 1 MFCD00002206 0.00 0.18 13 11 1 4
aNumber of charged atoms in a screened ligand [Eq. (9)]. bThe fraction of polar atoms in a screened ligand [Eq. (10)]. c,d,e,fUsing E
bind, Ebind⫹ Epharma, Ebind⫹ Eligpre, and Etot, respectively, for the scoring function. These energy terms are defined in Eq. (1). SELECTIVE ESTROGEN RECEPTOR MODULATORS
weight ligands, because the overall van der Waals interac-tion energy is summed over all pairs of ligand and target protein atoms within a specified cutoff distance. Figure 9(a) shows that ESA01 (blue) and EST03 (yellow) have a
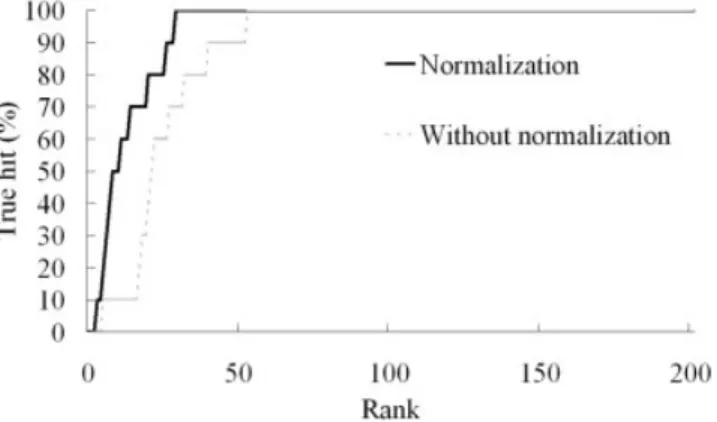
common group A, and that EST03 has an additional substructure group (side-chain B). The van der Waals force of a large ligand (e.g., EST03) is often larger than that of a small ligand (e.g., ESA01). In this case, EST03 acquires additional van der Waals force from side-chain B, as shown in Figure 9(b). For example, when using Ebindalone for docking a ligand into the reference protein (3ert), GEMDOCK yielded docking energies of⫺127.27 for EST03 and ⫺82.82 for ESA01. Figure 10 shows the true hits obtained by GEMDOCK when screening ER agonists without (dashed line) or with molecular weight normaliza-tion [solid line; defined in Eq. (11)]. When GEMDOCK applied molecular weight normalization and pharmacolog-ical preferences to screen ER agonists, the average hit rate was 45.66%, the average FP rate was 0.75%, and the GH score was 0.48. In contrast, these averages were 21.18%, 2.02%, and 0.29, respectively, when molecular weight normalization was not considered.
Figure 11 shows the true hits of GEMDOCK using the cleaned lists and the original data set proposed by Bissantz et al.14For each test case (ER antagonists and ER
ago-Fig. 10. The accuracy of GEMDOCK for screening ER agonists, assessed using scoring functions with molecular-weight normalization (solid line) and without molecular-weight normalization (dash line).
TABLE VIII. GEMDOCK Ranks Using Different Combinations of Pharmacological Preferences for Some Typical Ligands When Screening ER Antagonists on the Data Set Proposed by Bissantz et al.14
Ligand ID in ACD Ligand structure NAeleca rhbb
Pure bindingc Interaction preferenced Ligand preferencee Bothf MFCD00016941 0 0.35 8 2 661 260 MFCD00016787 0 0.32 51 8 747 319 MFCD00001218 6 0.34 13 17 954 937 MFCD00010009 0 0.00 88 430 5 57 MFCD00002371 0 0.13 40 19 16 12 MFCD00002206 0 0.18 37 30 46 20
aNumber of charged atoms in a screened ligand [Eq. (9)]. bFraction of polar atoms in a screened ligand [Eq. (10)]. c,d,e,fUsing E
nists), we prepared the cleaned list by filtering the original set in order to eliminate the ligands that violate the electrostatic (LPelec) or hydrophilic constraints (LPhb). These two cleaned lists, including the known active com-pounds, consist of 590 and 701 compounds for screening the ER antagonists and ER agonists, respectively. As shown in Figure 11, the true hits (gray lines) of GEM-DOCK using Ebind(C-Pure binding) and Ebind⫹ Epharma (C-Interaction preference) as the scoring functions on the cleaned lists are similar to those (black lines) of GEM-DOCK using Ebind ⫹ Eligpre (W-Ligand preference) and
Ebind⫹ Eligpre⫹ Epharma(W-Both) as scoring functions, on the original set, respectively. Using GEMDOCK on the cleaned sets, average GH scores were 0.82 (Interaction preference) and 0.66 (Pure binding) for ER antagonists, and average GH scores were 0.41 (Interaction preference) and 0.29 (Pure binding) for ER agonists. These experi-ments indicated that the pharmacological interaction pref-erences were able to improve the GH scores for both the cleaned lists and original set; moreover, the ligand prefer-ences might improve the screening accuracy of a scoring function and become the filters to prepare a ligand data-base.
In summary, we developed a near-automatic tool with a novel scoring function for VS by making numerous modifi-cations and enhancements to our original techniques. By integrating a number of genetic operators, each having a unique search mechanism, GEMDOCK seamlessly blends the local and global searches so that they work coopera-tively. The key aspect of the present work is that our new scoring function uses pharmacological interaction prefer-ences to select the ligand structures that form pharmaco-logical interactions with target proteins; furthermore, the scoring function applies ligand preferences to select ligand structures that are similar to known active ligands. Our scoring function is indeed able to enhance the accuracy during flexible docking and improves the screening utility by reducing the number of FPs during the postdocking analysis. Our results demonstrate the applicability and adaptability of GEMDOCK for virtual screening.
REFERENCES
1. Lyne PD. Structure-based virtual screening: an overview. Drug Discov Today 2002;7:1047–1055.
2. Shoichet BK, McGovern SL, Wei B, Irwin J. Lead discovery using molecular docking. Curr Opin Chem Biol 2002;6:439 – 446. 3. Doman TN, McGovern SL, Witherbee BJ, Kasten TP, Kurumbail
R, Stallings WC, Connolly DT, Shoichet BK. Molecular docking and high-throughput screening for novel inhibitors of protein tyrosine phosphatase-1B. J Med Chem 2002;45:2213–2221. 4. Kubinyi H. QSAR and 3-D QSAR in drug design: 1. Methodology.
Drug Discov Today 1997;2:457– 467.
5. Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A geometric approach to macromolecular-ligand interactions. J Mol Biol 1982;161:269 –288.
6. Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol 1997;267:727–748.
7. Kramer B, Rarey M, Lengauer T. Evaluation of the FlexX incremen-tal construction algorithm for protein–ligand docking. Proteins 1999;37:228 –241.
8. Ewing TJ, Makino S, Skillman AG, Kuntz ID. DOCK 4.0: search strategies for automated molecular docking of flexible molecule databases. J Comput Aided Mol Des 2001;15:411– 428.
9. Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and empirical binding free energy function. J Comput Chem 1998;19:1639 –1662.
10. Yang J-M, Chen C-C. GEMDOCK: a generic evolutionary method for molecular docking. Proteins 2004;55:288 –304.
11. Gohlke H, Hendlich M, Klebe G. Knowledge-based scoring func-tion to predict protein–ligand interacfunc-tions. J Mol Biol 2000;295: 337–356.
12. Weiner SJ, Kollman PA, Case DA, Singh UC, Ghio C, Alagona G, Profeta SJ, Weiner P. A new force field for molecular mechanical simulation of nucleic acids and proteins. J Am Chem Soc 1984;106: 765–784.
13. Gehlhaar DK, Verkhivker GM, Rejto P, Sherman CJ, Fogel DB, Fogel LJ, Freer ST. Molecular recognition of the inhibitor AG-1343 by HIV-1 protease: conformationally flexible docking by evolutionary programming. Chem Biol 1995;2:317–324.
14. Bissantz C, Folkers G, Rognan D. Protein-based virtual screening of chemical databases: 1. Evaluation of different docking/scoring combinations. J Med Chem 2000;43:4759 – 4767.
15. Stahl M, Rarey M. Detailed analysis of scoring functions for virtual screening. J Med Chem 2001;44:1035–1042.
16. Langer T, Krovat EM. Chemical feature-based pharmacophores and virtual library screening for discovery of new leads. Curr Opin Drug Discov Dev 2003;6:370 –376.
17. Fradera X, Knegtel RMA, Mestres J. Similarity-driven flexible ligand docking. Proteins 2000;40:623– 637.
18. Hindle SA, Rarey M, Buning C, Lengauer T. Flexible docking Fig. 11. The accuracy of GEMDOCK for screening (a) ER antagonists and (b) ER agonists, assessed using
the cleaned ligand sets (C-Pure binding and C-Interaction preference) and the ligand set proposed by Bissantz et al.14
(W-Ligand preference and W-Both).
under pharmacophore type constraints. J Comput Aided Mol Des 2002;16:129 –149.
19. Pegg SC-H, Haresco JJ, Kuntz ID. A genetic algorithm for structure-based de novo design. J Comput Aided Mol Des 2001;15: 911–933.
20. Muegge I, Martin YC, Hajduk PJ, Fesik SW. Evaluation of PMF scoring in docking weak ligands to the FK506 binding protein. J Med Chem 1999;42:2498 –2503.
21. Yang J-M. Development and evaluation of a generic evolutionary method for protein–ligand docking. J Comput Chem 2004;25:843– 857.
22. Good AC, Cheney DL, Sitkoff DF, Tokarski JS, Stouch TR, Bassolino DA, Krystek SR, Li Y, Mason JS, Perkins TD. Analysis and optimization of structure-based virtual screening protocols: 2. Examination of docked ligand orientation sampling methodology: mapping a pharmacophore for success. J Mol Graph Model 2003;22:31– 40.
23. Joseph-McCarthy D, Thomas BEI, Belmarsh M, Moustakas D, Alvarez JC. Pharmacophore-based molecular docking to account for ligand flexibility. Proteins 2003;51:172–188.
24. Miller CP. SERMs: evolutionary chemistry, revolutionary biology. Curr Pharm Des 2002;8:2089 –2111.
25. Dutertre M, Smith CL. Molecular mechanisms of selective estro-gen receptor modulator (SERM) action. J Pharmacol Exp Ther 2000;295:431– 437.
26. Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 1998;95:927–937.
27. van Lipzig MM, ter Laak AM, Jongejan A, Vermeulen NP, Wamelink M, Geerke D, Meerman JH. Prediction of ligand
binding affinity and orientation of xenoestrogens to the estrogen receptor by molecular dynamics simulations and the linear inter-action energy method. J Med Chem 2004;47:1018 –1030. 28. Warnmark A, Treuter E, Gustafsson JA, Hubbard RE, Brzozowski
AM, Pike AC. Interaction of transcriptional intermediary factor 2 nuclear receptor box peptides with the coactivator binding site of estrogen receptor alpha. J Biol Chem 2002;277:21862–21868. 29. Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T,
Engstroem O, Oehman L, Greene GL, Gustafsson JA, Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 1997;389:753–758.
30. Renaud J, Bischoff SF, Buhl T, Floersheim P, Fournier B, Halleux C, Kallen J, Keller H, Schlaeppi JM, Stark W. Estrogen receptor modulators: identification and structure-activity relationships of potent ER-alpha-selective tetrahydroisoquinoline ligands. J Med Chem 2003;46:2945–2957.
31. Gust R, Keilitz R, Schmidt K. Synthesis, structural evaluation, and estrogen receptor interaction of 2,3-diarylpiperazines. J Med Chem 2002;45:2325–2337.
32. Garg R, Kapur S, Hansch C. Radical toxicity of phenols: a reference point for obtaining perspective in the formulation of QSAR. Med Res Rev 2001;21:73– 82.
33. Fisher LS, Guner OF. Seeking novel leads through structure-based pharmacophore design. J Braz Chem Soc 2002;13:777–787. 34. Sadowski J, Gasteiger J, Klebe G. Comparison of automatic three-dimensional model builders using 639 X-ray structures. J Chem Inform Comput Sci 1994;34:1000 –1008.
35. Jain AN. Surflex: fully automatic flexible molecular docking using a molecular similarity-based search engine. J Med Chem 2003;46: 499 –511.