Review
Biosignaling of mammalian Ste20-related kinases
Pin Ling
a, Te-Jung Lu
b, Chiun-Jye Yuan
c, Ming-Derg Lai
d,e,⁎
aDepartment of Microbiology and Immunology, College of Medicine, National Cheng Kung University, Tainan, Taiwan, ROC
b
Department of Medical Technology, College of Medicine and Life Sciences, Chung-Hwa University of Medical Technology, Tainan, Taiwan, ROC
c
Department of Biological Science and Technology, National Chiao Tung University, Hsinchu, Taiwan, ROC
d
Center for Gene Regulation and Signal Transduction Research, National Cheng Kung University, Tainan, Taiwan, ROC
e
Department of Biochemistry and Molecular Biology, College of Medicine, National Cheng Kung University, Tainan, Taiwan, ROC Received 6 December 2007; accepted 18 December 2007
Available online 4 January 2008
Abstract
Sterile 20 (ste20) protein is an upstream ser/thr kinase in yeast, and several mammalian Ste20-like (MST) kinases have been identified. This review focuses on the signal transduction, interacting proteins, and potential biological function of MST1, 2, 3, and 4 kinases, since several novel signal pathways of these kinases have been characterized recently. MST1 and MST2 kinases play an important role in cell growth and apoptosis, and the signal pathways involves many important molecules including RAS, AKT, and FOXO3. MST3 and MST4 have similar kinase domain, but have opposite effects on apoptosis and transformation. The downstream signaling molecules of these two kinases are beginning to be elucidated. Based on the expression pattern and signal pathways, we will discuss the perspective biological functions of four MST family kinases in cancer, immune, cardiovascular, and brain function.
© 2007 Elsevier Inc. All rights reserved.
Keywords: ste20; Kinase; Signal pathway; MST; Apoptosis contents
Contents
1. Introduction . . . 1238
1.1. Characteristic properties of MST1–4: cloning, expression, isoform, structure, and kinase activity . . . 1238
1.1.1. Cloning . . . 1238 1.1.2. Expression . . . 1238 1.1.3. Isoform . . . 1239 1.1.4. Structure . . . 1239 1.1.5. Kinase activity . . . 1239 1.2. Autophosphorylation . . . 1239 1.3. Proteolysis . . . 1239 1.4. Dimerization . . . 1239 1.5. Cofactor requirement . . . 1239
2. Signal transduction of MST kinases . . . 1239
2.1. Mst1 . . . 1239
2.1.1. Upstream activator . . . 1239
2.1.2. MST1, JNK, and histone 2B . . . 1240
Cellular Signalling 20 (2008) 1237–1247
www.elsevier.com/locate/cellsig
⁎ Corresponding author. Department of Biochemistry and Molecular Biology, College of Medicine, National Cheng Kung University, Tainan, Taiwan, ROC. Tel.: +886 6 2353535x5549; fax: +886 6 2741694.
E-mail address:[email protected](M.-D. Lai).
0898-6568/$ - see front matter © 2007 Elsevier Inc. All rights reserved. doi:10.1016/j.cellsig.2007.12.019
2.1.3. MST1, ras, Nore1, RASSF1A, and CNK1 . . . 1240
2.1.4. MST1, hSav, Wts, and cyclin E. . . 1241
2.1.5. MST1, FOXO3, and AKT . . . 1241
2.2. Mst2 . . . 1241
2.3. Mst3 . . . 1242
2.4. Mst4 . . . 1243
3. Physiological function of MST kinases . . . 1243
3.1. MST kinases and cancer . . . 1243
3.2. MST kinases and immune system . . . 1244
3.2.1. MST1 regulates lymphocyte polarity and adhesion . . . 1244
3.2.2. MST1 is implicated in somatic hypermutation and class switching in B cells . . . 1244
3.2.3. Association of MST1 with apoptosis of eosinophils . . . 1245
3.3. MST kinases and cardiovascular function . . . 1245
3.4. MST kinases and brain function . . . 1245
4. Conclusion and future perspective. . . 1245
Acknowledgements . . . 1246
References . . . 1246
1. Introduction
Sterile 20 (ste20) is located upstream of MAPK kinase in the mating pathway, a yeast mitogen-activated protein kinase kinase kinase kinase (MAP4K) in Saccharomyces cerevisiae. Its homologues in various organisms are called ste20-related kinases
[1]. The mammalian Ste20 family consists of two structurally distinct subfamilies, including the P21-activated kinase family (PAK) and the germinal center kinase (GCK) family. The phylogenetic relationship between these ste20-related kinases has been described. PAK can be further subdivided into PAKI and PAK II; on the other hand, GCK is subdivided into GCK I to GCK VIII [2–5]. PAKs contains a kinase domain located in the COOH terminus and an N-terminal p21 GTPase-binding domain that mediates binding to small GTPases such as Cdc42. PAKs are well-known regulators of cell migration, growth and apoptosis[6]. Alterations of PAK expression have been detected in human cancer, and may be considered as pharmacological targets for cancer treatment [7]. GCKs are characterized by the presence of kinase domain at the NH2 terminus and lack of GTPase binding domains. GCKs are involved in many physiologic processes such as cell volume regulation and immune response [4,8]. In this review, we focus on four mammalian ste20-related (MST) kinases, since several novel signal pathways of these kinases have been identified recently. MST1 and MST2 are members of the GCK II subfamily. MST3 and MST4 belong to the GCK III subfamily.
1.1. Characteristic properties of MST1–4: cloning, expression, isoform, structure, and kinase activity
1.1.1. Cloning
Most MST kinases were cloned either by homologous screening with degenerate primers or digital screening of the EST database. Creasy and Chernoff cloned the MST1 gene by screening a lymphocyte cDNA library using degenerate PCR primers corresponding to the catalytic domains of serine/
threonine kinases[9]; MST2 was cloned by screening a Hela cell cDNA library with an MST1 probe [10]. MST2 was independently cloned[11]by searching for protein kinases that respond to outside stress, and was named as kinase responsive to stress 1 (KRS1). Similarly, Schinkmann and Blenis cloned MST3 by screening a Hela cell cDNA library using degenerate primers based on conserved regions of the kinase domains of STE20[12]. MST4 was obtained by searching an EST data base with six 15-mer peptide sequences derived from human PAK1 kinase, CDK kinase, ERK1 kinase, YAK1 kinase, PKA kinase and PKC kinase[13]. Interestingly, MST4 was independently cloned as an interacting partner for c-raf in yeast two-hybrid assay, although MST4 did not interact with c-raf in vivo[14]. MST4 was also cloned through digital cloning on the EST database, and named as MASK (Mst3 and SOK1-related kinase). Therefore, different acronyms have been used for the same kinase in previously published reports: MST1/KRS2/STK4, MST2/ KRS1/STK3, MST3/STK24, and MST4/MASK1. The desig-nated gene names and acronyms can be found on the HUGO nomenclature website (http://www.gene.ucl.ac.uk/nomenclature/ index.html).
1.1.2. Expression
The mRNA expression of MST1–4 kinases is generally ubiquitous in most tissues and in most cell lines examined. An approximately 7.0-kb MST1 transcript was detected in most tissues with Northern blot analysis; a 3.5 kb alternatively spliced mRNA was detected in kidney, skeletal muscle, and placental tissues[9]. A 5.5 kb MST2 mRNA transcripts was detected with Northern blot analysis, and high levels of expression was observed in adult kidney, skeletal and placental tissues[10,11]. A single 2-kilobase MST3 mRNA was detected by Northern blot analysis with highest levels in heart, skel-etal muscle, and pancreas[12]. Western blot analysis detected a 52-kD MST3 protein in most cell lines examined. One 3.6-kb band for MST4 was detected in all tissues with Northern blot analysis. Highest expression of MST4 was observed in pla-cental tissue[14].
1.1.3. Isoform
MST3 isoform MST3b was identified by searching an EST database using the conserved catalytic domain of STE20 as the probe[15]. The sequence of MST3b is identical to MST3 except for an additional coding region at 5′end of cDNA. Reverse transcription-polymerase chain reaction and Northern blot analysis with a probe derived from 5′ distinct sequence of MST3b revealed that expression of the 2.5 kb MST3b mRNA is restricted to the brain, in contrast to the ubiquitous expression of MST3. An alternatively spliced variant, MST4a, has been cloned from a fetal brain cDNA library[14]. MST4a lacks part of the kinase domain, and may be considered as a dominant negative MST4 in vivo. MST4a expression is abundant in brain tissue. 1.1.4. Structure
MST family members have highly homologous kinase domains. MST1 and MST2 have an almost identical kinase domain; MST3 and MST4 share almost 90% amino acid identity. On the other hand, MST1 and MST2 share only 60% homology in the C-terminal non-kinase domain. MST3 and MST4 had less than 20% shared identity in this region. MST3 and MST4 have opposite effects on cell growth, suggesting that the C-terminal non-kinase regulatory domain may be essential in dictating the final consequence of kinase function.
1.1.5. Kinase activity
Regulation of MST kinases occurs at least through two pathways: (1) autophosphorylation (2) caspase cleavage. Dimer-ization and cofactor preference may also regulate kinase activity. 1.2. Autophosphorylation
The kinase domain of ser/thr kinase can be subdivided into subdomains. The MST family of kinases have a consensus sequence around subdomains VII and VIII, KRNT(V/F)(I/V) GTPFWMAPEVI. Autophosphorylation of threonine residue in KRNT is essential for kinase activity of MST1–3 because alteration of threonine to alanine almost completely abolishes kinase activity [16–18]. Studies have shown that mutation of threonine to glutamic acid to mimic phosphorylated state did not enhance but reduced MST1 activity. In contrast, the same mutation was shown to enhance MST3 kinase activity, suggesting subtle differences between these two subfamilies[16,18]. The autophosphorylation site of MST4 has not yet been reported. Autophosphorylation of threonine in GTPF has also been demonstrated in MST1. Autophosphorylation on residues outside the kinase domain in the MST family does not alter kinase activity, but may affect kinase susceptibility to caspase[16]. 1.3. Proteolysis
The 63-amino acid region, spanning amino acids 331–394 of MST1, contains an inhibitory domain. Removal of this domain increases kinase activity up to 9-fold. Caspase cleavage at DEMD326 and TMTD349 can remove the inhibitory domain, and render MST1 active during apoptosis [19,20]. Proteoly-sis removes the inhibitory domain, and also induces nuclear
translocation of MST1 kinases. MST1 nuclear activity may cause chromatin condensation. Similar proteolysis and nuclear translo-cation has also been observed in MST3 [21]. The sequence requirement of nuclear translocation has been mapped[19]. The cleaved C-terminal MST3 may be myristolated and relocated from the catalytic domain of MST3 [22]. A similar potential caspase cleavage site was also identified in MST4; however, actual cleavage has not been reported during apoptosis.
1.4. Dimerization
Both MST1 and MST2 can form homodimers. The extreme C-terminal domain (a. a. 431–487) is required for MST1 dimerization, which may not be essential for kinase activity, since deletion of this domain does not affect kinase activity when using myelin basic protein as a substrate [23]. MST2 contains nearly identical sequences in these regions, indicating that similar inhibitory regulation and dimerization may be conserved, although the C-terminal domain of MST1 and MST2 shares only 60% amino acid identity. The MST2 autopho-sphorylation reaction is strongly dependent on enzyme concentration, but the dimerization-deficient MST2 lacking the C-terminal domain has the same level of autophosphoryla-tion. Therefore, it is perceivable that autophosphorylation occurs through intermolecular interaction between MST kinases, but is not related to dimerization [17]. Whether MST3 or MST4 can form homodimers remains to be investigated.
1.5. Cofactor requirement
MST3 has a unique cofactor preference for manganese ions
[12]. MST3 has a high kinase activity in the presence of manganese ions in vitro[18]. YSK1/SOK-1 also has the same ion preference in kinase reactions (unpublished observation). It is interesting to note that manganese plays an important role in regulating brain neurotransmitters and an excess of manganese may be related to many types of brain diseases, including Parkinson's disease [24,25]. Excess manganese may activate MST3 in its participation in apoptosis. MST3 can also use zinc ions and cobalt ions as cofactors in kinase reactions in vitro
[26]. The biological significance of the specific ion preference remains to be determined in living cells.
2. Signal transduction of MST kinases 2.1. Mst1
2.1.1. Upstream activator
Many stimuli, including calf serum, heat shock, and high salt, have been examined for effects on MST1 kinase activity; however, none of these stimuli affect endogenous MST1 kinase activity. Interestingly, EGF stimulation has been observed by in-gel kinase assay to cause a transient decrease of MST1 kinase activity; the kinase activity decreases up to two-fold within 1 min and returns to normal level 30 min after EGF treatment
[9]. The first indication of MST1 upstream stimulus comes from the observation that MST1 is specifically cleaved by a
caspase 3-like activity during apoptosis, induced either by cross-linking CD95/Fas [20], anticancer drugs [27], or by bipho-sphonates that induce apoptosis in osteoclasts [28]. The in-duction of MST1 activation cleavage is not restricted to specific types of inducers. For example, addition of staurosporine or withdrawal of both serum and M-CSF also induced activation cleavage of MST1[28]. The role of MST1 in apoptosis may vary in different cell types. Caspase cleavage and activation of MST1 correlates with eosinophil apoptosis, but not with neutrophil apoptosis[29].
2.1.2. MST1, JNK, and histone 2B
Overexpression of MST1 activates JNK and p38 MAPK, but not ERK1 in 293T cells[20]; additionally, activation of JNK can be blocked by cotransfection with kinase-dead MEKK1 [30]
and dominant-negative JNK inhibits MST1-induced apoptosis. In contrast, neither p38 inhibitor nor dominant-negative p38 affects MST1-induced apoptosis [31]. The appearance of cleaved active MST1 fragment was usually 2 to 6 h after the addition of an apoptosis-inducing drug. However, the first appearance of JNK activation (approximately 30 min after stimulus) was earlier than MST1 during apoptosis induced by the anticancer drug cytotrienin A [27], suggesting that early phase activation of JNK was not mediated by MST1. MST1 induces DNA fragmentation through caspase activation, but induces chromatin condensation and membrane blebbing independent of caspase activation. One important issue is how MST1 activates chromatin condensation. It has been demon-strated that expression of a dominant-negative JNK mutant inhibited MST1-induced chromatin condensation [31]. Enhancement of MST1 nuclear translocation by mutation of the nuclear exporting sequence in the C-terminal of MST1 increased its ability to induce chromatin condensation, suggesting that nuclear translocation of MST1 was important for chromatin condensation [32]. Allis's group has demon-strated that the mechanism of triggering chromatin condensa-tion was mediated by phosphorylating histone 2B in vertebrate and yeast[33,34]. Deletion of ste20 rendered yeast resistant to apoptosis and loss of histone 2B phosphorylation[34]. MST1 phosphorylated histone 2B in vitro and the kinetics of histone 2B phosphorylation was similar to that of MST1 cleavage in HL-60 cells. In addition, expression of a C-terminal truncated MST1 could phosphorylate histone 2B in Hela cells; however, the requirement of MST1 in inducing histone 2B phosphoryla-tion has not been demonstrated in vivo[33]. The role of MST1 in chromatin condensation has been questioned by showing that depletion of MST1 further enhanced histone 2B phosphoryla-tion during apoptosis induced by apoptotic chromatin con-densation inducer in the nucleus (acinus). Furthermore, PKC delta may mediate H2B phosphorylation downstream of acinus
[35]. It was interesting to note that the active form of acinus binded to MST1 during apoptosis and enhanced its kinase activity, and that the binding of acinus to MST1 was reduced by nerve growth factor treatment. Recently, activation of the c-Jun N-terminal kinase pathway has been shown to be essential and sufficient for induction of chromatin condensation by over-expression of MST1[36]. The discrepancy in these reports may
result from interactions of several parameters. The first is that other MST members, such as MST2, may replace the function of MST1 on chromatin condensation or histone 2B phosphoryla-tion during apoptosis in certain cellular condiphosphoryla-tions. Therefore, depletion of MST1 has minimal effects in some experiments. The second is that histone 2B phosphorylation may be a good marker for chromatin condensation, but not necessarily for all types of chromatin condensation. The third is that MST1 exists in different types of complexes, including acinus, RASSF1, NOR1, and death-associated protein 4 (DAP4). The equilibrium and balancing of these complexes may determine the final consequence of MST1. The interacting partner DAP4 was identified by affinity co-purification with FLAG-tagged MST1. Co-expression of DAP4 and a sub-optimal level of MST1 enhanced MST1-induced apoptosis. However, DAP4 was not phosphorylated by MST1, and did not enhance MST1 kinase activity. DAP4 may enhance the apoptotic effect through promoting the caspase-catalyzed cleavage of MST1. DAP4 was originally identified by its ability to block apoptosis induced by interferon-gamma (IFN-γ) in Hela cells; however, MST1 was not activated by IFN-γ in Hela cells. The biological implication of this interaction requires further investigation[37].
2.1.3. MST1, ras, Nore1, RASSF1A, and CNK1
In search of the potential biological function of a Ras effector, Nore1/Rassf5, MST1 was shown to directly interact with Nore1/ RASSF5. The complex between Nore1 and MST1 was insensitive to serum withdrawal. On the other hand, the complex between endogenous Ras and MST1 and Nore1 only occurred in the presence of serum. Interestingly, the complex of Ras and Nore1 with MST1 did not enhance MST1 kinase activity, suggesting that Ras controls MST1 signaling primarily by its recruitment of MST1 to a site regulating MST1 action, probably a membrane fraction[38].The complex may involve other RASSF members, such as RASSF1A. RASSF1A is frequently inactivated in lung cancer cells due to hypermethylation of a CpG island in its promoter[39]. RASSF1A can form homodimers or heterodimers with Nore1 [40]. The complex appears to contain many other proteins, e.g., human scaffold protein CNK1, a c-Raf1 binding protein. Cell death induced by CNK1 required the participation of MST1 or MST2, which was mediated indirectly through association of RASSF1 polypeptides [41]. Recombinant RASSF1A inhibited MST1 kinase activity in vitro, but appeared to increase MST1 kinase activity in vivo. It seems that the interaction is more than a simple association. Depletion of RASSF1A by RNA interference reduced MST1 activation and Fas ligation-induced apoptosis [42]. Therefore, the interaction between RASSF1A and MST1 indeed plays a role in mediating apoptosis. Treatment of K-ras-transformed cells with bortezomib resulted in nuclear translocation of MST1 and an increase of phosphorylated histone H2B. Knockdown of MST1 expression by RNA interference reduced bortezomib-induced apoptosis. These results suggest that MST1 is an essential mediator in apoptosis of K-ras transformed cells. Although Ras-Nore1-MST1 is known to be involved in mediating Ras-induced apoptosis, bortezomib has little effect on NORE1, suggesting that other molecular interactions in this complex may be altered[43].
2.1.4. MST1, hSav, Wts, and cyclin E
Drosophila MST1/2 homologue Hippo (hpo) binds to and phosphorylates a tumor suppressor protein Salvador (Sav), which is known to interact with Warts (Wts) protein kinase. Loss of Hippo results in increased expression of cyclin E and cell-death inhibitor diap1[44–48]. The human orthologue hSav (also called hWW45) can bind to and be phosphorylated by MST1 and 2. MST2 has a stronger stabilizing effect on hSav, and may play a more prominent role in this complex[49]. 2.1.5. MST1, FOXO3, and AKT
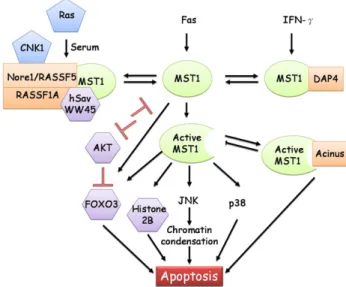
MST1 phosphorylates FOXO3 and disrupts its interaction with 14-3-3 proteins, promotes FOXO3 nuclear translocation, and induces apoptosis in neuronal cells. Knockdown of C. elegans MST1 ortholog CST-1 shortened life span and accelerated tissue aging, while overexpression of CST-1 promoted life span and delays aging[50]. The effect of MST1 is opposite to that of AKT. AKT phosphorylates FOXO3 and promotes its association with cytoplasmic 14-3-3 protein, preventing its transcriptional activity. Therefore, FOXO tran-scription factor can undergo inhibitory phosphorylation by AKT, and activating phosphorylation by MST1[51]. The signal transduction appears to be interconnected. AKT could also directly phosphorylate MST1 and inhibited its kinase activity towards FOXO3, but enhanced its kinase activity on the other substrate histone 2B [52]. In transgenic mice with constitu-tionally active p110-alpha subunit of PI3K, MST1 expression was elevated and the elevation could be blocked by the PI3K-specific inhibitor LY294002 [53]. These results suggest that AKT phosphorylates MST1 and prevents its action from promoting apoptosis. Furthermore, MST1/2 may be an important branch pathway of PI3K/AKT based on the observation that MST1 and AKT1 are localized to identical subcellular compartments in human prostate tumors. Both MST1 and its active cleavage form physically interact with AKT and act as direct inhibitors of AKT. Depletion of MST1 or MST2 with siRNA increased AKT activity, and depletion of both proteins further enhanced AKT activity [54]. The MST complex appears to contain many protein components, and the association or dissociation of one component in response to outside apoptotic stimulus may alter the activity of other components of this complex. A simplified scheme of MST1 signal transduction is shown inFig. 1.
2.2. Mst2
MST2 shares at least two similar pathways with MST1. The first is that MST1 and MST2 can phosphorylate AKT and inhibit AKT activation. In addition, they can cause activating phosphorylation of FOXO3. The second is that both proteins can be detected in a complex containing hSav, although the role of MST2 in this complex has been more clearly demonstrated. During apoptosis, MST2 was cleaved and undergone irreversible autophosphorylation, which was resistant to phosphatase [17]. Thyroid transcription factor-1 could be phosphorylated by rat MST2 in thyroid cells [55]; however, the role of phosphorylation in the regulation of thyroid
tran-scription factor-1 function was not investigated afterwards. Constitutionally active Ras can inhibit thyroid transcription factor-1 and thyroid differentiation [56]. MST2 also interacts with several Ras effector complexes including Raf-1 and AKT. Therefore, MST2 may play a role in mediating inhibitory effects of Ras on thyroid differentiation. MST2 can be inhibited by Raf-1 by forming a complex. Raf-1 inhibits MST2 independent of its kinase activity, probably by sequestering MST2 from its activation site[57].
The complex of hSav and MST2 also contains RASSF1A, Nore1 and LATS1. MST2 can be co-precipitated with LATS1 only in the presence of hSav, and furthermore, RASSF1A and hSav promote LATS phosphorylation by MST2 through this recruitment [58]. RASSF proteins have been suggested to participate in the Hpo-Sav-Wts pathway in mammalian cells in a manner dependent on a protein–protein interaction domain, SARAH, that is shared by Sav, RASSF, and Hpo[57].
MST2 is in the tumor suppressor pathway Hippo/Salvador/ Lats; MST2 direct phosphorylates LATS1 and LATS2 at their C-terminal catalytic domain. MST1 can perform similar kinase activity, but at a lower efficiency. In contrast, MST4 has almost no activity[59]. The interaction between raf-1 and MST2 was further extended to RASSF1A. RASSF1A causes the disruption of the inhibitory Raf1-MST2 complex, and enhances the released MST2 to phosphorylate its substrate, LATS1. The phosphorylated LATS1 releases the cytosol-sequestered transcription factor YAP1. YAP1 is the human homologue of Drosophila Yorkie, which connects the LATS1 Drosophila homologue Warts to the transcription of cyclin E gene[60]. The transcriptional regulator YAP1 translocates to the nucleus and associates with p73, resulting in transcription of the proapoptotic target gene, such as Puma[61]. An outline of MST2 signal pathways is illustrated in Fig.2.
Fig. 1. MST1 kinase signal transduction. MST1 has several interacting partners, including DAP4, acinus, and hSav/Nore1/RASSF1A. MST1 can be cleaved by caspase to remove inhibitory domain, and induce activation of JNK and p38 during apoptosis. The MST1 substrates include histone 2B, AKT, FOXO3, and hSav. MST1 causes activating phosphorylation of FOXO3 transcription factor, but introduces an inhibitory phosphorylation of AKT. AKT can also cause inhibitory phosphorylation of MST1. Interacting proteins are indicated by a rectangle; kinase substrates are indicated by a hexagon.
2.3. Mst3
MST3 was unable to activate ERK, JNK, and p38 MAPK kinase activity[12]. However, one report indicated that MST3, but not the isoform MST3b, showed activation of ERK in HEK 293 cells [15]. Overexpression of MST3 did not lead to activation of ERK in MDCK cells [unpublished observation]. Interestingly, protein kinase A appears to phosphorylate Thr-18 of MST3b isoform. Mutation of Thr-18 to alanine of MST3b enhances its activation of ERK. The role of ERK in wild-type MST3 downstream signaling remains to be investigated.
The first identified natural substrate for MST3 is NDR protein kinase[62].The nuclear Dbf2-related (NDR) family of Ser/Thr kinases consists of NDR and large tumor suppressor (LATS) kinase. Members of the NDR family regulate cell proliferation, tubulin cytoskeleton organization, cell spreading, morphogen-esis, polarized growth in C. elegans and D. elanogaster[63]. LATS/WARTS kinase is a tumor suppressor, negatively regulating cell cycles by cyclin-dependent kinase CDK1 and reduces cell proliferation and survival leading to growth arrest in the G2/M phase[63]. The function of Trc, the drosophila NDR kinase is required for the normal morphogenesis of polarized growth, including epidermal hairs, bristles, arista laterals and dendrites [64]. Casnorhabditis elegans NDR homolog SAX1 regulates neuronal cell shape and is involved in cell spreading, neurite initiation and dendritic tiling[65]. Genetic studies with yeast, C. elegans, and drosophila showed that many of the NDR members are highly conserved and phosphorylated by Ste20 family kinases [62]. From this genetic evidence, MST3 was identified to be able to phosphorylate and activate NDR protein kinase. NDR kinase was directly phosphorylated at Thr442/444
by MST3 when cells were treated with okadaic acid, a serine/ threonine phosphatase inhibitor 2A [62]. Therefore, the activated MST3 locates upstream of the NDR kinase signal pathway and controls cell shape and cell cycle. This pathway appears to be parallel to MST2 on LATS. The Ste20-like kinase, Hippo, phosphorylates WARTS/LATS kinase in S. cerevisiae. MST2 in mammals phosphorylates LATS1 and is involved in tumor suppressors pathways in mammals[57]. Cdc7 and Sid1, two other Ste20-like kinases, are upstream of NDR kinase homologue Sid2 in S. pombe, and are involved in cytokinesis
[66]. Recently, the svkA gene encoding severin kinase, a homolog of the human MST3, MST4 and YSK1 kinases, was knockout in Dictyostelium discoideum. SvkA-knockout cells showed dramatic defects in cytokinesis, leading to multi-nucleated cells[67]. MST3, MST4 and YSK1 share very high homology in the kinase domain. YSK1 was originally identified by the activation by oxidative stress and named as Ste20/oxidant stress response kinase-1 (SOK-1) [68,69]. The expression of MST3 was elevated by the oxidative stress, but not by the hormones released during labor such as prostaglandin E, in human term placenta. The hydrogen peroxide-induced apopto-sis of trophoblast cell was suppressed by overexpression of kinase-dead Mst3 or by knockdown of endogenous Mst3 with siRNA[70]. This result suggested that oxidative stress might promote apoptosis in part by upregulating MST3 expression.
Downregulation of endogenous MST3 with RNA interfer-ence was shown to enhance cellular migration in MCF-7 breast cancer cells. Aberrant protrusions were reduced by reconstitu-tion of RNAi-resistant MST3 in MST3-knockdown MCF7 cells
[18]. Overexpression of wild-type MST3 in MCF7 and MDCK cells leaded to tyrosine phosphorylation of Tyr-31 and Tyr-118 of paxillin. MST3 could keep paxillin tyrosine phosphory-lated to inhibit protrusion and migration in cells plating on fibronectin[18](Fig. 3).
MST3, a Ser/Thr kinase, can not act directly on the tyrosine residue of paxillin to control migration. Protein tyrosine Fig. 2. MST2 kinase signal transduction. MST2 substrates include FOXO3,
AKT, LATS1, and thyroid transcription factor-1. The interacting partner Raf-1 inhibits MST2 function. MST2 reacts similarly with MST1 in two aspects: (1) interaction with AKT and FOXO3, (2) activation by caspase cleavage. MST2 interacts with RASSF1A and hSav to phosphorylate LATS and regulate p73-target genes. The biological function of MST2 is inhibited by Raf-1 binding. Interacting proteins are indicated by a rectangle; kinase substrates are indicated by a hexagon.
Fig. 3. MST3 kinase signal transduction. MST3 can be cleaved by caspase to promote apoptosis. MST3 can phosphorylate NDR to regulate cell cycle and PTP-PEST to regulate migration. Kinase substrates are indicated by a hexagon.
phosphatase PTP-PEST binds with paxillin C-terminal LIM 3-4 domains as well as the N-terminal LD4 motif to phosphorylate tyrosine 31 and tyrosine 118 of paxillin[71]. The optimal level, localization or activity of PTP-PEST are important regulators that maintain the balance of promoting control cell motility in different contexts[72]. MST3 was shown to directly phosphor-ylate PTP-PEST but not PTP1B. The phosphorylation inactivated PTP-PEST and inhibited migration through tyr-31 and tyr-118 of paxillin[18]. The potential mechanism of PTP-PEST-dependent inhibition of migration by MST3 may involve other proteins such as Rac1. PTP-PEST can suppress the activation of Rac1 and impair membrane protrusion, cell spreading and hepatotaxis in CHOK1 cells [72]. It is possible that MST3 increases phosphorylation of paxillin and hence decreases Rac1 activity at the cadherin-containing junctions. At these sites the cadherin assembling increases and leads to reduction of cell migration. PTP-PEST is not only regulating cell migration, but also controlling cell growth and apoptosis. Expression of PTP-PEST sensitized cells to TNF-alpha-induced apoptosis, and PTP-PEST was cleaved by caspase 3 during apoptosis[73]. Therefore, MST3 may phosphorylate PTP-PEST and alter the scaffold function of PTP-PEST during apoptosis.
2.4. Mst4
MST4 was cloned by screening Raf-1 with yeast two-hybrid; however, Raf-1 did not interact with MST4 in mammalian cells. Furthermore, the C-terminal domain enhanced MST4 kinase activity in contrast to the inhibitory non-kinase domain of MST1[14]. MST4 did not activate ERK, JNK, or p38 in 293 cells, but MST4 could activate ERK via MEKK, but not the Ras/Raf pathway in Pheonix cells[14,74]. In contrast to other MST family kinases, MST4 did not activate JNK or p38. Overexpression of MST4 further enhanced transformation of phoenix cells as demonstrated by growth in soft agar. One conflicting report has suggested that overexpression of MST4 induced apoptosis in MCF-7 cells and 293 cells, and that kinase-dead MST4 did not have pro-apoptotic effects which indicating kinase activity was required[74]. The programmed cell-death 10 (PDCD10) gene has been implicated in mutations associated with Cerebral Cavernous Malformations (CCM). MST4 was shown to interact with PDCD10 with yeast two-hybrid and co-immunoprecipitation assay. Overexpression of either PDCD10 or MST4 enhances ERK kinase activity, and co-expression further enhanced ERK activity and cellular trans-formation[75](Fig. 4).
The orientation of Golgi matrix positions at wound edges, and polarizing the edge, coordinates the signal of the cytoskeleton and Rho family proteins. Activation of YSK1 or MST4 involves autophosphorylation at Thr174 in YSK1 and Thr178 in MST4. It may stabilize the binding of YSK1 and MST4 to GM130, a cis-Golgi protein, and restrict YSK1 and MST4 at the Golgi matrix to control cell migration. Interfering with YSK1 and MST4 function by siRNA disturbed the ordered localization of the Golgi matrix in the perinuclear region, and the Golgi was dispersed into the cell periphery. However, overexpressing YSK1 but not MST4 induced migration of cells
on collagen [76]. One possibility for the difference seen with YSK1 and MST4 is that they may have different substrates. YSK1, but not MST4, specifically phosphorylates 14-3-3ζ, which is localized in the Golgi matrix. Although MST3 is highly homologous to YSK1 and MST4, MST3 showed cyto-plasmic distribution and has no interaction with G130. Knock-down of MST3, unlike dominant negative YSK1, did not disturb the ordered localization of Golgi apparatus in the perinuclear region in MCF-7 cells (unpublished results). 3. Physiological function of MST kinases
3.1. MST kinases and cancer
It is well documented that MST1 and its close homolog MST2 play a key role in apoptosis in tissue culture cells. Several genetic analyses conducted in Drosophila have demonstrated the involvement of Hippo (Hpo), a MST2 homolog in Drosophila, in an emerging tumor suppressor pathway, which includes two other mediators, Salvador (Sav) and Warts (Wts)[44–48]. Sav and Wts, a scaffold protein and a ser/thr protein kinase res-pectively, are known to function together to regulate cell growth, proliferation, and death in Drosophila [77–78]. Like Sav and Wts, Hpo mutations result in the loss of growth restriction and impair apoptosis through elevated expression of cell cycle mediator cyclin E and the apoptosis inhibitor DIAP1. Biochem-ical analyses have further shown that Hpo interacts with and phosphorylates Sav. Also, the Hpo-Sav complex promotes Wts phosphorylation[44,48]. This evidence reveals the genetic and physical interactions among Hpo, Sav, and Wts to define a novel signaling pathway regulating cell cycles and apoptosis. More importantly, this tumor suppressor pathway seems evolutionally conserved in Drosophila and mammals [77,78]. Interestingly, results from Pan et al. have shown that MST2 is able to rescue the phenotype of aberrant cell growth in Drosophila Hpo mutants, suggesting a conserved role for Hpo and MST2 in growth control [48]. Moreover, cumulative evidence indicates deregulation of the Hpo-Sav-Wts pathway in human tumors. The counterparts of this pathway in mammals appear to function as tumor suppressors. However, obtaining direct evidence that Fig. 4. MST4 kinase signal transduction pathway. MST4 can activate ERK to induce cellular transformation under EGF stimulation. MST4 has two interacting partners, GM 130 and PDCD10. Interacting proteins are indicated by a rectangle.
MST1 or MST2 functions as a tumor suppressor in human tumors is still warranted.
Expression of MST1 can be upregulated by treating MDA-MB-435 human breast cancer cells with tissue inhibitor of metalloproteinases-1[79]. Similarly, elevated levels of TIMP-1 in tumors correlates with an adverse prognosis[80]. Although MST1 may be considered as a tumor suppressor gene, the expression level of MST1 does not correlate with disease prognosis. Surprisingly, a significantly increased risk for tumor-related death was found for patients with an unmethylated MST1 promoter[81]. Regarding the roles of kinase in diseases such as cancer, the kinase activity and the location are more crucial than the expression level to regulate cellular behavior. The MST1 kinase activity can be upregulated by nuclear translocation, the loss of cytoplasmic MST1 was found associating with higher N stage and shortened survival. In a multivariate analysis, loss of cytoplasmic MST1 was an independent adverse prognostic factor in this group of patients. Methylation analysis showed that the loss of cytoplasmic MST1 expression was not likely due to promoter methylation. The importance of loss of cytoplasmic MST1 awaits for further examination in a large number of human tumor samples[82]. MST1 levels declined progressively from clinically localized disease to hormone refractory disease, coinciding with an increase in AKT activation with transition from hormone naïve to hormone-resistant metastases[54]. Correlation of MST2 or MST3 with the status of tumor samples or cancer cell lines has not been established in previous reports. MST4 was not differentially expressed between tumor and normal samples; however, expression levels appeared to correlate with tumor-igenicity and androgen receptor status of prostate cancer samples and cell lines[83].
3.2. MST kinases and immune system
Mammalian ste20-like proteins, such as HPK1, play an important role in immune system function[2]. Much progress has been made to define the functions of the MST family of kinases in growth control, apoptosis, and cell migration. However, the roles of the MST family of kinases in the immune system remain mostly unexplored. A few studies have just started to reveal the multiple functions of MST1 in immune cells. 3.2.1. MST1 regulates lymphocyte polarity and adhesion
Cell migration is a key feature allowing lymphocytes to travel around the peripheral tissues to detect foreign antigens. Integrin-mediated adhesion plays a vital role in such lympho-cyte trafficking. Previous studies have revealed that the small GTPase Rap1 and its binding effector RAPL are critical for regulating the affinity and organization of integrins[84,85]. A recent work from Kinashi et al. has further demonstrated that MST1 functions as a key downstream effector of RAPL for regulating lymphocyte polarization and adhesion[86]. MST1 is associated with and activated by RAPL. Moreover, MST1 activation by TCR or chemokine CCL21 stimulation is abolished in RAPL-deficient T lymphocytes, suggesting that RAPL is essential for MST1 activation in T lymphocytes. In
siRNA approach, knocking down MST1 expression results in abrogating TCR-induced adhesion and immunosynapse forma-tion. In addition, RAPL and MST1 are co-localized at vesicular compartments and, upon stimulation, are translocated with integrins toward the leading edge. This study establishes the importance of the RAP1-RAPL-MST1 signaling pathway in the integrin-mediated adhesion and polarity of lymphocytes. Nevertheless, the mechanism by which MST1 regulates integ-rin-mediated lymphocyte adhesion remains to be elucidated. 3.2.2. MST1 is implicated in somatic hypermutation and class switching in B cells
In adaptive immunity, B and T cells can create a variety of immunoglobulins (from B cells) and T-cell receptors (from T cells) to sense the presence of foreign antigens. Genetic mecha-nisms have evolved for generating diverse immunoglobulins and T-cell receptors. The variable (V) regions of immunoglobulins and T-cell receptors are responsible for binding various antigens. V(D)J recombination, a process of somatic DNA rearrangement, is implicated in generating diverse V regions, and many V, D, and J gene segments are encoded in germ line DNA. During lymphocyte development, V(D)J recombination is initiated for randomly selecting gene segments from the V, D, and J groups to form a complete gene sequence for encoding a unique V region of the immunoglobulin or T-cell receptor. In addition to V(D)J recombination, two other genetic mechanisms, somatic hyper-mutation and class switching (also termed isotype switching), further contribute to the diversification of immunoglobulins in B cells. During B-cell activation by a foreign antigen, the V region DNA sequence of immunoglobulin undergoes somatic hyper-mutation, which leads to the generation of various immunoglo-bulins with higher affinity. Class switching also occurs during B-cell activation through recombination to delete the DNA sequence between the rearranged V region and the selected C region exon. This process facilitates B cells to generate different isotypes of antibodies with distinct effector functions.
It has been shown that histone modifications, such as acetylation and phosphorylation of histones, are involved in the regulation of V(D)J recombination and class switch recombina-tion. However, whether chromatin modifications play a role in somatic hypermutation is less defined. A study from Schatz et al. demonstrated that phosphorylation of histone H2B on serine 14 (H2BSer14P) is associated with somatic hypermutation and class switching[87]. Using the chromatin immunoprecipitation assay, H2BSer14Pis detected at the V region of immunoglobulin λ light chain (Igλ) from germinal center B cells, but not from naïve B cells. H2BSer14P at the Sμ region of immunoglobulin
heavy chain (IgH) is also observed in germinal center B cells. MST1 was previously shown to be implicated in H2BSer14P during apoptosis[33], which determined the potential implica-tion of MST1 in H2BSer14Pwith respect to somatic hypermuta-tion and class switching. Using chromatin immunoprecipitahypermuta-tion analysis, MST1 is detected at both the V region of Igλ and the Sμ region of IgH from germinal center B cells. This demonstrates the first link of MST1 in histone modifications associated with somatic hypermutation and class switching. Future studies, especially genetic analyses, are needed to
determine whether MST1 is required for these genetic processes in order to increase the antibody repertoire in B cells.
3.2.3. Association of MST1 with apoptosis of eosinophils A study using primary human eosinophils showed that cleaved and activated MST1 is present in apoptotic eosinophils
[29]. Meanwhile, treatment of primary eosinophils with pro-apoptotic agents (such as Fas activating antibody and dexamethosone) increases cleavage of MST1. Conversely, agents (such as IL-5 and GM-CSF) that suppress apoptosis in eosinophils can inhibit cleavage of MST1. Results from this work support the notion that MST1 is associated with eosinophil apoptosis. Further studies are needed to determine whether MST1 plays an active role in promoting eosinophil apoptosis or whether cleaved MST1 is just a byproduct of this cellular event.
3.3. MST kinases and cardiovascular function
The Sadoshima group generated a tissue-specific transgenic mouse model to determine the physiological role of MST1 in the cardiovascular system. They first observed that cardiac-specific transgenic mice expressing MST1 resulted in increased caspase 3 activation and apoptosis in cardiac myocytes. Dilated cardiomyopathy without compensatory hypertrophy was also found in these transgenic mice[88]. In contrast, cardiac-specific transgenic mice expressing kinase-defective MST1 displayed a dominant-negative effect to suppress endogenous MST1 activation and cardiac myocyte death induced by ischemic reperfusion [88]. Recently, using these dominant-negative MST1 transgenic mice, these researchers further addressed whether MST1 is also implicated in the development of heart-failure after myocardial infarction. Their results support a critical role for MST1 in regulating cardiac dilation, apoptosis, and fibrosis after myocardial infarction [89]. These findings reveal the activation of MST1 in promoting cardiac dysfunc-tion, and suggest that MST1 can be a therapeutic target for treating heart failure in the future.
MST1 functions as both an initiator of apoptosis and an inhibitor of hypertrophy in cardiac myocytes[88,89], and plays an important role in the induction of apoptosis of vascular smooth muscle cells, mediating the vascular remodeling process; as such MST1 may be a potential therapeutic target for vascular proliferative diseases[90]. Elevated expression of MST1 was detected during megakaryocyte differentiation to platelets using Northern blot analysis. MST1 kinase activity and downstream p38 MAPK activity were also elevated. Ectopic expression of MST1 increased DNA content per cell in the megakaryotic cell line, and the induced-polyploidization could be inhibited by p38MAPK inhibitor[91]. MST1 may regulate the thrombosis and hemostasis through platelets which are developed from megakaryocytes.
3.4. MST kinases and brain function
MST3 has a unique property enabling use of other metal ions besides Mg(+2) as cofactors in kinase reactions. Four metal ions
(Mg(+2), Mn(+2), Zn(2+), and Co(2+)) activate endogenous, exogenous, and baculovirus-expressed recombinant MST3 within the physiological concentration range [26]. Since Mn (+2) is abundant in brain tissue and Zn(2+) is important in maintaining brain function, MST3 may have a physiological role in the brain. MST3 has an isoform MST3b that contains an additional 5′end coding region at 5′ end of cDNA. The expression of MST3b mRNA is restricted to the brain[15]. This restricted expression further suggests potential brain activity of MST3 family. MST3b was activated in response to nerve growth factor in PC12 pheochromocytoma cells. Suppressing the function of MST3b, not MST3, blocked axon outgrowth. 6-thioguanine, a purine analogue that blocked axon outgrowth, inhibited the MST3b kinase activity. MST3b appears to be a purine-sensitive kinase, and a key regulator of axon outgrowth
[92]. MST4 also has an MST4a isoform, which is abundant in brain tissue. MST4a lacks part of the kinase domain, and may be a dominant negative MST4 in vivo. The role of MST4a in brain function remains to be investigated.
4. Conclusion and future perspective
Research in the last ten years has revealed several interacting proteins and downstream signal pathways of MST1 and MST2. MST1 kinase activates JNK and p38, but not ERK. MST1 induces chromatin condensation in response to apoptotic signals. The natural substrates for MST1 and MST2 include AKT, FOXO3 and histone 2B. Activation of JNK and phosphorylation on histone 2B are important for chromatin condensation during apoptosis. MST2 can directly phosphor-ylate thyroid transcription factor-1 and LATS1. LATS1 promotes the interaction between YAP1 and p73 to transactivate apoptosis genes. MST1 and MST2 may share similar signal pathways in certain signaling complexes; however, detailed analysis of the role of MST1/2 in each signal complex remains to be determined. MST1 and MST2 may exist in more than one complex. For example, one complex is involved in apoptosis, and a different one participates in differentiation, migration, or mitosis. On the other hand, the signaling complexes containing either MST3 or MST4 are much less understood. Several upstream signals for MST1 and MST2 have been identified, including inhibitory AKT phosphorylation and activation caspase cleavage. The only known activation stimulus for MST3 is apoptotic caspase cleavage. Oxidative stress may upregulate the expression of MST3 in certain cell types. MST3 phoshorylates NDR to regulate cell cycles, and PTP-PEST to regulate cell migration. In response to the apoptotic signal, MST1, 2, and 3 are cleaved by caspase to generate active fragments and to further promote the apoptosis cascade. In contrast to other members of the MST family, MST4 activates ERK and promotes cellular transformation. The direct activator for MST4 remains largely unknown. The roles of MST3 and MST4 in migration have been investigated, but the effect of MST1 and MST2 on cellular migration or invasion requires further study.
In the future, activation of MST kinases is better to be determined by the effects on their natural substrates, but not on the
general substrate such as myelin basic protein. Furthermore, the location and complex partners may be very important factors in determining the consequences of MST kinase activation. The physiological functions of MST1 and MST2 may be inferred from the signaling pathways of MST1 and MST2. However, the thorough and accurate understanding of their physiological functions must be completed with studies on animal and human diseases. The MST family kinases may be important in other types of diseases in addition to cancer and immune system, since their expression is ubiquitous. Finally, it will be interesting to compare the biological functions and interacting partners of MST3 and MST4. Both share high homology in the kinase domain but have dissimilar biological functions, which is probably due to different interacting partners and signal complexes.
Acknowledgements
This work is in part supported by the Grant NSC 95-2311-B-006-001 to Dr. Ming-Derg Lai from National Science Council, Taiwan, Republic of China.
References
[1] G. Manning, D.B. Whyte, R. Martinez, T. Hunter, S. Sudarsanam, Science 298 (2002) 1912.
[2] J.S. Boomer, T.H. Tan, J. Cell Biochem. 95 (2005) 34.
[3] I. Dan, N.M. Watanabe, A. Kusumi, Trends Cell Biol. 11 (2001) 220. [4] K. Strange, J. Denton, K. Nehrke, Physiology (Bethesda) 21 (2006) 61. [5] C.M. Pombo, T. Force, J. Kyriakis, E. Nogueira, M. Fidalgo, J. Zalvide,
Front. Biosci. 12 (2007) 850.
[6] C. Hofmann, M. Shepelev, J. Chernoff, J. Cell Sci. 117 (2004) 4343. [7] R. Kumar, A.E. Gururaj, C.J. Barnes, Nat. Rev. Cancer 6 (2006) 459. [8] J.W. Shui, J.S. Boomer, J. Han, J. Xu, G.A. Dement, G. Zhou, T.H. Tan,
Nat. Immunol. 8 (2007) 84.
[9] C.L. Creasy, J. Chernoff, J. Biol. Chem. 270 (1995) 21695. [10] C.L. Creasy, J. Chernoff, Gene 167 (1995) 303.
[11] L.K. Taylor, H.C. Wang, R.L. Erikson, Proc. Natl. Acad. Sci. U. S. A. 93 (1996) 10099.
[12] K. Schinkmann, J. Blenis, J. Biol. Chem. 272 (1997) 28695.
[13] J.L. Lin, H.C. Chen, H.I. Fang, D. Robinson, H.J. Kung, H.M. Shih, Oncogene 20 (2001) 6559.
[14] Z. Qian, C. Lin, R. Espinosa, M. LeBeau, M.R. Rosner, J. Biol. Chem. 276 (2001) 22439.
[15] T.H. Zhou, K. Ling, J. Guo, H. Zhou, Y.L. Wu, Q. Jing, L. Ma, G. Pei, J. Biol. Chem. 275 (2000) 2513.
[16] H. Glantschnig, G.A. Rodan, A.A. Reszka, J. Biol. Chem. 277 (2002) 42987. [17] Y. Deng, A. Pang, J.H. Wang, J. Biol. Chem. 278 (2003) 11760. [18] T.J. Lu, W.Y. Lai, C.Y. Huang, W.J. Hsieh, J.S. Yu, Y.J. Hsieh, W.T. Chang,
T.H. Leu, W.C. Chang, W.J. Chuang, M.J. Tang, T.Y. Chen, T.L. Lu, M.D. Lai, J. Biol. Chem. 281 (2006) 38405.
[19] W.S. Lee, C.Y. Hsu, P.L. Wang, C.Y. Huang, C.H. Chang, C.J. Yuan, FEBS Lett. 572 (2004) 41.
[20] J.D. Graves, Y. Gotoh, K.E. Draves, D. Ambrose, D.K. Han, M. Wright, J. Chernoff, E.A. Clark, E.G. Krebs, EMBO J. 17 (1998) 2224. [21] C.Y. Huang, Y.M. Wu, C.Y. Hsu, W.S. Lee, M.D. Lai, T.J. Lu, C.L. Huang,
T.H. Leu, H.M. Shih, H.I. Fang, D.R. Robinson, H.J. Kung, C.J. Yuan, J. Biol. Chem. 277 (2002) 34367.
[22] D.D. Martin, G.L. Vilas, J.A. Prescher, G. Rajaiah, J.R. Falck, C.R. Bertozzi, L.G. Berthiaume, FASEB J. 2007 [Electronic publication ahead of print]. [23] C.L. Creasy, D.M. Ambrose, J. Chernoff, J. Biol. Chem. 271 (1996) 21049. [24] J.R. Prohaska, Physiol. Rev. 67 (1987) 858.
[25] A. Barbeau, Neurotoxicology 5 (1984) 13.
[26] T.J. Lu, C.Y. Huang, C.J. Yuan, Y.C. Lee, T.H. Leu, W.C. Chang, T.L. Lu, W.Y. Jeng, M.D. Lai, J. Inorg. Biochem. 99 (2005) 1306.
[27] H. Kakeya, R. Onose, H. Osada, Cancer Res. 58 (1998) 4888.
[28] A.A. Reszka, J.M. Halasy-Nagy, P.J. Masarachia, G.A. Rodan, J. Biol. Chem. 274 (1999) 34967.
[29] P.M. De Souza, H. Kankaanranta, A. Michael, P.J. Barnes, M.A. Giembycz, M.A. Lindsay, Blood 99 (2002) 3432.
[30] J.D. Graves, K.E. Draves, Y. Gotoh, E.G. Krebs, E.A. Clark, J. Biol. Chem. 276 (2001) 14909.
[31] S. Ura, N. Masuyama, J.D. Graves, Y. Gotoh, Genes Cells 6 (2001) 519. [32] S. Ura, N. Masuyama, J.D. Graves, Y. Gotoh, Proc. Natl. Acad. Sci. U. S. A.
98 (2001) 10148.
[33] W.L. Cheung, K. Ajiro, K. Samejima, M. Kloc, P. Cheung, C.A. Mizzen, A. Beeser, L.D. Etkin, J. Chernoff, W.C. Earnshaw, C.D. Allis, Cell 113 (2003) 507.
[34] S.H. Ahn, W.L. Cheung, J.Y. Hsu, R.L. Diaz, M.M. Smith, C.D. Allis, Cell 120 (2005) 25.
[35] Y. Hu, Z. Liu, S.J. Yang, K. Ye, Cell Death Differ. 14 (2007) 2035. [36] S. Ura, H. Nishina, Y. Gotoh, T. Katada, Mol. Cell Biol. 27 (2007)
5514.
[37] Y. Lin, A. Khokhlatchev, D. Figeys, J. Avruch, J. Biol. Chem. 277 (2002) 47991.
[38] A. Khokhlatchev, S. Rabizadeh, R. Xavier, M. Nedwidek, T. Chen, X.F. Zhang, B. Seed, J. Avruch, Curr. Biol. 12 (2002) 253.
[39] R. Dammann, C. Li, J.H. Yoon, P.L. Chin, S. Bates, G.P. Pfeifer, Nat. Genet. 25 (2000) 315.
[40] S. Ortiz-Vega, A. Khokhlatchev, M. Nedwidek, X.F. Zhang, R. Dammann, G.P. Pfeifer, J. Avruch, Oncogene 21 (2002) 1381.
[41] S. Rabizadeh, R.J. Xavier, K. Ishiguro, J. Bernabeortiz, M. Lopez-Ilasaca, A. Khokhlatchev, P. Mollahan, G.P. Pfeifer, J. Avruch, B. Seed, J. Biol. Chem. 279 (2004) 29247.
[42] H.J. Oh, K.K. Lee, S.J. Song, M.S. Jin, M.S. Song, J.H. Lee, C.R. Im, J.O. Lee, S. Yonehara, D.S. Lim, Cancer Res. 66 (2006) 2562.
[43] F. Teraishi, W. Guo, L. Zhang, F. Dong, J.J. Davis, T. Sasazuki, S. Shirasawa, J. Liu, B. Fang, Cancer Res. 66 (2006) 6072.
[44] K.F. Harvey, C.M. Pfleger, I.K. Hariharan, Cell 114 (2003) 457. [45] J. Jia, W. Zhang, B. Wang, R. Trinko, J. Jiang, Genes Dev. 17 (2003) 2514. [46] S. Pantalacc, N. Tapon, P. Léopold, Nat. Cell Biol. 5 (2003) 921–927. [47] R.S. Udan, M. Kango-Singh, R. Nolo, C. Tao, G. Halder, Nat. Cell Biol. 5
(2003) 914.
[48] S. Wu, J. Huang, J. Dong, D. Pan, Cell 114 (2003) 445.
[49] B.A. Callus, A.M. Verhagen, D.L. Vaux, FEBS J. 273 (2006) 4264. [50] M.K. Lehtinen, Z. Yuan, P.R. Boag, Y. Yang, J. Villen, E.B. Becker, S. DiBacco,
N. de la Iglesia, S. Gygi, T.K. Blackwell, A. Bonni, Cell 125 (2006) 987. [51] H. Huang, D.J. Tindall, J. Cell Sci. 120 (2007) 2479.
[52] S.W. Jang, S.J. Yang, S. Srinivasan, K. Ye, J Biol Chem. 282 (2007) 30836.
[53] O. Renner, J. Fominaya, S. Alonso, C. Blanco-Aparicio, J.F. Leal, A. Carnero, Carcinogenesis 28 (2007) 1418.
[54] B. Cinar, P.K. Fang, M. Lutchman, D. Di Vizio, R.M. Adam, N. Pavlova, M.A. Rubin, P.C. Yelick, M.R. Freeman, EMBO J. 26 (2007) 4523. [55] L. Aurisicchio, R. Di Lauro, M. Zannini, J. Biol. Chem. 273 (1998) 1477. [56] G. De Vita, L. Bauer, V.M. da Costa, M. De Felice, M.G. Baratta, M.
De Menna, R. Di Lauro, Mol. Endocrinol. 19 (2005) 76.
[57] E.E. O'Neill, D. Matallanas, W. Kolch, Cancer Res. 65 (2005) 5485. [58] C. Guo, S. Tommasi, L. Liu, J.K. Yee, R. Dammann, G.P. Pfeifer, Curr.
Biol. 17 (2007) 700.
[59] E.H. Chan, M. Nousiainen, R.B. Chalamalasetty, A. Schafer, E.A. Nigg, H.H. Sillje, Oncogene 24 (2005) 2076.
[60] J. Huang, S. Wu, J. Barrera, K. Matthews, D. Pan, Cell 122 (2005) 421. [61] D. Matallanas, D. Romano, K. Yee, K. Meissl, L. Kucerova, D. Piazzolla,
M. Baccarini, J.K. Vass, W. Kolch, E. O'neill, Mol. Cell 27 (2007) 962. [62] M.R. Stegert, A. Hergovich, R. Tamaskovic, S.J. Bichsel, B.A. Hemmings,
Mol. Cell Biol. 25 (2005) 11019.
[63] R. Tamaskovic, S.J. Bichsel, B.A. Hemmings, FEBS Lett. 546 (2003) 73. [64] Y. He, K. Emoto, X. Fang, N. Ren, X. Tian, Y.N. Jan, P.N. Adler, Mol.
Biol. Cell 16 (2005) 4139.
[65] S. Seiler, N. Vogt, C. Ziv, R. Gorovits, O. Yarden, Mol. Biol. Cell 17 (2006) 4080.
[67] M. Rohlfs, R. Arasada, P. Batsios, J. Janzen, M. Schleicher, J. Cell Sci. 120 (2007) 4345.
[68] C.M. Pombo, J.V. Bonventre, A. Molnar, J. Kyriakis, T. Force, EMBO J. 15 (1996) 4537.
[69] C.M. Pombo, T. Tsujita, J.M. Kyriakis, J.V. Bonventre, T. Force, J. Biol. Chem. 272 (1997) 29372.
[70] H.Y. Wu, C.Y. Lin, T.Y. Lin, T.C. Chen, C.J. Yuan, Apoptosis (2007) [Electronic publication ahead of print].
[71] J.S. Jamieson, D.A. Tumbarello, M. Halle, M.C. Brown, M.L. Tremblay, C.E. Turner, J. Cell Sci. 118 (2005) 5835.
[72] S.K. Sastry, P.D. Lyons, M.D. Schaller, K. Burridge, J. Cell Sci. 115 (2002) 4305.
[73] M. Hallé, Y.C. Liu, S. Hardy, J.F. Théberge, C. Blanchetot, A. Bourdeau, T.C. Meng, M.L. Tremblay, Mol. Cell Biol. 27 (2007) 1172.
[74] I. Dan, S.E. Ong, N.M. Watanabe, B. Blagoev, M.M. Nielsen, E. Kajikawa, T.Z. Kristiansen, M. Mann, A. Pandey, J. Biol. Chem. 277 (2002) 5929. [75] X. Ma, H. Zhao, J. Shan, F. Long, Y. Chen, Y. Chen, Y. Zhang, X. Han, D. Ma,
Mol. Biol. Cell 18 (2007) 1965.
[76] C. Preisinger, B. Short, V. De Corte, E. Bruyneel, A. Haas, R. Kopajtich, J. Gettemans, F.A. Barr, J. Cell Biol. 164 (2004) 1009.
[77] K. Harvey, N. Tapon, Nat. Rev. Cancer 7 (2007) 182.
[78] L.J. Saucedo, B.A. Edgar, Nat. Rev. Mol. Cell Biol. 8 (2007) 613. [79] J.F. Porter, S. Sharma, D.L. Wilson, M.A. Kappil, R.P. Hart, D.T.
Denhardt, Breast Cancer Res. Treat. 94 (2005) 185.
[80] A.S. Schrohl, M.N. Holten-Andersen, H.A. Peters, M.P. Look, M.E. Meijer-van Gelder, J.G. Klijn, N. Brunner, J.A. Foekens, Clin. Cancer Res. 10 (2004) 289.
[81] C. Seidel, U. Schagdarsurengin, K. Blumke, P. Wurl, G.P. Pfeifer, S. Hauptmann, H. Taubert, R. Dammann, Mol. Carcinog. 46 (2007) 865. [82] P. Minoo, I. Zlobec, K. Baker, L. Tornillo, L. Terracciano, J.R. Jass, A.
Lugli, Mod. Pathol. 20 (2007) 331.
[83] V. Sung, W. Luo, D. Qian, I. Lee, B. Jallal, M. Gishizky, Cancer Res. 63 (2003) 3356.
[84] K. Katagiri, A. Maeda, M. Shimonaka, T. Kinashi, Nat. Immunol. 4 (2003) 741. [85] K. Katagiri, N. Ohnishi, K. Kabashima, T. Iyoda, N. Takeda, Y. Shinkai,
K. Inaba, T. Kinashi, Nat. Immunol. 5 (2004) 1045.
[86] K. Katagiri, M. Imamura, T. Kinashi. Nat. Immunol. 7 (2006) 919. [87] V.H. Odegard, S.T. Kim, S.M. Anderson, M.J. Shlomchik, D.G. Schatz,
Immunity 23 (2005) 101.
[88] S. Yamamoto, G. Yang, D. Zablocki, J. Liu, C. Hong, S.J. Kim, S. Soler, M. Odashima, J. Thaisz, G. Yehia, C.A. Molina, A. Yatani, D.E. Vatner, S.F. Vatner, J. Sadoshima, J. Clin. Invest. 111 (2003) 1463.
[89] M. Odashima, S. Usui, H. Takagi, C. Hong, J. Liu, M. Yokota, J. Sadoshima, Circ. Res. 100 (2007) 1344.
[90] H. Ono, T. Ichiki, H. Ohtsubo, K. Fukuyama, I. Imayama, Y. Hashiguchi, J. Sadoshima, K. Sunagawa, Arterioscler. Thromb. Vasc. Biol. 25 (2005) 1871.
[91] S. Sun, K. Ravid, J. Cell Biochem. 76 (1999) 44.
[92] N. Irwin, Y.M. Li, J.E. O'Toole, L.I. Benowitz, Natl. Acad. Sci. U. S. A. 103 (2006) 18320.