Efficient combination of multiple word models
for improved sequence comparison
Xiaoqiu Huang
1,∗, Liang Ye
1, Hui-Hsien Chou
1,2, I-Hsuan Yang
3and Kun-Mao Chao
31Department of Computer Science and2Department of Genetics, Development and Cell Biology, Iowa State University, Ames, IA 50011-1040, USA and3Department of
Computer Science and Information Engineering, National Taiwan University, Taipei, Taiwan
Received on January 5, 2004; revised on April 17, 2004; accepted on April 18, 2004 Advance Access publication April 29, 2004
ABSTRACT
Motivation: Studies of efficient and sensitive sequence com-parison methods are driven by a need to find homologous regions of weak similarity between large genomes.
Results: We describe an improved method for finding sim-ilar regions between two sets of DNA sequences. The new method generalizes existing methods by locating word matches between sequences under two or more word models and extending word matches into high-scoring segment pairs (HSPs). The method is implemented as a computer program named DDS2. Experimental results show that DDS2 can find more HSPs by using several word models than by using one word model.
Availability: The DDS2 program is freely available for academic use in binary code form at http://bioinformatics.-iastate.edu/aat/align/align.html and in source code form from the corresponding author.
Contact: [email protected]
INTRODUCTION
A number of fast comparison programs have been developed for analysis of genomic DNA sequences (Pearson and Lipman, 1988; Altschul et al., 1990, 1997; Gish, unpublished data; Huang et al., 1997; Delcher et al., 1999; Burkhardt et al., 1999; Kurtz and Schleiermacher, 1999; Zhang et al., 2000; Ning et al., 2001; Kent, 2002; Ma et al., 2002; Schwartz et al., 2003). The BLASTN program (Altschul et al., 1990) is widely used for finding homologous similarities between DNA sequences. It computes high-scoring segment pairs (HSPs) between sequences by locating exact word matches of certain length between sequences and extending each word match into an HSP. The PatternHunter program (Ma et al., 2002) enhances BLASTN, in sensitivity, by allowing base differences in word matches. Word matches are defined with respect to a word model. A word model of length k is specified
∗To whom correspondence should be addressed.
by a binary string of k bits. A position at which the model has a 1 bit is called a checked position. The number of checked positions in the model is the weight of the model. Two words of length k form a word match under a word model if bases at every checked position are identical. For example, the two words ACGTC and ATGAC form a word match under the word model of 10101 of length 5 and weight 3. Note that an exact word match of length k is a word match under a word model of length k and weight k, which is called a consecutive word model of length k. The BLASTZ program (Schwartz et al., 2003) takes the idea of PatternHunter further by allow-ing a transition (A–G, G–A, C–T or T–C) in any one of the checked positions.
The sensitivity of a model is the probability of generating a word match in a fixed-length region of a given percentage identity. An optimal word model of length 18 and weight 11 has a sensitivity value of 0.467 for HSPs of length 64 and 70% identity, whereas a consecutive word model of length 11 has a sensitivity value of 0.3 (Ma et al., 2002). More HSPs can be found by PatternHunter in different runs with differ-ent models. However, results from the differdiffer-ent runs have a large number of HSPs in common and a lot of time is spent on computing HSPs that are already computed in previous runs. In this paper, we describe an efficient algorithm for finding HSPs under a set of word models simultaneously. If an HSP contains a word match under one of the word models, then the HSP is reported and no additional time is spent on the HSP. In addition, HSPs that contain a transitive word match are computed and reported once. Words x and y form a transitive word match if there is a word z such that words x and z form a match under one model in the set, and words y and z form a match under another model in the set. The algorithm is implemented as a computer program named DDS2. Exper-imental results produced by DDS2 on sequences of human chromosome 21 and mouse chromosome 16 indicate that DDS2 can find more HSPs under a set of three word models than under one optimal word model.
METHODS
We describe an algorithm for computing HSPs between two sets of sequences under a set of word models. The sequences in one set are called query sequences and those in the other are called database sequences. The query and database sequences are concatenated together with a special boundary charac-ter inserted at each sequence boundary, where the query sequences are placed before the database sequences. The resulting sequence is called the combined sequence. The con-catenation of the two sets of sequences is a slightly efficient way to represent each of the query and database sequence itions by a unique identifier, which is the location of the pos-ition in the combined sequence. The unique representation of sequence positions is used by the algorithm to deal with sets of query and database sequence positions. An alternative method is to represent each sequence position by three identifiers: data set id, sequence id and position id. Assume that the first posi-tion of the combined sequence starts at 1, where the value 0 is used to indicate that a set of positions is empty. Any word of the combined sequence with the special boundary character or any irregular base is not considered in the following steps. An alphabet of size 4, corresponding to the four regular base types, is used to reduce the space requirement of the algorithm. Two positions of the combined sequence are equivalent under a word model of length k if the words of length k starting at the two positions consist only of regular bases and form a match under the model. Two positions p1and p2of the combined sequence are equivalent if there is a word model in the set such that the two positions are equivalent under the model, or there is a position p3of the combined sequence such that p1 and p3are equivalent, and p3and p2are equivalent. Assume that each position is equivalent to itself.
The algorithm for finding HSPs between query and database sequences under the set of word models consists of two major steps. In step 1, the sets of equivalent positions are computed. Then every query position is linked to a list of database posi-tions equivalent to the query position. In step 2, for each query sequence Q, HSPs between Q and the database sequences are computed as follows. For each position q of Q and for each position d in the list of database positions equivalent to q, if the pair of positions q and d is not covered by any HSP that is already computed, then a pair of words starting at the pair of positions is extended into an HSP and the HSP is saved if its score is greater than a cutoff. HSPs between Q and a database sequence are combined into high-scoring chains of HSPs (Wilbur and Lipman, 1983; Huang, 2002). Below we describe step 1 and parts of step 2 in detail.
In step 1, initially, each position of the combined sequence is a set by itself. Every word model wm in the set is considered to merge the sets of equivalent positions under model wm as follows. Let w and k be the weight and length of the current model wm. The value of a word of length k under model wm is computed by selecting the w checked positions from the k positions, forming a subword of length w with the bases at the
wpositions, and converting the subword into a base-4 integer of w digits (Huang, 2002). Note that two words of length k form a word match under model wm if and only if the two words have the same value under model wm.
For model wm, the positions of the combined sequence are considered one at a time. Let p be the current position of the combined sequence and let v be the value of a word of length kstarting at position p under model wm. We say that position pis of value v under model wm. To locate the set of previ-ous positions of value v under wm, a word table T of size 4wis constructed such that T[v] is a position in the set of all previous positions of value v under wm. Initially, before any position of the combined sequence is considered for model wm, table T is set to zero at every entry to indicate that the set is empty. The following steps are performed for position p. If T[v] = 0, then set T [v] to p. Otherwise, if the set contain-ing position T[v] is different from the set containing position p, then merge the two sets and set T[v] to a position in the resulting set.
The sets of positions are kept in a data structure that supports Union-Find operations (Aho et al., 1974). A set of positions is represented by a tree of nodes with each node corresponding to a position. Every child node points to its parent node. A position corresponding to the root node of the tree is used as the name of the set. The trees are implemented by an array Set of the size of the combined sequence. The array Set is set up as follows. Initially, for each position p, set Set[p] to p. The initial setting indicates that each position is a set by itself. To merge (or produce a union of ) two sets with names s1and s2, set Set[s1] to s2. The name of a set containing a position p is a position sisuch that s1= Set[p], s2= Set[s1], . . . , si = Set[si−1], and si = Set[si]. Two positions p1and p2are in the same set if and only if the name of a set containing position p1is identical to the name of a set containing position p2. See Aho et al. (1974) for details on the Union-Find data structure. After all word models are considered, a position array Pos of the size of the combined sequence is constructed to facilitate the generation of pairs of equivalent query and database positions. Every set of equivalent positions is pro-cessed as follows. Let d1, d2, . . . , dj be an ordered list of database positions in the current set. Then set Pos[d1] = d2, Pos[d2] = d3, . . . , Pos[dj−1] = dj, Pos[dj] = 0. For each query position q in the current set, set Pos[q] = d1. The array Pos is used to obtain a list of database positions equivalent to a given query position. The number of pairs of equivalent posi-tions produced for a query position is the number of database positions in the set.
A large number of database positions in a set results in a large number of pairs of equivalent positions for considera-tion in generaconsidera-tion of HSPs. Thus, it is desirable to limit the number of database positions in every set for efficiency. The construction of sets of equivalent positions is revised as fol-lows. Let t be a cutoff on the number of database positions in a set for model wm. Let p be a position of value v under
the current model wm. An additional requirement is placed on the merging of the set containing position T[v] and the set containing position p. If the number of database positions in each set is at most t , then merge the two sets and set T[v] to a position in the resulting set. Otherwise, if the number of database positions in the set containing position p is at most t, then set T[v] to p. Note that if one of the two sets has more than t database positions and the other set has at most tdatabase positions, then T[v] is set to the set with at most t database positions, because the set with more than t database positions cannot be merged with another set subsequently.
In step 2, a word match is extended into an HSP, an ungapped alignment with only base matches and mismatches. In the extension, any pair of bases involving an irregular base is treated as an ordinary mismatch, and any pair of bases involving the boundary character is treated as a special mis-match of negative infinite score, which is used to terminate the extension.
A pair of query and database positions is covered by an HSP if the HSP contains a base match or mismatch at the pair of positions. If a word match starts at a pair of positions covered by a previous HSP, then the new HSP produced by extending the word match is identical to or is contained in the previous HSP. Thus, it is not necessary to extend any word match starting at a pair of positions covered by a previously computed HSP.
We describe a simple method for deciding if a pair of positions is covered by any previously computed HSP. For an HSP h, let qstart(h) denote the start position of a query region in h and dstart(h) denote the start position of a data-base region in h. The diagonal of h is defined as diag(h) = dstart(h)− qstart(h). The query positions are considered one at a time in an increasing order for computation of HSPs from word matches. Let q be the current query position and d be a database position on the list starting with Pos[q]. If the pair of positions q and d is covered by an HSP h, then the pair of posi-tions q and d is on the diagonal of h, that is, d− q = diag(h), and h is the most recently computed HSP for the diagonal.
The simple method is based on the above observation. For each diagonal, the most recently computed HSP for the diag-onal is kept if it exists. For the current pair of equivalent positions q and d, if there is an HSP for the diagonal d− q and the pair is covered by the HSP, then no extension is performed for a word match starting at the current pair of positions. Otherwise, a word match starting at the current pair of positions is extended into an HSP. If the score of the HSP is greater than the cutoff, then the HSP is added to the set of saved HSPs and is also kept as the most recently computed HSP for the diagonal d− q. The method is implemented as follows. The set of saved HSPs is represented by an array of records, where there is one record for each HSP and the index of the record is used as an internal name of the HSP. An array named Ret is used to keep the most recently computed HSP for each diagonal. Initially, Ret is set to 0 at each entry,
indicating that no HSP is kept for each diagonal. When an HSP of score greater than the cutoff is saved with an internal name h, Ret[diag(h) + qlen] is set to h, where qlen is the largest query position and therefore diag(h)+ qlen is non-negative. Note that the array Set in step 1 and the array Ret in step 2 have no overlap in their life spans and can share the same physical memory.
RESULTS
The new algorithm is implemented as a computer program named DDS2, an improved version of the DDS (DNA–DNA Search) program by Huang et al. (1997). The DDS2 program handles only DNA sequences. The program takes as input two sets of sequences in FASTA format, where the first set is called a query set and the second set a database set. The memory requirement of DDS2 is reduced by processing the two sets in blocks. The query sequences are partitioned into a number of blocks. The total number of bases in each block is con-trolled by a parameter called a query block size. Every query sequence of length greater than or equal to the query block size is a block by itself. For the remaining query sequences of lengths less than the query block size, each block con-sists of a maximum number of query sequences with a total length less than or equal to the query block size. The database sequences are similarly partitioned into blocks according to a database block size. All pairs of query and database blocks are processed by DDS2 one at a time. For the current pair of query and database blocks, the query block in both orienta-tions is compared by DDS2 with the database block in given orientation.
The memory requirement of DDS2 is about 13–15 times the size s of the largest pair of query and database blocks. Two integer arrays of size s are used in DDS2. One is used for the word table T and the other for the array Set. After Set is con-structed, the physical memory for T is reused for the position array Pos. After Pos is constructed, the physical memory for Set is reused for the array Ret. Three character arrays of size s are used for query and database sequences in the current pair of blocks, their integer base codes and the number of equivalent positions in each set. Assume that each integer takes 4 bytes of memory and each character takes 1 byte of memory. Then the two integer arrays and three character arrays take a total of 11× s bytes. Additional memory is required to keep all HSPs between the current query sequence and database sequences. The space requirement for HSPs is usually between 2× s and 4× s.
The DDS2 program was evaluated in two experiments. In the first experiment, DDS2 was used to compare a set of three human chromosome 21 sequences with a total length of 37 Mb with a set of eight mouse chromosome 16 sequences with a total length of 99 Mb from the UCSC Genome Browser at http://www.genome.ucsc.edu. The human and mouse sequences were screened for repeats (Smit and Green, unpublished data). The masked sequences were used as input
to DDS2 in all runs described below. All the runs were per-formed on a Sun Blade 1000 server with 1 GB of memory. The query block size was set to 26 Mb and the database block size was set to 22 Mb so that the memory requirement of DDS2 did not exceed 1 GB. Each base match was given a score of 2 and each mismatch a score of−3. The extension in each direction for computing the HSP was terminated if the best score dropped by at least 40, as in the case of encountering 14 mismatches in a row. The HSP cutoff score was set to 25, the score of a 50 bp HSP of 70% identity, where 50 bp is a minimum length of most coding exons.
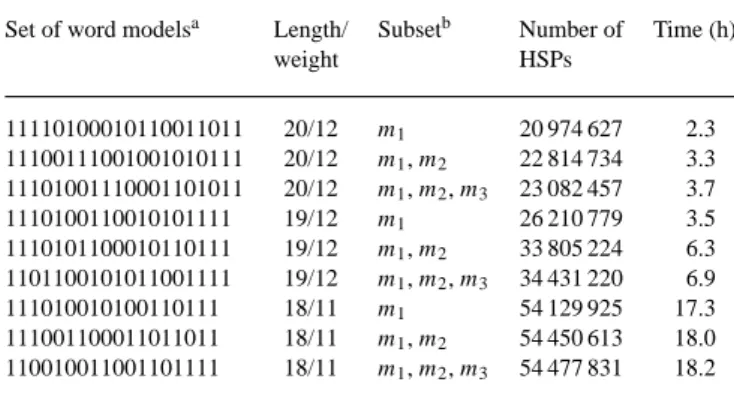
Three sets of word models, referred to as sets 1, 2 and 3, were selected to evaluate the performance of DDS2. The three sets of word models are shown in Table 1. Each set contains three word models referred to as models 1, 2 and 3. Set 1 was produced by a computer program (Yang et al., manuscript in preparation), whereas sets 2 and 3 were constructed by combining models from different sources. Model 1 in set 2 was a word model used by BLASTZ, and models 2 and 3 in set 2 were obtained by modifying models 2 and 3 in set 1. Model 1 in set 3 was a word model used by PatternHunter, and models 2 and 3 in set 3 were obtained by modifying models 2 and 3 in set 1. To see the effect of additional word models on the performance of DDS2, for each set of word models, three runs were made: the first run with only model 1, the second run with models 1 and 2, and the third run with models 1, 2 and 3. The number of HSPs produced by DDS2 and the time required by DDS2 in each run are shown in Table 1. The DDS2 program took at most 852 MB of main memory in each run.
For set 1, DDS2 was able to produce 10% more HSPs by using three models, at the cost of a 61% increase in running time. For set 2, DDS2 was able to produce 31% more HSPs by using three models, at the cost of a 97% increase in running time. For set 3, DDS2 was able to produce 1% more HSPs by using three models, at the cost of a 5% increase in running time. The results in Table 1 indicate that using one word model
Table 1. Performance of DDS2 for three sets of word models
Set of word modelsa Length/ Subsetb Number of Time (h)
weight HSPs 11110100010110011011 20/12 m1 20 974 627 2.3 11100111001001010111 20/12 m1, m2 22 814 734 3.3 11101001110001101011 20/12 m1, m2, m3 23 082 457 3.7 1110100110010101111 19/12 m1 26 210 779 3.5 1110101100010110111 19/12 m1, m2 33 805 224 6.3 1101100101011001111 19/12 m1, m2, m3 34 431 220 6.9 111010010100110111 18/11 m1 54 129 925 17.3 111001100011011011 18/11 m1, m2 54 450 613 18.0 110010011001101111 18/11 m1, m2, m3 54 477 831 18.2
aFor each set of three word models, the three models are shown on separate lines and are
referred to as m1, m2and m3.
bThe subset of models was used by DDS2 to compute HSPs between a set of human
chromosome 21 sequences and a set of mouse chromosome 16 sequences.
of smaller weight results in a dramatic increase in the sensit-ivity of DDS2 at the cost of a dramatic increase in its running time. On the other hand, using several word models of larger weight is an alternative way to obtain a modest increase in the sensitivity of DDS2 at the cost of a modest increase in its running time.
In the second experiment, the performance of DDS2 was compared with that of a local alignment program named SIM (Huang and Miller, 1991). The SIM program is a space-efficient implementation of the Smith–Waterman algorithm (Smith and Waterman, 1981), a standard in the area of DNA sequence comparison. The SIM program computes k best local alignments between two sequences. Because SIM is much slower than DDS2 and DDS2 is less sensitive than SIM on sequences of weak similarity, short sequences of weak similarity should be used in the comparison.
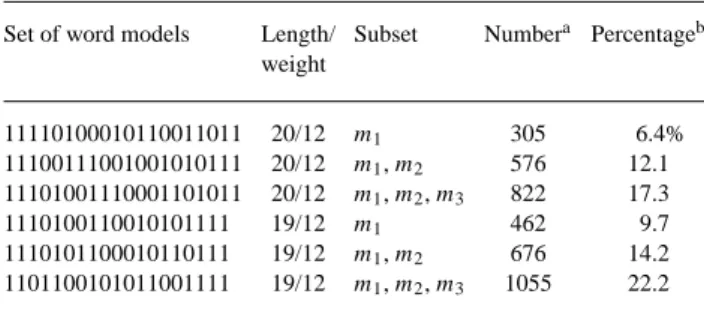
A major issue was how to find many pairs of short sequences of weak similarity. It was too slow to use SIM on the two large sets of human chromosome 21 and mouse chromosome 16 sequences. We decided to find pairs of regions of weak sim-ilarity between the two sets of sequences with DDS2 under model set 3, which is more sensitive than model sets 1 and 2. A total of 4587 pairs of regions of low similarity with a total length of 248 230 bp were selected for comparing the sensit-ivity of DDS2 under model sets 1 and 2 with the sensitsensit-ivity of SIM. The match and mismatch scores for both DDS2 and SIM were set to 2 and−3, respectively. The gap open and extension penalties for SIM were set to 20 and 5.
For each pair of regions, SIM was run to find all local align-ments of score at least 25 between the regions by setting the kparameter to a large value. A total of 4756 local alignments of score at least 25 were computed by SIM on all the pairs of regions. The performance of DDS2 on all the pairs of regions was evaluated with respect to model sets 1 and 2. For each of model sets 1 and 2, there were three subsets of word models, as in the first experiment. For each subset of word models, DDS2 was run under the subset on each pair of regions to compute HSPs of score at least 25 between the regions. The number and percentage of SIM alignments that were completely covered by HSPs from DDS2 under the subset of word models were calculated, where a SIM alignment was completely covered by HSPs from DDS2 if every match and mismatch of the SIM alignment was on one of the HSPs. The results, shown in Table 2, indicate that more SIM alignments were found by DDS2 under multiple word models. However, the low percent-ages of SIM alignments found by DDS2 suggest that model sets 1 and 2 are not sensitive enough to find the weak simil-arities between the sequences. Note that the weak similsimil-arities were found by DDS2 under model set 3.
DISCUSSION
The set of HSPs computed by our algorithm may not be identical to the set of HSPs produced by a simple method,
Table 2. The number and percentage of SIM local alignments found by
DDS2
Set of word models Length/ Subset Numbera Percentageb
weight 11110100010110011011 20/12 m1 305 6.4% 11100111001001010111 20/12 m1, m2 576 12.1 11101001110001101011 20/12 m1, m2, m3 822 17.3 1110100110010101111 19/12 m1 462 9.7 1110101100010110111 19/12 m1, m2 676 14.2 1101100101011001111 19/12 m1, m2, m3 1055 22.2
aThis column reports the number of SIM alignments that were completely covered
by HSPs from DDS2 under the subset of word models.
bThis column reports the percentage of SIM alignments that were completely covered
by HSPs from DDS2 under the subset of word models.
which first uses single word models one after another, and then produces the union of the sets of HSPs from each model. Our algorithm spends a significant amount of time on transit-ive word matches and is able to produce additional HSPs from some of those matches. On the other hand, the simple method spends a significant amount of time on HSPs that are already computed from other word models.
In the DDS2 program, a primitive method is used to extend a word match into an HSP, which allows only base matches and mismatches in the extension. Zhang et al. (2000) describes an advanced method called X-drop for extending a word match into an alignment, which allows insertions and deletions as well as matches and mismatches. We plan to replace the primitive method in DDS2 by the X-drop method in the future. The time requirement of our algorithm is significantly affected by the number of word models and their minimum weight w. The algorithm consists of three parts: construction of sets of equivalent positions, extension of word matches into HSPs and combination of HSPs into chains. The construction part always requires time proportional to the total length n of the input sequences. The combination part requires, on the average, time proportional to the total m number of HSPs, and in the worst case, time proportional to m2. The extension part is the most time-consuming part of the algorithm. If 4w > n and the number of word models is very small, then there are very few word matches for each query position and therefore the extension part requires time proportional to n, which is the best case. On the other hand, if 4w < nor the number of word models is large, then up to t word matches are con-sidered for each query position and therefore the extension part is, in the worst case, t times slower than in the best case. The parameter t was set to 250 in DDS2.
ACKNOWLEDGEMENTS
We thank Brian Haas for suggestions on improvements to DDS. We are grateful to the reviewer for constructive
suggestions and comments on the manuscript. X.H. and L.Y. are supported in part by NIH grants R01 HG01502 and R01 HG01676, USA. H.-H.C. is supported in part by NIH grant 4R33 GM066400, USA. K.-M.C. is supported in part by NSC grant 92-2213-E-002-059, Taiwan.
REFERENCES
Aho,A.V., Hopcroft,J.E. and Ullman,J.D. (1974) The Design and Analysis of Computer Algorithms. Addison-Wesley Publishing Company, Reading, MA.
Altschul,S.F., Gish,W., Miller,W., Myers,E.W. and Lipman,D.J. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410.
Altschul,S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Gapped blast and psi-blast: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402.
Burkhardt,S., Crauser,A., Lenhof,H.-P., Rivals,E., Ferragina,P. and Vingron,M. (1999) Q-gram based database searching using a suffix array. In Third Annual International Conference on Computational Molecular Biology. Lyon, France, April 1999, pp. 11–14.
Delcher,A.L., Kasif,S., Fleischmann,R.D., Peterson,J., While,O. and Salzberg,S.L. (1999) Alignment of whole genomes. Nucleic Acids Res., 27, 2369–2376.
Huang,X. (2002) Bio-sequence comparison and applications. In Jiang,T., Xu,Y. and Zhang,M. (eds), Current Topics in Compu-tational Molecular Biology. MIT Press, Cambridge, pp. 45–69. Huang,X., Adams,M.D., Zhou,H. and Kerlavage,A.R. (1997) A tool
for analyzing and annotating genomic sequences. Genomics, 46, 37–45.
Huang,X. and Miller,W. (1991) A time-efficient, linear-space local similarity algorithm. Adv. Appl. Math., 12, 337–357.
Kent,W.J. (2002) BLAT—the BLAST-like alignment tool. Genome Res., 12, 656–664.
Kurtz,S. and Schleiermacher,C. (1999) REPuter—fast computation of maximal repeats in complete genomes. Bioinformatics, 15, 426–427.
Ma,B., Tromp,J. and Li,M. (2002) PatternHunter: faster and more sensitive homology search. Bioinformatics, 18, 440–445. Ning,Z., Cox,A.J. and Mullikin,J.C. (2001) SSAHA: a fast search
method for large DNA databases. Genome Res., 11, 1725–1729. Pearson,W.R. and Lipman,D. (1988) Improved tools for
biolo-gical sequence comparison. Proc. Natl Acad. Sci., USA, 85, 2444–2448.
Schwartz,S., Kent,W.J., Smit,A., Zhang,Z., Baertsch,R., Hardison,R., Haussler,D. and Miller,W. (2003) Human–mouse alignments with BLASTZ. Genome Res., 13, 103–107.
Smith,T.F. and Waterman,M.S. (1981) Identification of common molecular subsequences. J. Mol. Biol., 147, 195–197.
Wilbur,W.J. and Lipman,D.J. (1983) Rapid similarity searches of nucleic acid and protein data banks. Proc. Natl Acad. Sci., USA, 80, 726–730.
Zhang,Z., Schwartz,S., Wagner,L. and Miller,W. (2000) A greedy algorithm for aligning DNA sequences. J. Comput. Biol., 7, 203–214.