行政院國家科學委員會專題研究計畫 成果報告
腎病症侯群的分子生物研究(3/3)
計畫類別: 個別型計畫 計畫編號: NSC92-2314-B-039-018- 執行期間: 92 年 08 月 01 日至 93 年 07 月 31 日 執行單位: 中國醫藥大學醫學研究部 計畫主持人: 張建國 報告類型: 完整報告 報告附件: 出席國際會議研究心得報告及發表論文 處理方式: 本計畫可公開查詢中 華 民 國 93 年 8 月 19 日
行政院家科學委員會專題研究計畫成果報告
計畫名稱:腎病症候群的分子生物研究
計畫編號:NSC 90-2320-B039-013
NSC 91-2314-B-039-007
NSC 92-2314-B-039-018
執行期限:90 年 08 月 01 日至 93 年 07 月 31 日
主 持 人:張建國
執行機構及單位名稱:中國醫藥大學 醫學研究所
聯絡方式:張建國
中國醫藥大學附設醫院 分子醫學科
404 台中市北區育德路 2 號
Tel:
(04)2205-2121 ext.7075
Fax:(04)2203-3295
E-mail:[email protected]
中文摘要 糖尿病及高血壓是造成腎衰竭的最 主要的原因之一,有相當多的因子可使糖 尿 病 或 高 血 壓 的 病 人 更 快 進 展 至 腎 衰 竭。在過去兩年我們探討了自由基代謝基 因及腎病症候群相關基因在腎衰竭所扮 演的角色。而本年度的計劃則主要探討藥 物代謝基因在腎衰竭發展過程所扮演的 角色,我們利用 PCR-RFLP 及直接定序的 方法,分析了 CYP1A1、CYP2E1、GSTM1、 GSTT1、MTHFR、APOV6 及 CCR5 等基因的變 異型與腎衰竭的關係,結果顯示,CYP2E1 的 Wild type 與高血壓進展至腎衰竭有 關,(p=0.007),而 GSTT1 與糖尿病進展 至腎衰竭有關(p=0.004)。 關鍵詞:高血壓、糖尿病、腎衰竭、藥物 代謝基因、變異型、PCR-RFLP 。 Abstract
DM and Hypertension play important roles in the development of chronic renal insufficiency(CRI).There are a number of factors which may promote the development of CRI in DM and Hypertension patients.In the past two years﹐ we focused on the studies of free radical metabolized-related genes and nephritic ryndrome-related genes to explore their roles in the development of CRI.In this study﹐we explored the relationshy’s letueen the dmg-metabolized-related t genes and direct sequencing methods.We analyzed the vaiant forms of CYP1A1 、 CYP2E1 、 GSTM1、GSTT1、MTHFR、APOV6、CCR5 and the results showed that CYP2E1 and GSTT1 play a role in the development of CRI from DM and Hypertension.
Keywords: DM、 Hypertension、 chronic
renal insufficiency 、
dmg-metabolized-related genes 、 vaiant forms、PCR-RFLP
Introduction
Reactive oxygen metabolites (ROS) are the product of partial reduction of oxygen in biological systems and include superoxide anions, hydrogen peroxides, hydroxyl radicals and hypochlorous acid (1-5). Many evidences have shown that ROS plays a role in the pathogenesis of a variety of diseases (1-6). In addition, ROS plays a role in neutrophil-dependent and macrophage-dependent or -independent glomerular injury (7-8), and also in the development of acute renal failure caused by endotoxin, glycerol, gentamicin, adriamycin, lupus nephritis, puromycin aminonucleoside, and the institution of ischemia (9-11). In addition to invading cells (macrophages, neutrophils), ROS may be produced by isolated glomeruli and by cultured mesangial cells in response to diverse stimuli.
Hyperglycermia has been shown to associate with the production of ROS in diabetic patients (12-14). The production of ROS is considered to be one of the major causes of diabetic complications, including nephropathy (15-16). There are a number of endogenous antioxidants that provide protection against the harmful effects of ROS. For instance, superoxide can be scavenged by superoxide dismutase (SOD), and hydrogen peroxide can be degraded by catalase or glutathione peroxidase, resulting in the formation of water (17-18). Glutathione S-transferases (GSTs) are a family of multifunctional enzymes that play an important role in the cellular detoxification and excretion of numerous physiological and xenobiotic substances (19-22). GSTs also work as antioxidants by catalyzing the conjugation of electrophilic compounds including carcinogens, cytotoxic drugs and organic hydroperoxides with reduced glutathione (19-21). Therefore, diminished expression of GSTs may result
2
in a reduced capacity of defense against oxidative stress, followed by the development of diabetic nephropathy.
Human GSTs contain at least five classes: α, π, µ, θ and κ (19,21,23,24). In human kidney, GST µ class is mainly localized in tubuli (25). Human µ class GSTs are thought to be products of M1, M2, M3, M4 and M5 gene loci (21, 26-27). The expression of M1 membrane of human GST
µ class (GSTM1) is detected only in about 50% of all populations. The genetic locus encoding human GSTM1 is polymorphic and the absence of GSTM1 has been ascribed to the homozygous deletion of the gene (null genotype) (28-30). GST θ class is highly expressed in liver and kidney, and there are two types: GSTT1 and GSTT2. The genetic locus encoding human GSTT1 is also polymorphic, and the absence of the GSTT1 has been found in 15%-30% of Caucasians and over 50% of Chinese (31-32).
In order to determine whether the GSTM1 and GSTT1 null genotypes are associated with the development of diabetic or hypertensive related end-stage renal disease (ESRD), we investigated the genetic polymorhisms in type 2 diabetic patients with ESRD and in hypertension patients with ESRD as well as patients without ESRD.
Materials and Methods
Patients
We recruited 119 type 2 DM patients with diabetic-related ESRD and 111 type 2 DM patients without diabetic ESRD, microalbuminuria or proteinuria. We also recruited 101 patients with hypertension-related ESRD and 374 patients of hypertension without ESRD. All patients were Taiwanese of Han-Origin. The diagnostic criterion for ESRD was chronic renal failure with serum creatinine >8.0 mg/dl or creatinine clearance <5 ml/min, need start dialysis. Ethnic approvals were obtained from the institutional review board of China Medical University Hospital and Changhua Christian Hospital, and informed consent was received from all participants.
GSTM1 and GSTT1 genotyping
Genomic DNA was extracted from peripheral white blood cells as in our previous studies (33). Genotyping of GSTM1 and GSTT1 was performed by multiplex polymerase chain reaction (PCR) using primers from the protocols of Comstock et al (34) and Pemble et al (35) with some modifications. The primers used were as follows: for detection of GSTM1, the forward primer was 5'-CTG CCC TAC TTG ATT GAT GGG-3' and the reverse primer was 5'-CTG GAT TGT AGC AGA TCA TGC; for detection of GSTT1, the forward primer was 5'-TTC CTT ACT GGT CCT CAC ATC TC-3' and the reverse primer was 5'-TCA CCG GAT CAT GGC CAG CA-3'. In order to confirm that the PCR had worked in subjects homozygous for the GSTM1 or GSTT1 gene deletion, one pair of primers was used as an internal control to amplify a 100 bp fragment of the
β-globin gene: the forward primer was 5'-ACA CAA CTG TGT TCA CTA GC-3', the reverse primer was 5'-CAA CTT CAT CCA CGT TCA CC-3'. The PCR mixture (100 µl) contained 200-500 ng of genomic DNA, 10 pmol of each primer, 2.5mM of each dNTP, 1.5mM MgCl2, 500 mM KCl,
10 mM Tris HCl (pH8.3) and 2.5 units of Taq polymerase (Protech, Taipei, Taiwan). Amplification was performed in a thermal cycler (Perkin Elmer, Foster City, CA, USA) for 35 cycles with steps of denaturing at 94 ℃ for 1min, annealing at 58℃ for 1 min and extension at 72℃ for 5 min. The PCR products were analyzed by electrophoresis in 3% agarose gels and visualized by UV light.
Statistical analysis
The differences in distribution of the GSTM1 and GSTT1 genotypes between ESRD patients with diabetes and ESRD patients with hypertension, and controls were determined by chi-square test. Probability values of <0.05 were regarded as statistically significant. Odds ratios (ORs) with a 95% confidence interval (CI) calculated using unconditional logistic regression and adjusted for age and gender
were computed to estimate the association between certain genotypes and diseases. All of the statistical analyses were performed by Statistical Analysis System software (SAS Institute, Cary, NC, USA).
Results
The clinical characteristics of the subjects in this study are shown in the Tables 1 and 2. The diabetic (119 with ESRD, 111 without ESRD) and
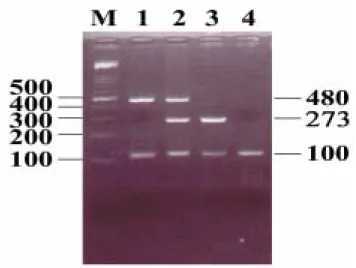
hypertensive (101 with ESRD, 374 without ESRD) groups were well-matched in regard to age, cholesterol, and triglyceride levels. As expected, serum creatine concentrations of patients with ESRD were significantly high. The results of the multiplex PCR analysis of GSTM1 and GSTT1 are shown in Fig. 1. The PCR product of the GSTM1 and GSTT1 genes are a fragment of 273 bp and a fragment of 480 bp, respectively. The amplican of the β-globin gene for internal control showed a 100 bp fragment. In total, 705 cases were analyzed.
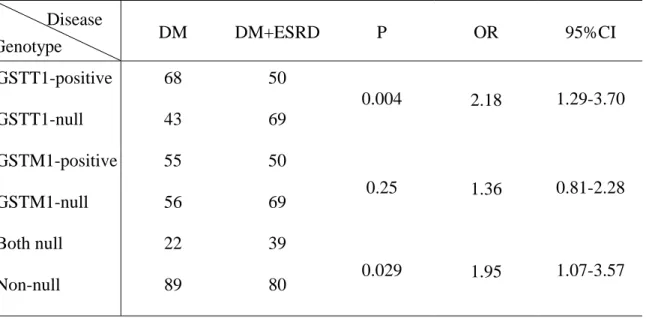
The genotypic frequencies of the GSTM1 and GSTT1 polymorphisms in DM patients with or without ESRD and in hypertension patients with or without ESRD are shown in Tables 3 and 4, respectively. The homozygous deletion of the GSTM1 gene was observed in 58.0% (69/119) of diabetic patients with ESRD versus 50.5% (56/111) of those without ESRD, and in 53.4% of hypertensive patients with ESRD versus 54.8% of those without ESRD. The presence of the GSTM1 gene was identified in 42.0% of diabetic patients with ESRD versus 49.5% of those without ESRD, and in 46.6% of hypertensive patients with ESRD versus 45.2% of those without ESRD. No significant difference in genotypic frequencies was found in these two groups (P=0.25 for the diabetic group, p=0.81 for the hypertensive group) (Table 3 and Table 4). The homozygous deletion mutation of the GSTT1 gene was observed in 58.0% of diabetic patients with ESRD versus 38.8% without ESRD, and in 40.6% of hypertensive patients with ESRD versus 36.6% without ESRD. The presence of the
GSTT1 gene was identified in 42.0% of diabetic patients with ESRD versus 61.2% without ESRD, and in 59.4% of hypertensive patients with ESRD versus 63.4% without ESRD. Genotypic frequencies were significantly different between the diabetic groups (P=0.004, OR=2.18, 95% CI=1.29-3.70), but no significant difference was observed between the hypertensive groups (P=0.47).
The frequencies of homozygous deletion of both GSTM1 and GSTT1 genes was much higher in diabetic patients (32.8%) than those without ESRD (19.9%). The difference in frequencies was statistically significant (Table 3). However, the homozygous deletion of both GSTM1 and GSTT1 genes was observed in 17.8% of hypertensive patients with ESRD versus 12.0% without ESRD. There was no statistical significance in this group (Table 4)
Discussion
ROS participates in the pathogenesis of various renal diseases, including inflammatory lesions such as glomerulonephritis and interstitial nephritis, ischemic reperfusion injury, hemolytic uremic syndrome, toxic nephropathies, and possibly chronic renal failure (7,9). Several studies have repected that antioxidants may improve renal function of the pationts with these diseases (17-19). Recent studies also demonstrated that vitamin E and glutathione can prevent the development of renal complications in diabetic animals and patients (36-37). These results suggest that the levels of endogenous antioxidants may be a determinant of the susceptibility of diabetic patients to renal complications in.
The activity of GSTM1, one of the endogenous antioxidant enzyme, is determined genetically (24,30). Several studies have demonstrated that the genotype of homozygous GSTM1 deletion GSTM1 activity (24, 29-30), and no association of this genotype with diabetic complications was found in the study by Fujita et al (38). Our data are consistent with their results.
4
However, another endogenous antioxidant, GSTT1, showed its effect on diabetic complications. The homozygous deletion genotype, which reduced the GSTT1 activity, was associated with diabetic ESRD.
Little has been known about the role of ROS in the development of ESRD in hypertensive patients. Our study revealed that lower activities of antioxidant enzymes GSTM1 and GSTT1 were not associated with the development of ESRD in hypertensive patients. Therefore, ROS may not play an important role in the development of ESRD in hypertensive patients.
The mechanism of development of ESRD in DM patients and hypertensive patients is different. DM may produce much more oxidative stress than hypertension so that it requires more antioxidant to reduce the stress. This is the reason for the association between GSTT1-null genotype and DM in patients with ESRD, but not in hypersensitive patients with ESRD. GSTM1 is also an important endogenous antioxidant enzyme, but it does not play an important role in the development of ESRD in DM or hypertensive patients. This suggests that GSTT1 plays a more important role than GSTM1 in the process of reducing oxidative stress in the kidney.
In conclusion, our results indicate that genetic variations of GSTT1 enzyme involving in free radical metabolism in DM are associated with the development of ESRD and may permit the targeting of preventative and early intervention strategies to high-risk individuals.
Acknowledgments
We thank Miss W. L. Chan for editing the manuscript. This study was supported in part by grants from National Science Council, Taiwan (NSC 90-2320-B039-013; NSC 91-2314-B-039-007; NSC 92-2314-B-039-018 for Chang, JG), a grant from China Medical University Hospital, Taiwan. (DMR-91-030), and a grant from Changhua Christian Hospital, Taiwan
(Grant no.9248). Reference:
1. Baud L and Ardaillou, R: Reactive oxygen species: Production and role in the kidney. Am J Physiol 251: F765-776, 1986.
2. McCord JM: Oxygen-derived free radicals in postischemic tissue injury. N Engl J Med 312: 159-163, 1985.
3. Fantone JC and Ward PA: Role of oxygen-derived free radicals and metabolites in leukocyte-dependent inflammatory reactions. Am J Pathol 107: 397-418, 1982.
4. Weiss SJ: Oxygen, ischemia and inflammation. Acta Physiol Scand 126: 9-37, 1986.
5. McCord JM and FridovichI: The biology and pathology of oxygen radicals. Ann Intern Med 89: 122-127, 1978.
6. Cross CE, Halliwell B, Borish ET, Pryor WA, Ames BN, Saul RL, McCord JM and Harman D: Oxygen radicals and human disease. Ann Intern Med 107: 526-545, 1987.
7. Shah S: Oxidant mechanisms in glomerulonephritis. Semin Nephrol 11: 320-326, 1999.
8. Li JZ, Sharma R, Dileepan KN and Savin VJ: Polymorphonuclear leukocytes increase glomerular albumin permeability via hypohalous acid. Kidney Int 46: 1025-1030, 1994.
9. Saulo K: Oxygen radicals and renal diseases. Miner Electrolyte Metab 23: 140-143, 1997.
10. Baliga R, Ueda N, Walker PD and Shah SV: Oxidant mechanisms in toxic acute renal failure. Am J Kidney Dis 29: 465-477, 1997.
11. Nath KA and Norby SM: Reactive oxygen species and acute renal failure. Am J Med 109: 655-678, 2000.
12. Dandona P, Thusu K, Cook S, Snyder B, Makowski J, Armstrong D and Nicotera T: Oxidative damage to DNA in diabetes mellitus. Lancet 346: 444-445, 1996.
and Carrascosa A:. Oxidative stress at onset and in early stages of type 1 diabetes in children and adolescents. Diabetes Care 21: 1736-1742, 1998. 14. Hinokio Y, Suzuki S, Hirai M, Chiba
M, Hirai A and Toyota T: Oxidative DNA damage in diabetes mellitus: its association with diabetic complications. Diabetologia 42: 995-998, 1999.
15. Baynes JW: Role of oxidative stress in development of complications in diabetes. Ddiabetes 40: 405-415, 1991. 16. Giugliano D, Paolisso G and Ceriello
A: Oxidative stress and diabetic vascular complications. Diabetes Care 19: 257-267, 1996.
17. Fridovich I: The biology of oxygen radicals. The superoxide radical is an agent of oxygen toxicity; superoxide dismutases provide an important defense. Science 201: 875-880, 1978. 18. Turi S, Nemeth I, Torkos A, Saghy L,
Varga I, Matkovics B and Nagy J: Oxidative stress and antioxidant defense mechanism in glomerular diseases. Free Radic Biol Med 22: 161-168, 1997.
19. Jakoby WB: The glutathione S-transferases: a group of multifunctional detoxification proteins. Adv Enzymol 46: 383-414, 1978.
20. ChasseaudLF: The role of glutathione and glutathione S-transferases in the metabolism of chemical carcinogens and other electrophilic agents. Adv Cancer Res 29: 175-274, 1979.
21. Wilce MCJ and ParkerMW: Structure and function of glutathione S-transferases. Biochem Biophys Acta 1205: 1-18, 1994.
22. Prohaska JR and Ganther HE: Glutathione peroxidase activity of glutathione S-transferases purified from rat liver. Biochem Biophsy Res Common 76: 437-445, 1997.
23. Lawrence RA and Burk RF: Species, tissue and subcellular distribution of non Se-dependent glutathione peroxidase activity. J Nutr 108: 211-215, 1978.
24. Prohaska JR: The glutathione peroxidase activity of glutathione
S-transferases. Biochem Biophys Acta 611: 87-98, 1980.
25. Harrison DJ, Kharbanda R, Cunningham DS, McLellan LI and Hayes JD: Distribution of glutathione S-transferase isoenzymes in human kidney: basis for possible markers of renal injury. J Clin Pathol 42: 624-628, 1989.
26. Zhong S, Suprr NK, Hayes JD and Wolf CR: Deduced amino acid sequence, gene structure and chromosomal location of a novel human class mu glutathione S-transferase, GSTM4. Biochem J 291: 41-50, 1993.
27. Takahashi Y, Campbell EA, Hirata Y, Takayama T and Listowsky I: A basis for differentiating among the multiple human mu glutathione S-transferases and molecular cloning of brain GSTM5. J Biol Chem 268: 8893-8898, 1993. 28. Seidegard J, Pero RW, Markowitz MM,
Roush G, Miller DG and Beattie EJ: Isoenzyme (s) of glutathione transferase (class Mu) as a marker for the susceptibility to lung cancer: a follow up study. Carcinogenesis 11: 33-36, 1990.
29. Zhong S, Wyllie AH, Barnes D, Wolf CR and Spurr NK: Relationship between the GSTM1 genetic polymorphism and susceptibility to bladder, breast and colon cancer. Carcinogenesis 14: 1821-1824, 1993. 30. Seidegard J, Vorachek WR, Pero RW
and Person WR: Hereditary differences in the expression of the human glutathione transferase active on trans-stilbene oxide are due to a gene deletion. Proc Natl Acad Sci U S A 85: 7293-7297, 1988.
31. Deakin M, Elder J, Hendrickse C, Peckham D, Baldwin D, Pantin C, Wild N, Leopard P, Bell DA, Jones P, Duncan H, Brannigan K, Alldersea J, Fryer AA and Strange RC: Glutathione S-transferase GSTT1 genotypes and susceptibility to cancer: studies of interactions with GSTM1 in lung cancer, oral, gastric and colorectal cancers.
6
Carcinogenesis 17: 881-884, 1996. 32. Garte S, Gaspari L, Alexandrie AK, et
al: Metabolic gene polymorphism frequencies in control populations. Cancer Epidemiol Biomarkers Prev 10: 1239-1248, 2001.
33. Lee HH, Chang JG, Lin SP, Chao HT, Yang ML and Ng HT: Rapid detection of trisomy 21 by homologous gene quantitative PCR (HGQ-PCR). Hum Genet 99: 364-367, 1997.
34. Comstock KE, Sanderoon BJS, Claflin G and Henner WD: GST1 gene deletion determined by polymerase chain reaction. Nucleic Acids Res 18: 3670, 1990.
35. Pemble S, Schroeder KR, Spencer SR, et al: Human glutathione S-transferase theta (GSTT1): cDNA cloning and the characterization of a genetic polymorphism. Biochem J 300: 271-276, 1994.
36. Keegan A, Walbank H, Cotter MA and Cameron NE: Chronic vitamin E treatment prevents defective endotheliun-dependent relaxation in diabetic rat aorta. Diabetologia 38: 1475-1478, 1995.
37. Bravenboer B, Kappelle AC, Hamers FP, van Buren T, Erkelens DW and Gispen WH: Potential use of glutathione for the prevention and treatment of diabetic neuropathy in the streptozotocin-induced diabetic rat. Diabetologia 35: 813-817, 1992.
38. Fujita H, Narita T, Meguro H, et al: No association of glutathione S-transferase M1 gene polymorphism with diabetic nephropathy in Japanese type 2 diabetic patients. Renal Fail 22: 479-486, 2000.
Fig. 1. The results of representative PCR analysis of GSTM1 and GSTT1 genes are shown.
M: marker. Lane 1: GSTM1 positive (273 bp fragment) with GSTM1 deletion. Lane 2:
GSTT1 (480 bp fragment) and GSTM1 positive. Lane 3: GSTM1 positive with GSTT1
deletion. Lane 4: Both GSTM1 and GSTT1 deletion. The 100 bp fragment is the product of
8
Table 1. The clinical data of type2 DM with or without ESRD
DM (n=111) DM with ESRD (n=119) P-value
Gender (M/F) 53/58 57/62 Age (years) 62.51±12.03 62.50±11.94 0.92 BMI (kg/m2) 23.86±4.06 23.09±3.65 0.13 Cholesterol (mg/dL) 190.06±51.07 181.54±51.39 0.33 Triglyceride (mg/dL) 189.94±131.71 176.57±185.04 0.73 Creatinine (mg/L) 1.092±0.49 11.258±5.46 <0.05 HbA1C (%) 7.86±1.85 7.08±2.19 <0.02
Data are means±SD. BMI: body mass index.
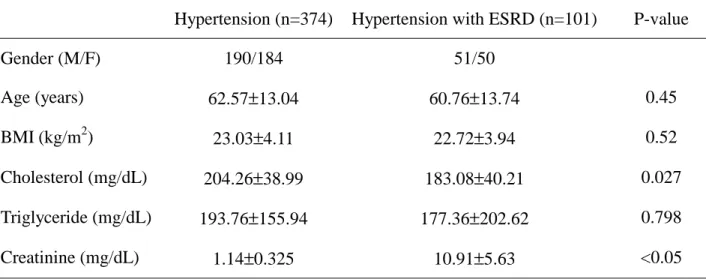
Table 2. The clinical data of hypertension with or without ESRD
Hypertension (n=374) Hypertension with ESRD (n=101) P-value
Gender (M/F) 190/184 51/50 Age (years) 62.57±13.04 60.76±13.74 0.45 BMI (kg/m2) 23.03±4.11 22.72±3.94 0.52 Cholesterol (mg/dL) 204.26±38.99 183.08±40.21 0.027 Triglyceride (mg/dL) 193.76±155.94 177.36±202.62 0.798 Creatinine (mg/dL) 1.14±0.325 10.91±5.63 <0.05
Table 3. The genotype frequencies of GSTM1 and GSTT1 polymorphisms in DM with or without ESRD DM DM+ESRD P OR 95%CI GSTT1-positive 68 50 GSTT1-null 43 69 0.004 2.18 1.29-3.70 GSTM1-positive 55 50 GSTM1-null 56 69 0.25 1.36 0.81-2.28 Both null 22 39 Non-null 89 80 0.029 1.95 1.07-3.57
Table 4. The genotype frequencies of GSTM1 and GSTT1 polymorphism hypertension with
or without ESRD
Hypertension Hypertension+ESRD P OR 95%CI
GSTT1-positive 237 60 GSTT1-null 137 41 0.47 1.18 0.75-1.85 GSTM1-positive 169 47 GSTM1-null 205 54 0.81 0.95 0.61-1.47 Both null 45 18 Non-null 329 83 0.21 1.46 0.82-2.49 Disease Genotype Disease Genotype