© 2002 Kluwer Academic Publishers. Printed in the Netherlands. 1
Synthesis and Electroluminescence of Side-chain Liquid Crystalline
Polyacrylates and Polyoxiranes Containing Bistolane Side Groups
Shu-Wen Chang and Chain-Shu Hsu
∗Department of Applied Chemistry, National Chiao Tung University, Hsinchu, Taiwan 30050
(∗Author for correspondence; Tel.: +886-3-5712121 ext 56523; Fax: +886-3-5723764; E-mail: [email protected])
Received 21 August 2001; accepted in revised form 12 November 2001
Key words: electroluminescence (EL), liquid crystals (LC), polyacrylates, polymer light emitting diodes, polyoxiranes
Abstract
The synthesis and characterization of side-chain liquid crystalline (LC) polyacrylates and polyoxiranes containing bistolane side-groups are presented. The phase behavior of the prepared monomers and polymers was characterized by differential scanning calorimetry and optical polarizing microscopy. All of the obtained monomers and polymers reveal an enantiotropic nematic phase. The birefringences of the LC monomers are in the range from 0.35–0.6 depending on the measuring wave-length. The photoluminescence (PL) and electroluminescence (EL) properties of the obtained monomers and polymers are also reported.
Introduction
Side-chain liquid crystalline polymers (side-chain LCPs) are of both theoretical and practical interest because they com-bine the anisotropy of liquid crystals (LCs) with properties of the polymeric backbone. Some new applications based on these materials have been reported [1–3]. Recently, we syn-thesized some high birefringent side-chain LCPs containing tolane and diphenyldiacetylene side groups [4, 5]. These side-chain LCPs have potential applications in non-linear optics and reflective polarizers [6].
Progress in the field of polymer light emitting diodes (PLED) has been developing rapidly since 1990 [7]. Conju-gated polymers can be used for both single layer and multi-layer PLEDs [8] by a spin coating process [9]. Recently, some main-chain LCPs were also used for the application of PLED [10–14]. These LCPs produced linearly polar-ized electroluminescence based on the self organization of polymer chains. In 2000, we synthesized several series of bistolane LC compounds [15] which posses an extra ordinar-ily high birefringence and low melting points. These LCs are particularly useful for polymer dispersed LC displays [16], cholesteric displays [17] and infrared special light modula-tors for nondestructive missile seeker simulations [18]. This bistolane LCs with an extended long conjugation length are also useful for light emitting diodes.
The goal of this study is to present the synthe-sis and characterization of side-chain LC polyacrylates and polyoxiranes containing bistolane side groups. Both the monomers and polymers were characterized by dif-ferential scanning calorimetry and optical polarizing mi-croscopy. The wavelength-dependent birefringences of the obtained monomers are reported. The photoluminescence
(PL) and electroluminescence (EL) properties of the ob-tained monomers and polymers are also discussed.
Experimental
Materials
Tetrahydrofuran (THF) was distilled over sodium. Dichloro-methane was distilled over calcium hydride. Ethyl acetate and hexane was distilled before use. Initiator 2,2’-azo-bisisobutyronitrile (AIBN) was recrystallized by methanol. Bis(triphenylphosphine)palladium (II) chloride, triphenyl-phosphine, 2-methyl-3-butyn-2-ol, copper (I) iodide, m-chloroperoxybenzoic acid (MCPBA) and all other rea-gents were purchased from Aldrich and used as received. 1-[4-(6-Hydroxyhexan-1-yloxy)phenyl]acetylene was pre-pared according to a study [5] reported by our lab.
Characterization Techniques
NMR spectra (300 MHz) were recorded on a Bruker-300 spectrometer. Thermal transitions and thermodynamic parameters were determined by using a Seiko SSC/5200 differential scanning calorimeter equipped with a liquid nitrogen-cooling accessory. Heating and cooling rates were 10◦C min−1. Transition temperatures reported here were collected during the first cooling and second heating scans. A Carl-Zeiss Axiphot optical polarizing microscope equipped with a Mettler FP 82 hot stage and a FP 80 cen-tral processor was used to observe the thermal transitions and analyze the anisotropic textures. The molecular weights of the obtained polymers were determined by GPC using a
Viscotek T50A differential viscometer and LR125 laser re-fractometer with THF as eluent solvent and with polystyrene standards.
Synthesis of Monomers and Polymers
The synthetic procedures of monomers 1M∼ 4M and poly-mers 1P∼ 4P are outlined in Schemes 1 and 2.
4-{[4-(6-Hydroxyhexan-1-yloxy)phenyl]ethynyl}-3-ethyl-4’-n-propyltolane (1)
1-[4-(6-Hydroxyhexan-1-yloxy)phenyl]acetylene (6.20 g, 28.0 mmol), 3-ethyl-4-iodo-4’-n-propyltolane (12.10 g, 32.3 mmol), bis(triphenylphosphine) palladium (II) chloride (0.21 g, 0.3 mmol), triphenylphosphine (0.55 g, 2.1 mmol), copper (I) iodide (0.29 g, 1.5 mmol) were dissolved in 450 mL of triethylamine. The solution was heated to 50◦C for 24 h, then cooled to room temperature. The solvent was then removed in a rotatory evaporator and the crude prod-uct was treated with saturated ammonium chloride solution. The product was extracted into diethyl ether. The ether so-lution was washed with water, saturated NaCl soso-lution and dried over anhydrous MgSO4. After ether was removed,
the crude product was purified by column chromatography (silica gel, ethyl acetate/n-hexane= 1/3 as eluent) to yield: 8.01 g (61.9%) of yellow crystals. mp= 61.0◦C. 1H NMR (CDCl 3, TMS) δ = 0.94 [t, 3H, Ph–(CH2)2– CH3], 1.31 (t, 3H, Ph–CH2–CH3), 1.43–1.79 [m, 10H, Ph–O–CH2–(CH2)4– and Ph–CH2–CH2–CH3], 2.59 (t, 2H, Ph–CH2–CH2–CH3), 2.87 (q, 2H, Ph–CH2–CH3), 3.64 (t, 2H, –CH2–OH), 3.95 (t, 2H, Ph–O–CH2–), 6.85–7.46 (m, 11 aromatic protons). Synthesis of Monomer 1M 4-{[4-(6-Hydroxyhexan-1-yloxy)phenyl]ethynyl}-3-ethyl-4’-n-propyltolane (8.10 g, 17.0 mmol) was dissolved in a mixture of dried dichloromethane (200 mL) and triethyl-amine (10 mL, 70.0 mmol). After the solution was cooled in an ice-water bath to 0◦C, acryloyl chloride (3.20 g, 35.0 mmol) was added dropwise. The reaction mixture was allowed to warm slowly to room temperature and stirred overnight. The product was isolated by pouring the solution into water and filtering it. The crude product was purified by column chromatography (silica gel, ethyl acetate/n-hexane = 1/3 as eluent) to yield 7.43 g (84.1%) of yellow crys-tals. M+ = 518, elemental analysis: calculated: C: 83.4%, H: 7.4%, found: C: 83.1%, H: 7.4%. 1H NMR (CDCl 3, TMS) δ = 0.92 (t, 3H, Ph–(CH2)2– CH3), 1.30 (t, 3H, Ph–CH2–CH3), 1.41–1.77 (m, 10H, Ph–O–CH2–(CH2)4– and Ph–CH2–CH2–CH3), 2.56 (t, 2H, Ph–CH2–CH2–CH3), 2.80 (q, 2H, Ph–CH2–CH3), 3.89 (t, 2H, ph–O–CH2–), 4.14 (t, 2H, –COO–CH2–), 5.79 (d, 1H, H–CH=CH–COO–), 6.11 (m, 1H, CH2=CH–), 6.36 (d, 1H, H–CH=CH–COO–), 6.84–7.45 (m, 11 aromatic protons). 4-(6-Hydroxyhexan-1-yloxy)-3’-ethyl-4’-aminotolane (2)
Compound 2 was prepared by Cadiot-ChodKiewicz cou-pling [17] of 4-(6-hydroxyhexan-1-yloxy)phenylacetylene with 4-iodo-2-ethylaniline according to similar synthetic procedures used for compound 1. Yield: 61.4%, mp = 84.7◦C. 1H NMR (CDCl 3, TMS) δ= 1.24 (t, 3H, Ph–CH2–CH3), 1.30–1.85 [m, 8H, Ph–O–CH2–(CH2)4–], 2.46 (q, 2H, Ph– CH2–CH3), 3.61 (t, 2H, –CH2OH), 3.92 (t, 2H, Ph–O– CH2–), 6.58–7.42 (m, 7 aromatic protons). 4-(6-Hydroxyhexan-1-yloxy)-3’-ethyl-4’-iodotolane (3)
Compound 2 (3.50 g, 10.0 mmol) was dissolved in a mixture of THF (10 mL) and hydrochloric acid (4.1 mL, 50.0 mmol), which was kept below 5◦C. The aqueous solution (6.0 mL) of sodium nitrite (2.10 g, 31.0 mmol) was added dropwise. The resulting mixture was cooled to 0◦C and stirred for 10 min. An aqueous solution (20 mL) of sodium iodide (14.50 g, 100.0 mmol) was then added dropwise. The re-action mixture was stirred at 0◦C for 3 h. The product was extracted into ethyl acetate. The collected ethyl acetate so-lution was washed with 10% sodium thiosulfate aqueous solution, water, and dried over anhydrous MgSO4. After the
ethyl acetate was removed, the crude product was purified by column chromatography (silica gel, ethyl acetate/n-hexane = 1/5 as eluent) to yield 2.90 g (65.2%) of brown crystals. mp= 49.0◦C.
1H NMR (CDCl
3, TMS) δ= 1.23 (t, 3H, Ph–CH2–
CH3), 1.42–1.79 [m, 8H, Ph–O–CH2–(CH2)4–], 2.69
(q, 2H, Ph–CH2–CH3), 3.65 (t, 2H, –CH2–OH), 3.96 (t, 2H,
Ph–O–CH2–), 6.85–7.75 (m, 7 aromatic protons).
4-{[4-(6-Hydroxyhexan-1-yloxy)phenyl]ethynyl}-2-ethyl-4’-n-propyltolane (4)
Compound 4 was prepared by Cadiot-ChodKiewicz cou-pling of 1-(1-ethynyl)-4-propylbenzene with compound 3 according to similar synthetic procedures used with com-pound 1. Yield: 84.7%, mp= 66.8◦C. 1H NMR (CDCl 3, TMS): δ (ppm) = 0.95 (t, 3H, Ph– CH2–CH2–CH3), 1.32 (t, 3H, Ph–CH2–CH3), 1.45–1.81 (m, 10H, Ph–O–CH2–(CH2)4–, Ph–CH2–CH2–CH3), 2.61 (t, 2H, Ph–CH2–CH2–CH3), 2.89 (q, 2H, Ph–CH2–CH3), 3.66 (t, 2H, –CH2–OH), 3.97 (t, 2H, Ph–O–CH2–), 6.88– 7.48 (m, 11 aromatic protons). Synthesis of Monomer 2M
Monomer 2M was prepared by the esterification of acryloyl chloride with compound 4 according to similar synthetic procedures used with compound 1M. Yield: 97.1%, M+ = 518, elemental analysis: calculated: C: 83.4%, H: 7.4%, found: C: 83.4%, H: 7.5%. 1H NMR (CDCl 3, TMS): δ (ppm) = 0.94 (t, 3H, Ph– CH2–CH2–CH3), 1.31 (t, 3H, Ph–CH2–CH3), 1.45–1.81 (m, 10H, Ph–O–CH2–(CH2)4–), Ph–CH2–CH2–CH3), 2.60 (t, 2H, Ph–CH2–CH2–CH3), 2.87 (q, 2H, Ph–CH2–CH3), 3.96 (t, 2H, Ph–O–CH2–), 4.17 (t, 2H, (O)CO–CH2–), 5.80 (d, 1H, H–CH=CH–COO–), 6.15 (m, 1H, CH2=CH–), 6.40
(d, 1H, H–CH=CH–COO–), 6.86–7.48 (m, 11H, aromatic proton).
4-{[4-(4-Penten-1-yloxy)phenyl]ethynyl}-3-ethyl-4’-n-propyltolane (5)
Compound 5 was prepared by Cadiot-ChodKiewicz cou-pling of 1-[(penten-1-yloxy)phenyl]acetylene with 4-iodo-3-ethyl-4’-n-propyltolane according to similar syn-thetic procedures used for compound 1. Yield: 98.0%, mp= 57.9◦C. 1H NMR (CDCl 3, TMS): δ (ppm) = 0.94 (t, 3H, Ph– CH2–CH2–CH3), 1.29 (t, 3H, Ph–CH2–CH3), 1.66 (m, 2H, Ph–CH2–CH2–CH3), 1.88 (m, 2H, Ph–O–CH2–CH2–), 2.23 [m, 2H, Ph–O–(CH2)2–CH2–], 2.59 (t, 2H, Ph–CH2–CH2– CH3), 2.86 (q, 2H, Ph–CH2–CH3), 3.98 (t, 2H, Ph–O– CH2–), 5.03 (m, 2H, CH2=CH–), 5.85 (m, 1H, CH2=CH–), 6.85–7.45 (m, 11 aromatic protons). Synthesis of Monomer 3M 4- {[4-(4-Penten-1-yloxy)phenyl]ethynyl}-3-ethyl- 4’-n-pro-pyltolane (0.15 g, 0.35 mmol) was dissolved in the dried dichloromethane (10 ml). m-Chloroperoxybenzoic acid (MCPBA) (0.18 g, 1.0 mmol, 50%) was added slowly at room temperature. The mixture was stirred for 24 hours. After solvent was removed, the crude product was puri-fied using column chromatography (silica gel; hexane/ethyl acetate= 2 : 1) to yield 0.75 g (48.1%) of yellow crys-tals. M+= 448, elemental analysis: calculated: C: 85.7%, H: 7.2%, found: C: 85.6%, H: 7.4%. 1H NMR (CDCl 3, TMS): δ( ppm) = 0.84 (t, 3H, Ph– (CH2)2–CH3), 1.22 (t, 3H, Ph–CH2–CH3), 1.50–1.90 (m, 6H, Ph–CH2–CH2–CH3 and Ph–O–CH2–(CH2)2–), 2.41 (m, 1H, epoxide proton), 2.49 (t, 2H, Ph–CH2–CH2–), 2.67 (m, 1H, epoxide proton), 2.76 (q, 2H, Ph–CH2–CH3),
2.89 (m, 1H, epoxide proton), 3.91 (t, 2H, Ph–O–CH2–),
6.77–7.37 (m, 11 aromatic protons).
4-(4-Penten-1-yloxy)-3’-ethyl-4’-aminotolane (6)
Compound 6 was prepared by Cadiot-ChodKiewicz cou-pling of 1-[(penten-1-yloxy)phenyl]acetylene with 4-iodo-2-ethylaniline according to similar synthetic proce-dures used for compound 1 to yield 3.21 g (90.9%) of brown oil. 1H NMR (CDCl 3, TMS): δ (ppm) = 1.25 (t, 3H, Ph– CH2–CH3), 1.85–1.91 (m, 2H, Ph–O–CH2–CH2–), 2.21– 2.25 (m, 2H, Ph–O–CH2–CH2–CH2–), 2.48 (q, 2H, Ph– CH2–CH3), 3.75 (s, broad, 2H, NH2), 3.96 (t, 2H, Ph–O– CH2–), 4.89–5.10 (m, 2H, H2C=CH–), 5.75–5.95 (m, 1H, H2C=CH–), 6.60–7.43 (m, 7H, aromatic protons). 4-(4-Penten-1-yloxy)-3’-ethyl-4’-iodotolane (7)
Compound 7 was prepared by iodonation of compound 6 with sodium iodide according to similar synthetic pro-cedures used for compound 3 to yield 2.93 g (70.1%) of yellow oil. 1H NMR (CDCl3, TMS): δ (ppm)= 1.24 (t, 3H, Ph–CH2–CH3), 1.87 (m, 2H, Ph–O–CH2–CH2–), 2.54 [m, 2H, Ph–O–(CH2)2–CH2–], 2.71 (q, 2H, Ph– CH2–CH3), 3.96 (t, 2H, Ph–O–CH2–), 5.00–5.11 (m, 2H, H2C=CH–), 5.78–5.92 (m, 1H, H2C=CH–), 6.86–7.76 (m, 7H, aromatic protons). 4-{[4-(4-Penten-1-yloxy)phenyl]ethynyl}-2-ethyl-4’-n-propyltolane (8)
Compound 8 was prepared by Cadiot-ChodKiewicz cou-pling of 1-(4-n-propylphenyl)acetylene with compound 7 ac-cording to similar synthetic procedures used for compound 1. Yield: 88.0%, mp = 105.0◦C. 1H NMR (CDCl 3, TMS): δ (ppm)= 0.93 (t, 3H, Ph– CH2–CH2–CH3), 1.29 (t, 3H, Ph–CH2–CH3), 1.63 (m, 2H, Ph–CH2–CH2–CH3), 1.88 (m, 2H, Ph–O–CH2–CH2–), 2.22 [m, 2H, Ph–O–(CH2)2–CH2–], 2.59 (t, 2H, Ph–CH2– CH2–), 2.87 (q, 2H, Ph–CH2–CH3), 3.97 (t, 2H, Ph–O– CH2–), 4.95–5.10 (m, 2H, CH2=CH–), 5.75–5.92 (m, 1H, CH2=CH–), 6.85–7.46 (m, 11 aromatic protons). Synthesis of Monomer 4M
Monomer 4M was prepared by epoxidation of compound 8 with MCPBA according to similar synthetic procedures used for compound 3M. Yield: 30.4%, M+ = 448, elemental analysis: calculated: C: 85.7%, H: 7.2%, found: C: 85.5%, H: 7.2%. 1H NMR (CDCl 3, TMS): δ (ppm)= 0.93 (t, 3H, Ph– CH2–CH2–CH3), 1.30 (t, 3H, Ph–CH2–CH3), 1.59–1.95 (m, 6H, Ph–CH2–CH2–CH3, Ph–O–CH2–(CH2)2–), 2.50 (m, 1H, epoxide proton), 2.59 (t, 2H, Ph–CH2–CH2–), 2.77 (m, 1H, epoxide proton), 2.85 (q, 2H, Ph–CH2–CH3),
2.98 (m, 1H, epoxide proton), 4.00 (t, 2H, Ph–O–CH2–),
6.85–7.47 (m, 11 aromatic protons).
Synthesis of Polymers 1P and 2P
Both polymer 1P and 2P were prepared by free radical poly-merization of monomers 1M and 2M using AIBN as an initiator. The synthesis of 1P is described below.
Monomer 1M (1.00 g) and initiator AIBN (0.01 g) were dissolved in THF (1 ml). The reaction bottle was sealed under vacuum, and the reaction proceeded at 60◦C for 24 hours. The product was precipitated in methanol three times.
1P: yield: 82.0%, elemental analysis: calculated: C: 83.4%, H: 7.4%, found: C: 83.1%, H: 7.5%.
2P: yield: 70.0%, elemental analysis: calculated: C: 83.4%, H: 7.4%, found: C: 83.1%, H: 7.4%.
Synthesis of Polymers 3P and 4P
Both polymers 3P and 4P were prepared by cationic poly-merization of monomers 3M and 4M using borontrifluoro etherate as an initiator. The synthesis of 3P is described below.
Compound 3M (0.50 g) was dissolved in dichloro-methane (0.5 ml) under a nitrogen atmosphere at 0◦C. Borontrifluoro etherate (2.0 µml) was added into the so-lution, and the solution was stirred at room temperature for 24 hours. The product was precipitated in hexane three times. 1H NMR and GPC were used to check for the ab-sence of monomer 3M. The results of the polymerization
Table 1. Phase transition temperatures and corresponding enthalpy changes of monomers 1M∼ 4M and compounds
5 and 8
Compound Phase transition temperatures (◦C) and enthalpiesa(Kcal mol−1)
1M K 43.8 (8.14) N 92.3 (0.14) I I 89.5 (0.18) N < 50◦CbK 2M K 50.9 (8.05) N 93.8 (0.27) I I 90.8 (0.31) N < 50◦CbK 3M K 82.1 (3.86) N 158.1 (0.29) I I 144.9 (0.10) N 40.6 (2.12) K 4M K 93.8 (11.22) N 162.3 (0.40) I I 157.4 (0.47) N –1.6 (4.88) K 5 K 88.9 (8.37) N 150.1 (0.37) I I 144.9 (0.34) N 87.6 (7.23) K 8 K 105.3 (8.71) N 156.9 (0.36) I I 153.5 (0.35) N 40.9 (6.15) K aK: crystalline, N: nematic, I: isotropic.
bThe sample was cooled to−50◦C and the crystallization peak did not appear.
Table 2. Polymerization results of monomer 1M∼ M4
Polymers Yield Mn Mw Mm/Mw (%) (×10−3) (×10−3) 1P 82.0 38.3 41.3 1.18 2P 70.6 33.7 44.1 1.31 3P 72.7 9.8 21.0 2.15 4P 68.3 8.6 16.4 1.91
of all monomers are summarized in Table 2. As shown in Table 2, all of the polymers gave reasonable yields in the polymerization. The molecular weight distributions were less than 2.15.
3P: yield: 72.0%, elemental analysis: calculated: C: 85.7%, H: 7.2%, found: C: 85.6%, H: 7.2%.
4P: yield: 68.0%, elemental analysis: calculated: C: 85.7%, H: 7.2%, found: C: 85.6%, H: 7.2%.
Results and Discussion
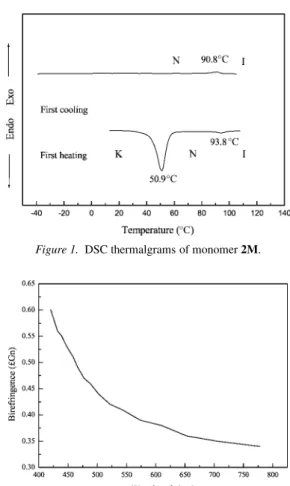
The synthesis of acrylate monomers 1M and 2M and epox-ide monomers 3M and 4M is outlined in Schemes 1 and 2. All monomers gave reasonably high yields. The purity of the obtained monomers was determined via chromatography to be high after purification. Figure 1 shows the representative DSC thermalgrams of monomer 2M. The heating scan re-vealed a melting point at 50.9◦C and a nematic to isotropic phase transition at 93.8◦C. On the cooling scan, only an isotropic to nematic transition was present at 90.8◦C. The sample did not crystallize even if it was cooled to−50◦C at a cooling rate of 1◦C/min. The crystallization of acry-late monomers 1M and 2M is a kinetic controlled process. Both monomers were crystallized after they were stored in a
refrigerator (0◦C) for several days. The reason for this crys-tallization could be due to the lateral ethyl side group and acrylate head group that obstruct the crystallization of the liquid crystals. Table 1 summarizes the phase transitions of monomers 1M∼ 4M and intermediate compounds 5 and 8. Comparing the chemical structures of monomer 1M and 2M, the only difference that can be noticed is in the position of lateral ethyl group. The ethyl group in monomer 1M is close to the acrylate head, while the ethyl group in monomer 2M is close to the terminal propyl group. In general, this lateral ethyl group is introduced to increase the lateral width of the mesogenic core. Its presence decreases the packing density of liquid crystal and results in a lowering decrease of the phase transition temperatures. In our previous report [7], we reported the synthesis of a series of asymmetric bistolane liquid crystals with different alkyl chain length on both ends. Their chemical structures were as follows:
We found that when the lateral ethyl group was close to a longer alkyl chain end, it resulted in a dramatical decrease in the phase transition temperatures. Monomers 1M and 2M contain a propyl group on the right-hand side and a hexy-loxy group together with an acrylate head on the left-hand side. The alkyl chain length on the left-hand side is longer than that on the right-hand side; therefore, monomer 1M reveals lower phase transition temperatures. Similar results can be observed for monomers 3M and 4M and compounds 5 and 8. Furthermore, if we compare three kinds of LC com-pounds, their melting points show the following tendencies: 1M, 2M < 3M, 4M < compound 5, 8. Again this can be explained by the molecular packing of the LC compounds. Compounds 5 and 8 have perfect linear rigid rod structures and higher melting points. On the other hand, monomers 1M and 2M contain a relatively bulky acrylate head and exhibit lower melting points. All six compounds reveal very wide temperature ranges in the nematic phase. Figure 2 shows the birefringence dispersion of monomer 1M at 25◦C. The birefringence of monomer 1M decreases as the wavelength increases. Its birefringence is about 0.4 when the measuring wavelength is 550 nm.
Scheme 3 outlines the polymerization of monomers 1M∼ 4M. Both monomers 1M and 2M were polymer-ized by a free radical polymerization mechanism while monomers 3M and 4M were polymerized by a cationic ring-opening polymerization process. Table 2 reports the poly-merization results. Both polymers 1P and 2P were achieved with number average molecular weights higher than 3.3× 104. However 3P and 4P were obtained with much lower number average molecular weights (> 8× 103). Table 3 summarizes the thermal transitions of polymers 1P∼ 4P. To our surprise, all polymers exhibit an enan-tiotropic nematic phase. Since the polymers contain six methylene units as spacers and a long rigid rod-like bistolane mesogen, they should reveal smectic phases. We believe that the reason why all polymers show a nematic phase is
Figure 1. DSC thermalgrams of monomer 2M.
Figure 2. The wavelength-dependent birefringence of monomer 1M at 25◦C.
because the introduction of the lateral ethyl group. Introduc-ing the lateral ethyl group into the bistolane core decreases the length to width ratio of the mesogen and prevents from forming layer smectic structures. A comparison of the ther-mal transition of both series of polymers shows their glass transition temperatures to be very close. However the LC temperature ranges of polymers 3P and 4P are wider than those of polymers 1P and 2P. This could be due to the more flexible polyoxirane backbone. According to the lit-erature [2], a more flexible polymer backbone allows its mesogenic side chains to form a more stable mesophase and hence it exhibits a wider mesomorphic temperature range.
Figure 3 shows the optical spectra of monomer 1M mea-sured in THF and in its solid state. The UV-vis absorption peak of monomer 1M are about 330 nm, and both solu-tion and film samples show very similar absorpsolu-tion spectra. The PL emissions of monomer 1M in THF and in solid state are 378.8 and 427.4 nm, respectively. A bathochromic is observed in the neat film when the solution spectra are compared. This might due to the aggregation of the liquid crystal molecules in the neat film. Table 4 summarizes the optical data of monomers 1M∼ 4M. All monomers present almost the same UV absorption and PL emission data. The results demonstrate that the absorption and emission peaks of all monomers are attributed to the π − π∗transition of the bistolane core. Figure 4 presents the optical spectra of polymer 1P measured in THF and in the solid state. Basi-cally, the spectra are similar to those of monomer 1M. This
Table 3. Phase transition temperatures and corre-sponding enthalpy changes of polymers 1P∼ 4P
Phase transition temperatures (◦C) and enthalpies (Kcal mol−1)
1P G 10.7 N 96.7 (0.32) I I 85.9 (0.30) N 5.3 G 2P G 5.4 N 70.2 (0.32) I I 68.8 (0.31) N –0.3 G 3P G 2.6 N 145.0 (0.42) I I 110.0 (0.21) N –6.9 G 4P G 5.4 N 141.5 (0.52) I I 116.5 (0.17) N –3.0 G N: nematic, I: isotropic, G: glassy.
Figure 3. UV-vis absorption and photoluminescence spectra of monomer
1M in THF and in films.
is reasonable because the absorption and emission of poly-mer 1P are attributed to the mesogenic core only. Figure 5 shows the EL spectrum of polymer 1P that was made as a single layer device (ITO/1P/Al). This EL spectrum is very similar to its PL spectrum. The current-voltage (J-V) and the luminance-voltage (L-V) curves are shown in Figure 6. The threshold voltage is 3.0 V. The power efficiency of the device is 1.0× 10−2lm/W at 9.0 V.
Table 4. UV-vis absorption and photoluminescence spectra of monomers 1M∼ 4M and polymers 1P ∼ 4P in THF and in films
UV absorption (λmax, nm) PL emission (λmax, nm) Solution Film Solution Film
1M 330.0 332.0 378.8 427.4 2M 330.0 334.0 378.2 426.8 3M 331.0 333.6 378.6 427.2 4M 331.0 333.9 378.6 425.2 1P 326.8 327.0 378.8 426.8 2P 328.4 332.0 378.8 426.8 3P 331.5 342.5 378.8 427.1 4P 331.5 334.4 378.8 427.6
Figure 4. UV-vis absorption and photoluminescence spectra of polymer 1P in THF and in films.
Figure 5. EL emission spectrum of an ITO/1P/Al device.
Conclusion
Two kinds of side-chain LC polyacrylates and polyoxi-ranes were synthesized. All monomers and polymers exhibit an enantiotropic nematic phase. Both polyoxiranes exhibit wider temperature ranges in the nematic phase than poly-acrylates because a polyoxirane backbone is more flexible than a polyacrlate backbone. The polymers emit blue light at 427 nm.
Acknowledgement
The authors are grateful to the National Science Council of the Republic of China for financial support of this work.
Figure 6. The J-V (1) and L-V (!) curves of an ITO/PEDOT/1P/Al device.
References
1. L. L. Chapoy, Recent Advances in Liquid Crystalline Polymers, Elsevier, London, 1985.
2. C. B. McArdle, Side Chain Liquid Crystalline Polymers, Blackie, Glasgow, 1989.
3. C. S. Hsu and Y. H. Lu, Macromolecules, 28, 1673 (1995). 4. Y. H. Lu, K. T. Tsay, C. S. Hsu and H. L. Chang, Mol. Cryst. Liq.
Cryst., 250, 85 (1994).
5. C. J. Hsieh, S. H. Wu, G. H. Hsiue and C. S. Hsu, J. Polym. Sci., Polym. Chem., 32, 1077 (1994).
6. G. H. Hsiue, L. H. Wu, C. J. Hsieh and R. J. Jeng, Liq. Cryst., 19, 189 (1995).
7. J. H. Burroughes, D. D. C. Bradley, A. R. Broom, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burn and A. B. Holms, Nature, 347, 539 (1990).
8. M. Grell, D. D. C. Bradley, M. Inbasekaran and E. P. Woo, Adv. Mater., 9, 798 (1997).
9. D. Braun and A. J. Heegerm, Appl. Phys. Lett., 58, 1982 (1991). 10. J. Oberski, R. Festag, C. Schmidt, G. Lussem, J. H. Wendorff and
A. Greiner, Macromolecules, 28, 8676 (1995).
11. G. Lussem, R. Festag, A. Greiner, C. Schmidt, C. Unterlechner, W. Heitz, J. H. Wendorff, M. Hopmier and J. Feldmann, Adv. Mater.,
2, 923 (1995).
12. G. Lussem, F. Geffarth, A. Greiner, W. Heitz, M. Hopmier, M. Ob-serski, C. Unterlechner and J. H. Wendorff, Liq. Cryst., 21, 903 (1996).
13. A. P. Davey, R. G. Howard and W. J. Blan, J. Mater. Chem., 7, 417 (1997).
14. M. Ozaki, T. Fujisawa, A. Fuji, L. Tong, K. Yoshino, M. Kijima, I. Kinoshita and H. Shirakawa, Adv. Mater., 12, 587 (2000). 15. C. S. Hsu, K. F. Shyn, Y. Y. Chuang and S. T. Wu, Liq. Cryst., 27, 283
(2000).
16. J. L. Fergason, SID Tech. Dig., 16, 68 (1985).
17. D. K. Yang, J. W. Doane, Z. Yaniv and J. Glasser, Appl. Phys. Lett.,
64, 1905 (1994).
18. S. T. Wu, U. Efron, J. Grinberg and L. D. Hess, SID Tech. Dig., 16, 262 (1985).