Ab Initio Study of the ClO + NH
2Reaction: Prediction of the Total Rate Constant and
Product Branching Ratios
†R. S. Zhu‡and M. C. Lin*,‡
Department of Chemistry, Emory UniVersity, Atlanta, Georgia 30322 ReceiVed: October 31, 2006; In Final Form: February 11, 2007
The mechanism for ClO + NH2 has been investigated by ab initio molecular orbital and transition-state theory calculations. The species involved have been optimized at the B3LYP/6-311+G(3df,2p) level and their energies have been refined by single-point calculations with the modified Gaussian-2 method, G2M(CC2). Ten stable isomers have been located and a detailed potential energy diagram is provided. The rate constants and branching ratios for the low-lying energy channel products including HCl + HNO, Cl + NH2O, and HOCl +3NH (X3Σ
-) are calculated. The result shows that formation of HCl + HNO is dominant below 1000 K; over 1000 K, Cl + NH2O products become dominant. However, the formation of HOCl + 3NH (X3Σ-) is unimportant below 1500 K. The pressure-independent individual and total rate constants can be expressed as k1(HCl + HNO) ) 4.7 × 10-8(T-1.08) exp(-129/T), k2(Cl + NH2O) ) 1.7 × 10-9(T-0.62) exp(-24/T), k3(HOCl + NH) ) 4.8 × 10-29(T5.11) exp(-1035/T), and ktotal ) 5.0 × 10-9(T-0.67) exp(-1.2/T),
respectively, with units of cm3molecule-1s-1, in the temperature range of 200-2500 K.
1. Introduction
ClOx species are known to play a central role in the O3
destruction process by Freons in the earth’s stratosphere.1They are also believed to be the key chain carriers in the combustion of ammonium perchlorate (AP) in its incipient stages.2,3 The fact that high concentrations of NOxare formed in the initial
stages of AP combustion suggests that the oxidation of NHx(x ) 2, 3) by ClOy(y ) 0-4) can occur rapidly. These oxidation
reactions may generate HNO, NH2O, and HONO, which can be further decomposed or oxidized to NO and NO2. There are no existing experimental or computational kinetic data on these reactions in the literature.
In present work, the ClO + NH2 reaction is chosen for investigation; we attempt to map out a detailed potential energy surface (PES) for the system and study the temperature and pressure effects on rate constants for the low-lying energy pathways. For this radical-radical process, the reaction is expected to take place primarily by exothermic association producing ClONH2 and possibly OClNH2 followed by their isomerization and fragmentation, similar to the analogous reactions involving ClO and NOx (x ) 1 and 2), which were
investigated earlier in this laboratory.4,5The results of our studies on the two reactions by means of the ab initio MO/statistical theories using the G2M method in conjunction with the multichannel Rice-Ramsperger-Kassel-Marcus (RRKM) cal-culations allowed us to account fully for the observed temper-ature and pressure effects. The results of this study are presented herein.
2. Computational Methods
The geometric parameters of the reactants, products, inter-mediates, and transition states of the title reaction have been
fully optimized by using the hybrid density functional B3LYP method (Becke’s three-parameter nonlocal exchange func-tional6,7with the correlation functional of Lee, Yang, and Parr8) with the 6-311+G(3df,2p) basis set. All of the stationary points have been identified for local minima and transition states by vibrational analysis. Intrinsic reaction coordinate analyses9have been performed to confirm the connection between transition states and designated reactants, products, or intermediates. On the basis of the optimized geometries, higher-level single-point energy calculations of the stationary points were performed by the G2M(CC2) method.10All of the electronic structure calcula-tions were performed with Gaussian 03.11
The rate constants were calculated with the transition state theory (TST) for the direct abstraction channel and the RRKM theory12,13for reactions occurring via long-lived intermediates. For the barrierless processes, the number of a variational transition quantum states, N†EJ, was given by the variationally determined minimum in NEJ(R), as a function of the bond length along the reaction coordinate R, which was evaluated according to the variable reaction coordinate flexible transition state theory.14-17The basis of these methods involves a separation of the vibrational modes into conserved and transitional modes. With this separation, one can evaluate the number of states by Monte Carlo integration for the convolution of the sum of vibrational quantum states for the conserved modes with the classical phase space density of states for the transitional modes. A step size of 1.00 cm-1was used for the convolution of the conserved mode-vibrations, and a step size of 50.00 cm-1was used for the generation of the transitional-mode number of states. The VariFlex code18was used for rate calculations. The PES computed by the G2M(CC2)10method was used in the calcula-tion.
3. Results and Discussion
A. Potential Energy Surfaces and Reaction Mechanism. The structures of the intermediates optimized at the B3LYP/
†Part of the special issue “James A. Miller Festschrift”.
* To whom correspondence should be addressed. E-mail: [email protected]. NSC Distinguished Visiting Professor at National Chiao Tung University, Hsinchu, Taiwan.
‡Emory University.
10.1021/jp067178d CCC: $37.00 © 2007 American Chemical Society Published on Web 04/19/2007
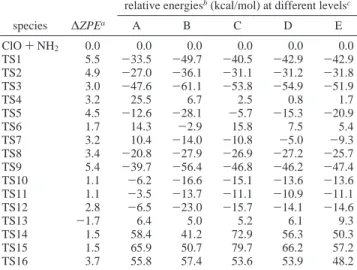
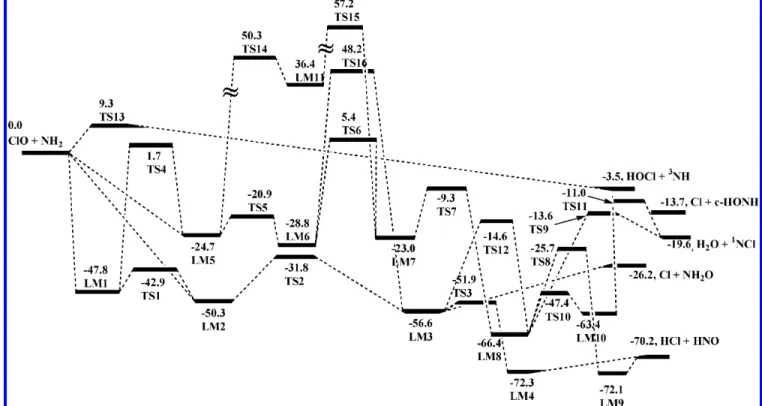
6-311+G(3df,2p) level are shown in Figure 1; those of the transition states optimized at the same levels are displayed in Figure 2a,b. On the basis of the optimized structures calculated at the B3LYP/6-311+G(3df,2p) level, the relative energies for the intermediates and products calculated at B3LYP/6-311+G-(3df,2p), MP2/6-311+GB3LYP/6-311+G-(3df,2p), PMP4/6-311+G(d,p), PMP4/ 6-311G(2df,p), and G2M(CC2) levels are summarized in Table 1. Table 2 displays the relative energies of the transition states involved in this reaction. The values in the two tables show that for most of the species, the B3LYP/6-311+G(3df,2p) method overestimated the energies, but at the MP2/6-311+G-(3df,2p) level, the energies are underestimated. The energies at the PMP4/6-311+G(d,p), PMP4/6-311G(2df,p), and G2M(CC2) levels are much closer. Because the “higher level correction”10 was included in the G2M(CC2) energies, in the rest discussion, only the energies obtained at this level are cited. The potential energy diagram obtained at the G2M level is presented in Figure 3. The vibrational frequencies and rotational constants for the intermediates and transition states are summarized in the tables of the electronic Supporting Information ESI I and ESI II, respectively.
On the basis of the predicted PES, the reaction of ClO with NH2 is expected to take place primarily by the association/ isomerization-decomposition mechanism. The formation of major products via different key low-energy paths is discussed below.
HCl + HNO and Cl + NH2O. There are two possible
pathways to form HCl + HNO as shown in Scheme 1 and Figure 3. For the first pathway, the O atom in the ClO radical attacks the N atom in NH2from different directions to form LM1 (ClONH2) and LM2 (ClONH2′). The Cl-O and N-O bond lengths in LM1 are 0.128 Å shorter and 0.073 Å longer
than those in LM2, respectively. The typical differences are the ClONH1 and ClONH2 dihedral angles. They are 120.3 and -120.3°in LM1 and 66.4 and -66.4°in LM2, respectively. LM1 lies 47.8 kcal/mol below the reactants (ClO + NH2), and LM2is more stable than LM1 by 2.5 kcal/mol. TS1 connects LM1 and LM2 by rotation of the NH2group around the NO bond. It lies 4.9 kcal/mol above LM1. The Cl atom in LM2 can migrate to N via TS2 to form LM3 [ClN(H2)O]. TS2 lies 18.5 kcal/mol above LM2, and LM3 is 6.3 kcal/mol more stable than LM2. One of the H atoms in LM3 migrates to the Cl atom via a three-center transition state, TS3, to form a van der Waals complex LM4, ClH‚‚‚N(H)O, followed by decomposition to HCl + HNO. LM4 lies below the reactants by 72.3 kcal/mol, which is 2.1 kcal/mol below the final products, HCl + HNO.
For the second pathway, as shown in Scheme 1 and Figure 3, LM1 (ClONH2) can isomerize to LM5 (OClNH2) via a three-center transition state, TS4, which involves the N-O bond breaking and the N-Cl bond formation. This process takes a large amount of energy (49.5 kcal/mol). LM5 can also be regarded as an intermediate formed by the interaction of the Cl atom in ClO and the N atom in NH2. A similar isomer, LM6, lying 4.1 kcal/mol lower than LM5 was located. The structural differences between LM5 and LM6 are similar to those between LM1 and LM2. LM5 and LM6 lie 24.7 and 28.8 kcal/mol below the reactants, respectively. TS5 connects LM5 and LM6, corresponding to the NH2group rotation around the ClN bond. The barrier of TS5 is 3.8 kcal/mol above LM5. LM6 isomerizes to LM7 (HOClNH) via a four-center transition state, TS6, with a 34.2 kcal/mol barrier above LM6. The OH group in HOClNH (LM7) migrates from Cl to the N atom to form LM8 [t-HON-(H)Cl] via TS7 with a 32.3 kcal/mol barrier height. LM7 and LM8 lie below the reactants by 23.0 and 66.4 kcal/mol, Figure 1. Optimized geometry of the intermediates involved in the reaction of ClO + NH2, computed at the B3LYP/6-311+G(3df,2p) level. The
bond lengths are given in angstroms (Å); angles in degrees (°).
Figure 2. (a, b) Optimized geometry for the transition states involved in the reaction of ClO + NH2, computed at the B3LYP/6-311+G(3df,2p)
level. The bond lengths are given in angstroms (Å), and the angles are given in degrees (°).
respectively. LM8 isomerizes to a loose complex, ClH‚‚‚ONH (LM9), via a four-center transition state, TS8, with a 40.7 kcal/ mol barrier. The stability of ClH‚‚‚ONH (LM9) is similar to that of ClH‚‚‚N(O)H (LM4), lying 1.9 kcal/mol below the final products (HCl + HNO).
As shown in Scheme 1, Cl + NH2O are formed by the barrierless decomposition of ClN(H2)O (LM3).
The calculated heats of reaction and available experimental values are summarized in Table 3. On the basis of the experimental heats of formation19of ClO (24.2 kcal/mol), NH2 (46.2 ( 1.5 kcal/mol), HCl (-22.0 kcal/mol), and HNO (24.5 kcal/mol), the heat of reaction for ClO + NH2f HCl + HNO, -67.9 ( 1.5 kcal/mol, is close to the predicted value, -70.2
kcal/mol, in this work. It needs to be mentioned that the G2M method has average absolute deviations in the range of 0.88 to ∼1.28 kcal/mol.10 The heat of formation for NH2O is not experimentally available. In this work, it is determined by combining the computed heat of reaction (∆rH0°) of the following isodesmic reaction
with the experimental∆fH°(0 K) values for CH3O (5.5 ( 0.7 kcal/mol),20NH2(46.2 ( 1.5 kca/mol),19and CH3(35.6 ( 0.2 kcal/mol).19On the basis of∆
rH°for reaction 1 at the G2M-(CC2) level, 1.9 kcal/mol, the heat of formation∆fH°(0 K) for NH2O is predicted to be 17.1 kcal/mol, which is in good agreement with the most recently reported value, 17.3 ( 0.8 kcal/mol, based on the average atomization energy obtained by Dixon et al.21at the CCSD(T)/aug-cc-pV5Z//CCSD(T)/aug-cc-pVTZ level. If the value obtained by the isodesmic reaction is used with the experimental heats of formation of other species involved in the reaction, the heat of reaction for ClO + NH2f Cl + NH2O is -24.7 ( 2.4 kcal/mol, which may be compared with our predicted value, -26.2 kcal/mol.
H2O + NCl. As shown in Figure 3 and Scheme 2, LM8 can
directly decompose to H2O + HCl via TS9 with a 52.8 kcal/ mol barrier. It can also isomerize to LM10 via TS10, and LM10 further decomposes to H2O + NCl via TS11. Meanwhile, LM8 can isomerize to LM3 via TS12 (Figure 3). LM10 lies -63.4 kcal/mol below the reactants, and TS9, TS10, TS11, and TS12 lie -13.6, -47.4, -11.1, and -14.6 kcal/mol below the reactants, respectively. The heat of reaction for this channel is -19.6 kcal/mol.
Cl + c-HONH (t-HONH). t-HONH is 5.2 kcal/mol more stable than c-HONH. As shown in Scheme 3, the HONH isomers can be produced without barriers from the decomposi-tion of t-HON(H)Cl (LM8) and c-HON(H)Cl (LM10), respec-tively.
The heats of reaction for these processes are -13.7 and -18.8 kcal/mol. If the experimental heats of formation for ClO, NH2, Cl and the heat of formation for HONH based on the average atomization energy obtained by Dixon et al.21at the CCSD(T)/ aug-cc-pV5Z//CCSD(T)/aug-cc-pVTZ level are used,∆rH0°is -17.0 ( 2.3 kcal/mol, which can be compared to our calculated value, -18.8 kcal/mol (Table 3).
HOCl +3NH (X3Σ-). The O atom in ClO can directly abstract
one of the H atoms in NH2via a C1symmetry triplet transition state TS13 to form HOCl +3NH (X3Σ-), as shown in Scheme 4 and Figure 3. TS13 has 9.3 kcal/mol barrier and this process has 3.5 kcal/mol exothermicity. On the basis of the experimental heats of formation19of ClO (24.2 kcal/mol), NH2(46.2 ( 1.5 kcal/mol), HOCl (-17.1 kcal/mol), and3NH (90.0 ( 4 kcal/ mol), the exothermicity for this process is 2.5 ( 5.5 kcal/mol. As one can see that the experimental heat of formation for3NH has a larger error, a more accurate value can be obtained by the following isodesmic reaction.
On the basis of the heat of reaction (∆rH°) of reaction 2, 17.7 kcal/mol, obtained at the G2M(CC2) level in this work and the experimental19heats of formation of CH3(35.6 ( 0.2 kcal/mol), CH2(93.2 kcal/mol), and NH2(46.2 ( 1.5 kcal/mol), the heat formation of3NH is determined to be 86.1 ( 1.7 kcal/ mol, which is close to the value, 85.9 ( 0.1 kcal/mol, based on the average atomization energy calculated at the CCSD(T)/aug-cc-pV5Z//CCSD(T)/aug-cc-pVTZ level by Dixon et al.22If the
TABLE 1: Relative Energies of the Reactants,
Intermediates, and Products for the Reaction of ClO with NH2Calculated at Different Levels of Theory Based on the Geometries Optimized at the B3LYP/6-311+G(3df,2p) Level
relative energiesb(kcal/mol) at different levelsc
species ∆ZPEa A B C D E ClO + NH2 0.0 0.0 0.0 0.0 0.0 0.0 LM1 5.5 -40.0 -54.4 -46.9 -47.8 -47.8 LM2 5.8 -44.6 -56.9 -50.4 -51.5 -50.3 LM3 6.5 -50.9 -68.1 -58.7 -59.6 -56.6 LM4 1.6 -60.4 -79.9 -77.4 -75.3 -72.3 LM5 5.0 -16.7 -32.9 -7.9 -18.1 -24.7 LM6 5.3 -20.7 -37.3 -13.3 -23.8 -28.8 LM7 4.6 -14.6 -33.4 -14.4 -20.2 -23.0 LM8 6.2 -57.0 -74.3 -67.2 -66.9 -66.4 LM9 1.4 -59.8 -79.5 -77.1 -75.0 -72.1 LM10 6.1 -53.9 -71.3 -62.9 -63.4 -63.4 HCl + HNO -0.2 -58.4 -77.2 -75.3 -72.4 -70.2 Cl + NH2O 3.4 -25.1 -32.2 -34.6 -29.5 -26.2 Cl + c-HONH 3.3 -10.2 -19.3 -22.1 -17.3 -13.7 Cl + t-HONH 3.8 -15.3 -24.6 -28.8 -23.1 -18.9 H2O + NCl 1.5 -8.1 -19.2 -15.1 -11.1 -19.6 HOCl +3NH -0.2 2.9 -9.9 -9.9 -6.6 -3.5 aThe relative zero-point energies were calculated at the B3LYP/ 6-311+G(3df,2p) level in units of kcal/mol.bThe ZPEs are included in the relative energies.cDifferent methods are represented by: A, B3LYP/6-311+G(3df,2p); B, MP2/6-311+G(3df,2p); C, PMP4/ 6-311+G(d,p); D, PMP4/6-311G(2df,p); E, G2M(CC2).
TABLE 2: Relative Energies of the Transition States for the Reaction of ClO with NH2Calculated at Different Levels of Theory Based on the Geometries Optimized at the B3LYP/ 6-311+G(3df,2p) Level
relative energiesb(kcal/mol) at different levelsc
species ∆ZPEa A B C D E ClO + NH2 0.0 0.0 0.0 0.0 0.0 0.0 TS1 5.5 -33.5 -49.7 -40.5 -42.9 -42.9 TS2 4.9 -27.0 -36.1 -31.1 -31.2 -31.8 TS3 3.0 -47.6 -61.1 -53.8 -54.9 -51.9 TS4 3.2 25.5 6.7 2.5 0.8 1.7 TS5 4.5 -12.6 -28.1 -5.7 -15.3 -20.9 TS6 1.7 14.3 -2.9 15.8 7.5 5.4 TS7 3.2 10.4 -14.0 -10.8 -5.0 -9.3 TS8 3.4 -20.8 -27.9 -26.9 -27.2 -25.7 TS9 5.4 -39.7 -56.4 -46.8 -46.2 -47.4 TS10 1.1 -6.2 -16.6 -15.1 -13.6 -13.6 TS11 1.1 -3.5 -13.7 -11.1 -10.9 -11.1 TS12 2.8 -6.5 -23.0 -15.7 -14.1 -14.6 TS13 -1.7 6.4 5.0 5.2 6.1 9.3 TS14 1.5 58.4 41.2 72.9 56.3 50.3 TS15 1.5 65.9 50.7 79.7 66.2 57.2 TS16 3.7 55.8 57.4 53.6 53.9 48.2
aThe relative zero-point energies were calculated at the B3LYP/ 6-311+G(3df,2p) level in units of kcal/mol.bThe ZPEs are included in the relative energies.cDifferent methods are represented by A, B3LYP/6-311+G(3df,2p); B, MP2/6-311+G(3df,2p); C, PMP4/6-311+G(d,p); D, PMP4/6-311G(2df,p); and E, G2M(CC2).
CH3O + NH2f CH3+ NH2O (1)
CH3+3NH f3CH2+ NH2 (2)
value obtained by the isodesmic reaction is used, the heat of reaction is given to be -1.5 ( 1.7 kcal/mol, which may be compared with our predicted value of -3.5 kcal/mol.
Other Higher-Energy Isomerization Pathways. Aside from the above channels, there are some high-energy pathways. As
shown in Figure 3 and Scheme 5, OClNH2(LM5) can isomerize to HNCl(H)O (LM11) via TS14 and LM11 isomerizes to LM7 via TS15. TS14, LM11, and TS15 lie above the reactants by 50.3, 36.4, and 57.2 kcal/mol, respectively. In addition, the isomerization transition state TS16 between LM6 and LM3 lies Figure 3. Schematic energy diagram (kcal/mol) of the ClO + NH2system computed at the G2M(CC2)//B3LYP/6-311+G(3df,2p) level.
SCHEME 1
TABLE 3: Comparison of the Calculated Heats of Reaction at the G2M(CC2) Level and the Available Experimental Dataa reactions ∆rH0(0 K) exptl (0 K) ClO + NH2f HCl + HNO -70.2 -67.9 ( 1.5 ClO + NH2f Cl + NH2O -26.2 -24.7 ( 2.4 ClO + NH2f NCl + H2O -19.6 ClO + NH2f Cl + c-HONH -13.7 ClO + NH2f Cl + t-HONH -18.8 -17.0 ( 2.3 ClO + NH2f HOCl +3NH (X3∑-) -3.5 -1.5 ( 1.7
aThe experimental values are obtained on the basis of the
experi-mental heats of formation of the related species as mentioned in the text. Values are in units of kcal/mol.
SCHEME 2
SCHEME 3
SCHEME 4
SCHEME 5
above the reactants by 48.2 kcal/mol. Apparently, these reaction paths are not competitive in the whole reaction.
B. Rate-Constant Calculations. The rate constants for the low-lying energy pathways forming HCl + HNO, Cl + NH2O, and HOCl +3NH have been calculated. For the association of ClO + NH2f ClONH2and the decomposition of ClN(H2)O f Cl + NH2O, the barrierless association/dissociation potential-energy curves were computed from the equilibrium structures of ClONH2and ClN(H2)O to cover the O-N and Cl-N bonds with separations from 1.387 to 3.987 Å and 2.164 to 5.164 Å, respectively, with an interval step size of 0.2 Å at the UB3LYP/ 6-311+G(3df,2p) level. Other geometric parameters were fully optimized. The Morse potential function E(R) ) De[1 - exp-(-β(R - Re))]2was employed to approximate the minimum-energy path for the system in our rate-constant calculation. In the above equation, R is the reaction coordinate (i.e., the distance between the two bonding atoms; the O-N and Cl-N bonds in this work), Deis the bond energy excluding zero-point energy, and Reis the equilibrium value of R. The computed potential energies could be fitted reasonably to the Morse potential function with the parameterβ ) 2.885 and 1.513 Å-1for these processes. The calculated and fitted curves for the association process are plotted in ESI V for reference. The Morse potentials with the above-mentioned parameters, the Lennard-Jones pairwise potential, and the anisotropic potential are added together to form the final potential for the variational rate constant calculation by the VariFlex code. The estimation of the transitional mode contribution to the transition-state number of states for a given energy is evaluated via Monte Carlo integration with 20 000 configuration numbers. The energy-transfer process was computed on the basis of the exponential down model with the 〈∆E〉 down value (the mean energy transferred per collision) of 400 cm-1 (in Ar). To achieve convergence in the integration over the energy range, an energy grain size of 100 cm-1 was used. This grain size provides numerically converged results for all temperature studies with the energy range spanning from 19 789.6 cm-1 below to 60 210.4 cm-1above the threshold. The total angular momentum J covered the range from 1 to 250 in steps of 10 for the E- and J-resolved calculation. The numbers of the states for the tight transition states involved in this calculation are evaluated according to the rigid-rotor harmonic-oscillator assumption implemented in the VariFlex code.18
In the calculation, because the energy difference between LM1 and LM2 is only 2.5 kcal/mol and the isomerization from
LM1 to LM2 has only a 4.9 kcal/mol barrier, TS1 was ignored, and the two complexes were treated as one with a hinder rotor. The quantum tunneling effects were included in the related cases with the Eckart tunneling model.23
Figure 4 presents the effect of temperature on the product-branching ratios covering the entire temperature range studied. The results show that formations of HCl + HNO, Cl + NH2O, and HOCl +3NH are pressure independent below 100 atm in the temperature range of 200-2500 K. Among these channels, formation of HCl + HNO is dominant below 1000 K; over 1000 K, Cl + NH2O products become dominant; for the products of HOCl + NH, below 1500 K, the branching ratio is less than 2%, and at 2500 K, the ratio reaches around 20%.
The predicted-total and individual rate constants in units of cm3molecule-1s-1in the temperature range of 200-2500 K can be presented by
4. Conclusions
The mechanisms for the ClO + NH2reaction over the singlet and triplet potential-energy surfaces have been elucidated at the G2M(CC2)//6-311+G(3df,2p) level of theory. Three low-lying energy product channels, (1) HCl + HNO, (2) Cl + NH2O, and (3) HOCl +3NH, have been identified. The rate constants for the formation of these products have been calculated in the temperature range of 200-2500 K using variational RRKM theory with the VariFlex code. The predicted results show that the rate constants of these channels are pressure independent; the formation of HCl + HNO is dominant below 1000 K; at T > 1000 K, Cl + NH2O and HCl + HNO product channels become competitive.
Acknowledgment. This work was supported by the Office of Naval Research under grant no. N00014-02-1-0133. MCL gratefully acknowledges support from Taiwan’s National Sci-ence Council for a distinguished visiting professorship at the Center for Interdisciplinary Molecular Science, National Chiao Tung University, Hsinchu, Taiwan.
Supporting Information Available: Tables of vibrational frequencies, rotational constants, and Cartesian coordinates and a graph of relative decomposition energies vs the O-N bond length. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
(1) Wayne, R. P. Chemistry of Atmospheres, 2nd ed.; Clarendon Press: Oxford, U.K. 1991.
(2) Jacobs, P. W. M.; Whitehead, H. M. Chem. ReV. 1969, 69, 551. (3) Politzer, P.; Lane, P. J. Mol. Struct.: THEOCHEM 1998, 454, 229. (4) Zhu, R. S.; Lin, M. C. Chem. Phys. Chem. 2005, 6, 1514. (5) Zhu, R. S.; Lin, M. C. Chem. Phys. Chem. 2004, 5, 1864. (6) Becke, A. D. J. Chem. Phys. 1993, 98, 5648.
(7) Becke, A. D. J. Chem. Phys. 1992, 96, 2155. Becke, A. D. J. Chem. Phys. 1992, 97, 9173.
(8) Lee, C.; Yang, W.; Parr, R. G. Phys. ReV. 1988, B37, 785. (9) Gonzalez, C.; Schlegel, H. B. J. Phys. Chem. 1989, 90, 2154. (10) Mebel, A. M.; Morokuma, K.; Lin, M. C. J. Chem. Phys. 1995, 103, 7414.
(11) Frisch, M. J.; Trucks, G. W.; Head-Gorden, M.; Gill, P. M. W.; Wong, M. W.; Foresman, J. B.; Johnson, B. G.; Schlegel, H. B.; Robb, M. Figure 4. Effect of temperature on the product branching ratios
covering the range of 200-2500 K.
ktotal) 5.0 × 10-9T-0.67exp(-1.2/T) k1(HCl + HNO) ) 4.7× 10-8(T-1.08) exp(-129/T)
k2(Cl + NH2O) ) 1.7× 10-9(T-0.62) exp(-24/T) k3(HOCl + NH) ) 4.8× 10-29(T5.11) exp(-1035/T)
A.; Replogle, E. S.; Gomperts, R.; Andres, J. L.; Rahavachari, K.; Binkley, J. S.; Gonzalez, C.; Martin, R. L.; Fox, D. J.; Defrees, D. J.; Baker, J.; Stewart, J. J. P.; Pople, J. A. Gaussian 92; Gaussian, Inc.: Pittsburgh, PA, 1992.
(12) Gilbert, R. G.; Smith, S. C. Theory of Unimolecular and Recom-bination Reactions; Blackwell Scientific: Carlton, Australia, 1990.
(13) Holbrook, K. A.; Pilling, K. J.; Robertson, S. H. Unimolecular Reactions; Wiley: Chichester, U.K., 1996.
(14) Wardlaw, D. M.; Marcus, R. A. Chem. Phys. Lett. 1984, 110, 230. (15) Wardlaw, D. M.; Marcus, R. A. J. Chem. Phys. 1985, 83, 3462. (16) Klippenstein, S. J. J. Phys. Chem. 1994, 98, 11459.
(17) Klippenstein, S. J. Chem. Phys. Lett. 1990, 170, 71.
(18) Klippenstein, S. J.; Wagner, A. F; Dunbar, R. C.; Wardlaw, D. M.; Robertson, S. H. VariFlex, version 1.00; 1999.
(19) Chase, M. W., Jr. NIST-JANAF Thermochemical Tables, 4th ed.; Woodbury, NY, 1998.
(20) Mazyar, O. A.; Baer, T. J. Phys. Chem. A 1999, 103, 1221 (21) Dixon, D. A.; Francisco, J. S.; Alexeev, Y. J. Phys. Chem. A 2006, 110, 185.
(22) Dixon, D. A.; Feller, D.; Peterson, V. J. Chem. Phys. 2001, 115, 2576.
(23) Baer, T.; Hase, W. L. Unimolecular Reaction Dynamics (Interna-tional Series of Monographs on Chemistry); Oxford University Press: New York, 1996.