Inhibition of the ERK phosphorylation plays a role in terbinafine-induced
p21 up-regulation and DNA synthesis inhibition in
human vascular endothelial cells
Pei-Yin Ho
a, Sung-Po Hsu
b, Yu-Chih Liang
c, Min-Liang Kuo
d,
Yuan-Soon Ho
c, Wen-Sen Lee
e,f,g,⁎
aGraduate Institute of Cell and Molecular Biology, Taipei Medical University, Taipei, Taiwan bInstitute of Physiology, College of Medicine, National Taiwan University, Taiwan c
Graduate Institute of Biomedical Technology, Taipei Medical University, Taipei, Taiwan
d
Institute of Toxicology, College of Medicine, National Taiwan University, Taiwan
e
Graduate Institute of Neuroscience, Taipei Medical University, Taipei, Taiwan
f
Graduate Institute of Medical Sciences, Taipei Medical University, 250 Wu-Hsing Street, Taipei 110, Taiwan
g
Department of Physiology, Medical College, Taipei Medical University, Taipei, Taiwan Received 5 September 2007; revised 17 December 2007; accepted 28 December 2007
Available online 17 January 2008
Abstract
Previously, we showed that terbinafine (TB) induces cell-cycle arrest in cultured human umbilical vein endothelial cells (HUVEC) through an
up-regulation of the p21 protein. The aim of this study is to delineate the molecular mechanisms underlying TB-induced increase of p21 protein. RT-PCR
analysis demonstrated that the mRNA levels of p21 and p53 were increased in the TB-treated HUVEC. The p21 promoter activity was also increased by
TB treatment. Transfection of HUVEC with p53 dominant negative (DN) abolished the TB-induced increases of p21 promoter activity and protein level,
suggesting that the TB-induced increase of p21 is p53-dependent. Western blot analysis demonstrated that TB decreased the levels of phosphorylated
extracellular signal-regulated kinase (ERK). Over-expression of mitogen-activated protein kinase (MEK)-1, the immediate upstream activator kinase of
ERK, abolished the TB-induced increases of p21 and p53 protein and decrease of thymidine incorporation. The ERK inhibitor (PD98059) enhanced the
TB-induced inhibition of thymidine incorporation into HUVEC. Taken together, these data suggest that the decrease of ERK activity plays a role in the
TB-induced up-regulation of p21 in HUVEC. On the other hand, pretreatment of the cells with geranylgeraniol (GGOH), farnesol (FOH), or Ras inhibitor
peptide did not affect the TB-induced decrease of thymidine incorporation. Taken together, our results suggest that TB might cause a decrease of MEK,
which in turn up-regulates p53 through the inhibition of ERK phosphorylation, and finally causes an increase of p21 expression and cell-cycle arrest.
© 2008 Elsevier Inc. All rights reserved.
Keywords: Terbinafine; p53; p21; ERK; DNA synthesis
Toxicology and Applied Pharmacology 229 (2008) 86–93
www.elsevier.com/locate/ytaap
Abbreviations: CDK, cyclin-dependent kinase; DEPC, diethyl-pyrocarbonate; DMSO, dimethylsulfoxide; DN, dominant negative; DTT, dithiothreitol; ECL, enhanced chemiluminescence; ECs, endothelial cells; ECGS, endothelial cell growth supplement; ERK, extracellular signal-regulated kinase; FBS, fetal bovine serum; FTI, farnesyl transferase inhibitor; FOH, farnesol; GGTI, geranylgeranyl transferase I inhibitor; GGOH, geranylgeraniol; GST, Glutathione-S-Transferase; HUVEC, human umbilical vein endothelial cells; M199, medium 199; MEK, mitogen-activated protein kinase; TB, terbinafine; PMSF, phenylmethyl sulfonyl fluoride; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis; HRP, horseradish peroxidase; RT-PCR, reverse transcription-polymerase chain reaction; Hb, hemoglobin.
⁎ Corresponding author. Graduate Institute of Medical Sciences, Taipei Medical University, 250 Wu-Hsing Street, Taipei 110, Taiwan. Fax: +886 2 27391775. E-mail address:[email protected](W.-S. Lee).
0041-008X/$ - see front matter © 2008 Elsevier Inc. All rights reserved. doi:10.1016/j.taap.2007.12.028
Introduction
Angiogenesis, the process of generating new capillary blood
vessels from pre-existing ones, is essential for embryogenesis,
but happens rarely in the adult with few exceptions (e.g., female
reproductive system and wound healing). A pathological
imbal-ance in this process contributes to numerous malignant, ischemic,
infectious, inflammatory and immune disorders (
Carmeliet, 2005
).
It is believed that the major steps in angiogenesis include: (a)
degradation of basement membrane by proteases; (b) migration of
endothelial cells (ECs) into the interstitial space and sprouting; (c)
ECs proliferation at the migrating tip; (d) differentiation into new
vessels; and (e) the stabilization and maturation of vessels by
mediator molecules that recruit mesenchymal cells to vessel walls
(
Bussolino et al., 1997; Gupta and Qin, 2003
). A useful approach
for the treatment of pathological angiogensis might exist in the
further development of novel anti-angiogenic drugs.
Angiogenesis is controlled by a number of growth factors.
Through their transmembranal receptors, growth factor signals
activate signaling pathways leading to an array of responses.
Mitogen-activated protein kinase (MEK) and ERK isoforms,
which are involved in activating endothelial cell proliferation
(
Nakagami et al., 2000, 2001
), appear to be the principal signaling
pathways in growth factor signaling. It has been reported that ERK
inhibitor caused cell apoptosis, which was accompanied by
in-creased p53 accumulation (
Kim et al., 2002; Kwon et al., 2004
).
By elevating levels of p21 and decreasing levels of cyclins A and
B, wild type p53 can prevent abrogation of arrest (
Eastman, 2004
).
Terbinafine (TB), an inhibitor of fungal ergosterol
biosynth-esis, is a newly synthesized oral antimycotic drug (
Petranyi et al.,
1984
). Previous reports have indicated that TB has relative few
drug interactions and is safe for clinical uses (
Abdel-Rahman and
Nahata, 1997
). The cream form and oral tablet of TB have been
approved for clinical uses in the United States (
Gupta and Shear,
1997
), and the oral formulation has been on the market in various
countries (
Gupta et al., 1998
). Previously, we demonstrated that
TB could inhibit the proliferation and migration of cultured
HUVEC, the formation of capillary-like tube, and sprouting of
capillaries (
Ho et al., 2004
). The TB-induced cell-cycle arrest in
HUVEC occurred when the cyclin-dependent kinase 2 (CDK2)
was inhibited, just as the protein level of p21 was increased.
Although the elevation of p21 protein levels has been
demon-strated to be responsible for TB-induced cell-cycle arrest, the
molecular mechanism underlying the TB-induced increase of p21
expression is still unclear. In the present study, we found that the
ERK-mediated pathway may contribute to the TB-induced
up-regulation of p53 protein, which is responsible for regulating the
p21 protein level in HUVEC.
Materials and methods
Cell culture
HUVEC were harvested from human umbilical vein by enzymatic dissociation as described (Moraes and Stastny, 1977). The cells were grown in M199 medium containing 10% fetal bovine serum (FBS), sodium heparin (100 U/ mL), endothelial cell growth supplement (ECGS, 0.03 mg mL−1, Biomedical Technologies Inc., MA, USA) and antibiotics in gelatin-coated plates, and incubated at 37 °C in 5% CO2in air. Cells from passages 5–9 were used.
Western blot analyses
To determine the protein levels in HUVEC, the total proteins were extracted and Western blot analyses were conducted as previously described. Briefly, HUVEC were cultured in 15 cm petri dishes. After reaching subconfluence, the cells were rendered quiescent and then treated with various concentrations of TB (Novartis Pharmaceuticals Corporation, East Hanover, New Jerseym USA) and incubated at 37 °C. At different time points, the cells were washed with phosphate buffer saline (NaCl 0.8%, Na2HPO40.144%, KH2PO40.024%, KCl 0.02%, pH
7.4), incubated with extraction buffer (Tris 50mM, pH 7.5, NaCl 150mM, PMSF 1mM, NP-40 1%, and 0.1% SDS) on ice, and then centrifuged at 12,000 × g for 30 min. The cell extract was then boiled in a ratio of 3:1 with sample buffer (Tris– HCl 250 mM, pH 6.8, glycerol 40%, dithiothreitol 400 mM, SDS 8% and bromophenol blue 0.2%). Electrophoresis was carried out using 12% SDS-polyacrylamide gel (2 h, 110 V, 40 mA, and 50μg protein per lane). Separated proteins were transferred to PVDF membranes (1 h, 400 mA), treated with 5% fat-free milk powder (Anchor, Manurewa, NZ) to block the nonspecific IgGs, and incubated for 1h with mouse anti-human antibody specific for p21, p53, p-ERK, ERK or GAPDH (Jackson ImmunoResearch Laboratories, PA, USA). The blots were then incubated with anti-mouse IgG (Jackson ImmunoResearch Labora-tories, PA, USA) linked to HRP (1:1000) for 1 h. Subsequently, the blots were developed using the ECL (enhanced chemiluminescence) system (Amersham Biosciences, UK). The bands were quantified by densitometry, using Image Pro-Plus 4.5 Software (Media Cybernetcis, MD, USA).
Immunoprecipitation and kinase activity assay
As previously described (Liang et al., 1999), equal amounts of total cellular lysate were immunoprecipitated with anti-JNK1 antibody (2 mg/L) and protein A/G-PLUS agarose for 15 h at 4 °C. Kinase assay was carried out in 45μL of kinase buffer (40 mmol/L Tris–NaOH pH 7.5, 0.5 mol/L NaCl, 0.1% NP-40, 6 mmol/L EDTA, 6 mmol/L EGTA, 10 mmol/L b-glycerophosphate, 10 mmol/L NaF, 10 mmol/L PNPP, 300μmol/L sodium orthovanadate, 1 mmol/L benzamidine, 2 mmol/L PMSF, 10 g/L aprotinin, 1 g/L leupeptin, and 1 mmol/L DTT) containing 5 mmol/L cold ATP, 10 mCi [γ-32P] ATP (Amersham Biosciences, UK), and 1 mg
GST-c-Jun fusion protein (Santa Cruz Biotechnology, CA, USA) as substrate, and incubated for 20 min at 25 °C. Each sample was mixed with 8μL of 5×Laemmli's loading buffer to stop the reaction. The samples were analyzed by 8% SDS-PAGE, and the gel was then dried and subjected to autoradiography.
[
3H]thymidine incorporation
The [3H]thymidine incorporation was performed as previously described
(Lin et al., 2002). Briefly, HUVEC were applied to 24-well plates in growth medium (M199 plus 10% FBS and ECGS). After the cells had grown to 70– 80% confluence, they were rendered quiescent by incubation for 24 h in M199 containing 2% FBS. M199 supplemented with 10% FBS and 0.05% DMSO (control) or TB was added to the cells and the cultures were allowed to incubate for 24 h. During the last 3 h of the incubation without or with TB, [3H]thymidine
(Amersham Biosciences, UK) was added at 1 μCi/mL (μCi=37 kBq). Incorporated [3H]thymidine was extracted in 0.2 N NaOH and measured in a liquid scintillation counter.
RT-PCR
Total cellular RNA was extracted from HUVEC with Trizol (Invitrogen Cor-poration, CA, USA) according to the manufacturer's protocol (Uchima et al., 2004). The RNA pellet was washed in 70% cold ethanol, air-dried and redissolved in 20μL of diethyl-pyrocarbonate (DEPC)-treated water (Arkell and Jackson, 2003). Two microliters was used in a total of 20μL reaction volume as a template for PCR amplification. PCR was performed under standard conditions in 20μL; 10 mM Tris, pH 8.3, 40 mM KCl, 1.5 mM MgCl2, 250μM dNTP, 10 pM each primer (sense and
antisense) and 1 U Taq DNA polymerase (Lee et al., 2006). The experimental conditions were as follows: 95 °C for 1 min; 55 °C for 1 min; and 72 °C for 1min for 35 cycles. The following primers were used: p53 5 ′-ATGGAGGAGCCGCAGT-CAG-3′ and 5′-TCAGTCTGAGTCAGGCCCTTC-3′; p21 5′-AGGAGGCGC-CATGTCAGAAC C-3′ and 5′-TCCTGTGGGCGGATTAGGGCT-3′; p27 5′-CC-ACGAAGAGTTAACCCGGG-3′ and 5′-GTCTGCTCCACAGAACCGGC-3′;
GA3PDH 5′-CCACCATGGAGAAGGCTGGGGCTCA-3′ and 5′-ATCACGCCA-CAGTTTCCCGGAGGGG-3′. The PCR products were electrophoresed on 1.8% agarose in 1× Tris-acetate-EDTA (TAE) buffer, and stained with ethidium bromide solution.
Preparation of plasmid constructs
p21 promoter. The pWWP-luc plasmid construct, containing promoter region of human p21/WAF1 between positions−2300 and +8 was a gift from Dr. B. Vogelstein (Johns Hopkins University, Baltimore, MD) (el-Deiry et al., 1993). p21 promoter was subcloned into pGL3-basic luciferase reporter vector (Promega Corporation, WI, USA) at the unique HindIII site, and the sequence identity was confirmed by ABI PRISM 377 DNA Analysis System (Perkin-Elmer, MA, USA). p53 dominant negative (DN) construct. For amplification of the hot-spot mutant (V143A) of p53 (Wong et al., 1999), recombinant PCR was performed. Synthesized cDNA (SuperScript. III First-Strand Synthesis System for RT-PCR; Invitrogen Corporation, CA, USA) from HUVEC was used as a template and Phusion high-fidelity DNA polymerase (Finnzymes Oy, Espoo, Finland) with 3′→ 5′ exonuclease activity was used in subsequent PCR. In the first round PCR, forward (5 ′-ATGGA-GGAGCCGCAGTCAG-3′) and reverse (5′-TCAGTCTGAGTCAGGCCCTTC-3′) primers were used to amplify full-length wild type p53 (WT-p53). Reactions were treated in a thermal cycle machine to incubate at 98 °C for 30 s followed by 35 cycles of: 98 °C for 10 s, 66 °C for 30 s, 72 °C for 30 s and a final incubation of 72 °C for 7 min. PCR product of WT-p53 was cleaned up and used for the next PCR. In the second round PCR, two primers set 1 and 2 were used to synthesize fragment A and
fragment B. Primer set 1: forward (5′-CTTAAGCTTGCCGCCATGGAGGAG-3′), and reverse (5′-CACAGATGCGCAGGGCAGGT-3′). Primer set 2: forward (5′-ACCTGCCCTGCGCAGCTGTG-3′) and reverse (5′-GTGCTGGATATCTCAGT-CTGAGTC-3′). Two-step PCR reactions were applied to synthesize fragment A: a first cycle of 40 s at 98 °C and 72 °C for 1 min followed by 98 °C for 10 s, 72 °C for 1 min for 34 cycles and a final incubation of 72 °C for 7 min. Standard PCR for fragment B was treated at 98 °C for 30 s followed by 35 cycles of: 98 °C for 10 s, 67 °C for 30 s, 72 °C for 30 s and a final incubation of 72 °C for 7 min. For each fragment, the amplified PCR product was gel purified and used as a mixed template for synthesizing p53V143Ain the PCR reaction: a first cycle of 40 s at 98 °C, 67 °C for
30 s and 72 °C for 1 min followed by 98 °C for 10 s, 67 °C for 30 s and 72 °C for 1 min for 34 cycles and a final incubation at 72 °C for 7 min. The synthesized PCR product of p53V143Awas purified, digested with restriction enzyme HindIII and EcoRV, and
cloned into pcDNA3.1+ (Invitrogen Corporation, CA, USA). The p53V143Asequence
fidelity was confirmed by ABI PRISM 377 DNA Analysis System (Perkin-Elmer, MA, USA).
Constitutive active MEK-1 construct. MEK-1 cDNA was a gift from Prof. Ruey-Hwa Chen (Institute of Biological Chemistry, Taipei, Taiwan) (Mansour et al., 1994; Chen et al., 2005).
Cell transfection
Transfection was performed using jetPEI-HUVEC transfection reagent (Poly-plus Transfection, Bioparc, France) and following the manufacturer's protocol. Briefly, jetPEI-HUVEC/DNA mixture was added drop-wise onto the DMEM-Glutamax medium (Invitrogen Corporation, CA, USA) containing 2% FBS,
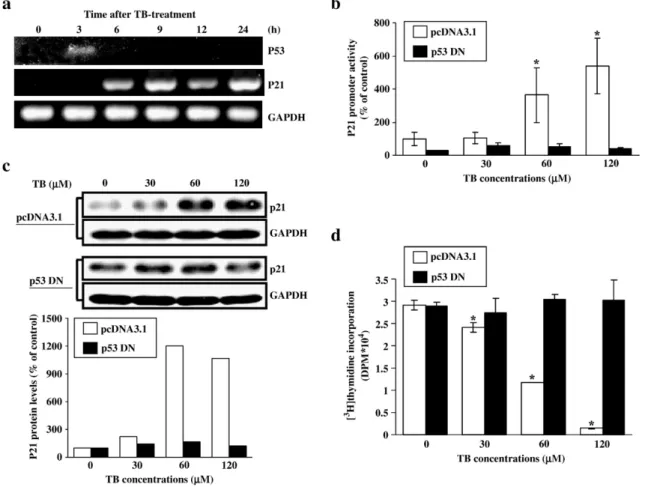
Fig. 1. Involvement of p53 in TB-induced up-regulation of p21 in HUVEC. (a) TB increases the levels of p21 and p53 mRNA in HUVEC. The increased p53 mRNA level was observed at 3 h after TB treatment, whereas the increased p21 mRNA was observed at 6 h after treatment. RT-PCR products of GAPDH were used as an internal control. (b) Transfection of the cells with p53DN abolished the TB-induced increases of p21 promoter activity. Four samples were analyzed in each group, and the values shown represent the means ± s.e.mean. ⁎pb0.05 different from control (cells transfected with pcDNA 3.1 without TB treatment). (c) Transfection of the cells with p53DN abolished the TB-induced increases of p21 protein level. The bottom panel shows the quantified results of p21 protein levels, which were adjusted with GAPDH protein level and expressed as percentage of control. (d) Transfection of the cells with p53DN abolished the TB-induced decreases of thymidine incorporation into HUVEC. Four samples were analyzed in each group, and values represent the means of percent of control ± s.e.mean. ⁎pb0.05 different from control. TB: terbinafine; p53DN, p53 dominant negative DNA.
mixed gently, and incubated in a humidified 37 °C incubator for 4 h. The transfected cells were incubated in fresh HUVEC growth medium for addi-tional 24 h. M199 supplemented with 10% FBS and TB was added to the cells and the cultures were allowed to incubate for the indicated hours. In these experiments, cells transfected with empty vector were served as a control.
Luciferase assay
After terbinafine treatment, the cells were washed twice with ice-cold phosphate buffer saline and lysed by lysis reagent (Promega Corporation, WI, USA). Luciferase activities were recorded in a 20/20n luminometer (Turner Biosystems, CA, USA) using the dual luciferase assay kit (Promega Corporation, WI, USA) according to the manufacturer's instructions. Each experiment was performed at least three times.
Matrigel angiogenesis assay
The Matrigel angiogenesis assay was performed as previously described with minor modifications (Passaniti et al., 1992). A 500μL portion of a liquid mixture of Matrigel (350 μL, BD Biosciences, CA, USA), M199 medium (150μL) and VEGF (50 μg/L) (Inoki et al., 2002) with or without terbinafine
was injected subcutaneously into 7-week-old C57/BL6 mice. Matrigel rapidly formed a single, solid gel plug. The plugs allowed VEGF to stimulate angio-genesis. On the 7th day, mice were euthanized, and the Matrigel plugs were excised. Vascularization of Matrigel plugs was quantified by measuring hemo-globin (Hb) content using Drabkin reagent (Sigma-Aldrich, MO, USA) ac-cording to the instructions of the manufacturer.
Statistics
All data were expressed as the mean value ± s.e.mean. Comparisons were subjected to one way analysis of variance (ANOVA) followed by Fisher's least significant difference test. Significance was accepted at pb0.05.
Results
p53-dependence of TB-induced p21 up-regulation
Previously, we demonstrated that TB exerts an
anti-pro-liferative effect in HUVEC through induction of p21 expression,
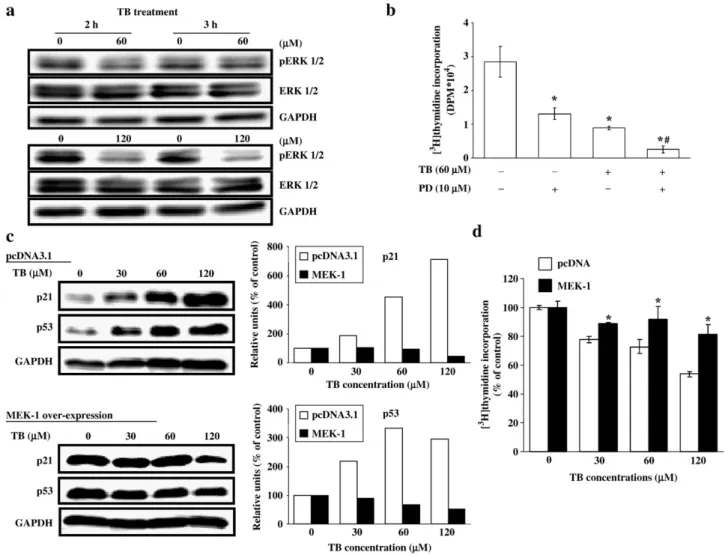
Fig. 2. Involvement of ERK in the TB-induced increases of p53 and p21 protein. After treatment with TB for 2–3 h, the cells were processed for total protein extraction and Western blot analysis. (a) Treatment of the cells with TB (60 and 120μM) for 2 or 3 h decreased the levels of phosphorylated ERK protein. (b) The TB-induced decrease of thymidine incorporation was enhanced by pretreatment of the cells with PD98059 (an ERK inhibitor). HUVEC were pre-treated with PD98059 for 1 h followed by TB (60μM) for 24 h. [3H]thymidine incorporation was done to examine the DNA synthesis. Four samples were analyzed in each group, and values
represent the means ± s.e.mean. ⁎pb0.05 different from control.#pb0.05 different from TB-treated. (c) Over-expression of MEK-1 abolished the increase of p21 and
p53 expression induced by TB. The right panels showed the quantified results of p21 and p53 protein levels, which were adjusted with GAPDH protein level and expressed by percentage of control. (d) Over-expression of MEK-1 abolished the TB-induced decrease of thymidine incorporation. HUVEC were transiently transfected with control vector (pcDNA 3.1) or vector coding for constitutive active MEK-1 followed by TB treatment for 24 h. Four samples were analyzed in each group, and values represent the means ± s.e.mean. ⁎pb0.05 different from corresponding control (pcDNA-transfected cells).
which in turn inhibits the CDK2 activity (
Ho et al., 2004
). To
examine whether the TB-induced increase of p21 expression is
regulated by p53, we performed RT-PCR to examine the
expression of p21 and p53 mRNA at different time points after
TB treatment (60
μM). As illustrated in the
Fig. 1
a, TB-induced
increases of p53 and p21 mRNA level in HUVEC began at 3h
and 6h, respectively. The RT-PCR products of GAPDH were
used as an internal control to monitor amounts of specimen
loading.
Fig. 1
b further demonstrates that TB
concentration-dependently increased the p21 promoter activity in HUVEC.
Transfection of the cells with p53DN abolished the TB-induced
increases of p21 promoter activity (
Fig. 1
b) and protein level
(
Fig. 1
c) and decrease of thymidine incorporation (
Fig. 1
d) in
HUVEC.
Involvement of ERK in the TB-induced increases of p21 and
p53 expression in HUVEC
Since the ERK pathway has been implicated to be involved in
the regulation of endothelial cell proliferation, we further
examined the levels of total and phosphorylated ERK protein
in the TB-treated HUVEC. As illustrated in
Fig. 2
a, TB
de-creased the levels of phosphorylated ERK in HUVEC.
More-over, treatment of the cells with PD98059, an ERK inhibitor,
decreased the DNA synthesis and accentuated the TB-induced
inhibition of DNA synthesis in HUVEC (
Fig. 2
b). To further
investigate the involvement of ERK in cell-cycle alternations
caused by TB, we over-expressed the MEK-1, the immediate
upstream activator kinase of ERK, in HUVEC. Over-expression
of MEK-1 abolished the TB-induced increases of p21 and
p53 protein (
Fig. 2
c) and decrease of thymidine incorporation
(
Fig. 2
d).
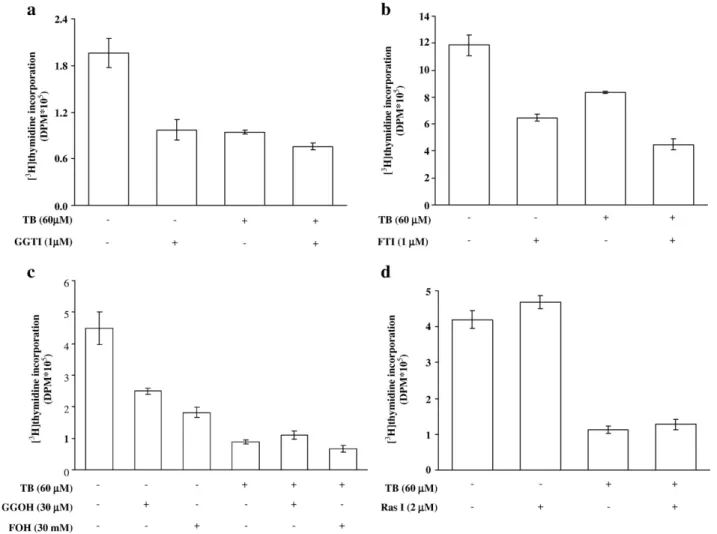
Isoprenylation might not be involved in the TB-induced growth
arrest
To examine whether prenylation was involved in the
TB-induced inhibition of proliferation in HUVEC, the cells were
pre-treated with isoprenoid pyrophosphate precursors (FOH and
GGOH) or isoprenoid inhibitors (FTI and GGTI) followed by TB
treatment. The results showed that treatment of the cells with
isoprenoid inhibitors (
Fig. 3
a and b) accentuated the TB-induced
inhibition of DNA synthesis. However, isoprenoid pyrophosphate
precursors (
Fig. 3
c) did not affect the TB-induced inhibition of
Fig. 3. Ras and protein isoprenylation might not be involved in TB-induced anti-proliferative effects in HUVEC. Pretreatment of the cells with the inhibitors of isoprenoid pyrophosphate, GGTI (a) and FTI (b), accentuated the TB-induced inhibition of DNA synthesis. However, neither the precursors of isoprenoid pyrophosphate, GGOH and FOH (c), nor Ras inhibitior (d) affected the TB (60μM)-induced anti-proliferation effects. Four samples were analyzed in each group, and values represent the means ± s.e.mean. Ras I, Ras inhibitor.
DNA synthesis in HUVEC. We also investigated the involvement
of Ras in the TB-mediated growth inhibition in HUVEC. Since
isoprenylation is important for subcellular distribution and
functions of Ras, we further examined the involvement of Ras
protein in regulating the HUVEC proliferation. As illustrated in
Fig. 3
d, Ras inhibitor did not affect the TB-induced inhibition of
DNA synthesis in HUVEC.
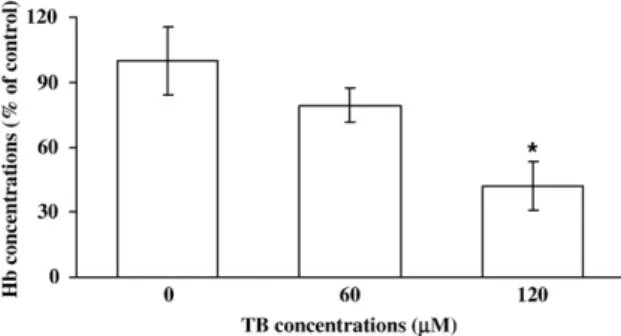
TB inhibits angiogenesis in vivo
Previously, we applied the capillary-like tube formation assay
and the chick embryo chorioallantoic membrane (CAM) assay to
demonstrate the anti-angiogenic activity of TB (
Ho et al., 2004
).
However, the results from in vitro study could not always be
replicated in an in vivo setting. To confirm the in vivo
anti-angiogenic effect of TB, we applied the Matrigel angiogenesis
assay. As illustrated in
Fig. 4
, TB (0, 60 and 120
μM)
concentration-dependently decreased Hb content, which
repre-sents the blood vessel formation, in the plug.
Discussion
Previously, we demonstrated that TB-induced cell-cycle
arrest at the G0/G1 phase in HUVEC through an up-regulation of
the CDK inhibitor, p21 protein (
Ho et al., 2004
). The findings of
the present study demonstrate the TB-induced p21 and p53
mRNA expression. Transfection of the cells with p53DN
abol-ished the TB-induced increases of promoter activity and protein
levels of p21. Treatment of HUVEC with TB decreased the
phosphorylated ERK protein levels (
Fig. 2
a). On the other hand,
over-expression of MEK-1 abolished the TB-induced increases
of p21 and p53 protein. Apparently, this is the first
demonstra-tion that TB induces increases of p21 mRNA and protein in
HUVEC through a p53-dependent pathway mediated by
in-hibition of ERK phosphorylation.
It has been suggested that p21 is a transcriptional target of the
tumor suppressor gene p53 and might promote p53-dependent
cell-cycle arrest or apoptosis (
Mendoza-Rodriguez and Cerbon,
2001; Jerry et al., 2002; Yu and Zhang, 2005
). However, p21 can
also be activated through a p53-independent pathway (
Marchetti
et al., 1996
). Our previous study in the COLO-205 cells showed
that the protein level of p53 and the binding between p53 protein
and p53 consensus binding site in p21 promoter DNA probe
were increased by TB treatment, and TB treatment caused an
increase in the level of p53 protein, which in turn up-regulated
the p21 protein, and finally caused cell-cycle arrest (
Lee et al.,
2003
). In the HUVEC, our previous data showed that TB
treatment resulted in an increase in the protein levels of p21 and
p53 (
Ho et al., 2004
). In this study, we demonstrated that TB
induced an increase in the mRNA levels of p21 and p53 in
HUVEC, suggesting that transcriptional regulation is involved in
the TB-induced increases in p21 and p53 protein levels. p53 has
been identified as a host protein interacting with the SV40 large T
(
Lane and Crawford, 1979
). It is essential for preventing
improper cell proliferation and maintaining the genome integrity
following genotoxic stress. Following DNA damage, hypoxia,
oncogene over-expression, and nucleotide depletion, p53
under-goes extensive post-translational changes which result in the
increased binding of p53 to DNA and transcriptional activation of
its target genes (
Vogelstein et al., 2000; Yu and Zhang, 2005
).
These targets regulate a number of cellular processes; among
which the best studied is a block in the cell-division cycle. The
p53 protein directly stimulates the expression of p21. In the
present study, RT-PCR results showed that the induction of p53
mRNA expression induced by TB treatment was observed prior
to p21 mRNA induction (
Fig. 1
a). In the p53DN-transfected
HUVEC, the increased p21 promoter activity and protein levels
induced by TB were abolished (
Fig. 1
b and c). Taken together,
our results suggest that TB increased p21 expression through p53
transactivation on the p21 promoter.
Mitogen-activated protein kinase (MAPK) cascades have
been shown to participate in a diverse array of cellular programs,
including cell division. In mammalian systems, four MAPK
modules, ERK1/2, JNK and p38 MAPK have been identified.
Activation of the MAPK pathway has been commonly
associated with cell-cycle progression. For example, activation
of the oncogenic MEK1 caused transformation of the NIH3T3
cells (
Okazaki and Sagata, 1995
), and injection of the
constitutive active form of MEK1 to the NIH3T3 cells induced
DNA replication (
Yuen et al., 2001
). In response to a wide
variety of extracellular stimuli (such as growth factors,
cyto-kines, and environmental stresses), ERK become
phosphory-lated and activated through a three-kinase signaling cascade
(
Marshall, 1995
). The ERK are vital in regulating diverse
cellular activities, including cell proliferation, cell-cycle
pro-gression, differentiation, apoptosis, and tumor invasion (
Mar-shall, 1995; Kolch, 2000
). ERK inhibitor has been reported to
increase p53 accumulation (
Kim et al., 2002; Kwon et al., 2004
).
In the present study, we showed that TB inhibited the
phos-phorylation of ERK (
Fig. 2
a), and over-expression of MEK-1
abolished the TB-induced increases of p53 and p21 protein level
(
Fig. 2
c). These findings suggest that ERK inactivation might be
involved in the TB-induced increases of p53 and p21 expression.
In the cholesterol synthesis pathway, squalene epoxidase
converts squalene to 2,3-oxidosqulene. TB, an inhibitor of
squa-lene epoxidase, might alter the protein prenylation by changing the
expression of farnesyl pyrophosphate (FPP) and geranylgeranyl
Fig. 4. Effect of TB on angiogensis in vivo. Angiogenesis was assessed by using the Matrigel plug assay. Matrigel mixed with 50 × 10− 6g/L VEGF and phos-phate buffer saline or TB (60 or 120μM) was injected s.c. into C57BL/6J mice. On the 7th day, the plugs were excised, and Hb levels were measured. The mean Hb concentration of plugs excised from animals treated with vehicle without TB is 100%. Seven or eight samples were analyzed in each group, and values represent the means ± s.e.mean. ⁎pb0.05 different from control.
pyrophosphate (GGPP), which are the upstream molecules of
squalene and might regulate protein post-translational
modifica-tions. It has been identified that membrane localization of the
small G proteins, such as Ras, is critical to their functions. The
membrane localization of the small G proteins is dependent on a
post-translational modification that results in attachment of
hydrophobic prenyl groups that anchor the small G proteins to
plasma membranes (
Magee and Marshall, 1999
). Activation of
Ras triggers a phosphorylation cascade that successively
acti-vates MEK1/2 and the ERK-MAP kinases, which are
translo-cated to the nucleus and activate transcription factors involved in
cell proliferation. Previously, we showed that TB
concentration-dependently decreased the protein levels of Ras in HUVEC and
Ras inhibitor peptide abolished the FOH-induced prevention of
TB-induced migration inhibition in HUVEC (
Ho et al., 2006
).
However, our data from the present study showed that
pre-treatment of the cells with Ras inhibitor did not significantly
affect the TB-induced inhibition of DNA synthesis (
Fig. 3
d).
These data suggest that Ras might be involved in the TB-induced
inhibition of endothelial cell migration, but not proliferation.
In the present study, our results showed that pretreatment of
the cells with FTI accentuated the TB-induced inhibition of
DNA synthesis (
Fig. 3
b); and this has led us conjecture the
involvement of farnesylation in TB-induced growth arrest.
Unexpectedly, FOH did not reverse TB-inhibited DNA
synth-esis. FTIs have been reported to induce hypophosphorylation of
retinoblastoma protein (pRb), a key player in the transition from
the G1 to S phase (
Sepp-Lorenzino and Rosen, 1998; Edamatsu
et al., 2000
), and increase expression of p21 at the transcriptional
level in the p53-independent pathway (
Feldkamp et al., 1999
).
Besides Ras, Rheb, which is required for cell-cycle progression
and cell growth, is also modified by farnesylation. We speculate
that FTI and TB might inhibit the DNA synthesis of HUVEC
through different pathways.
Previously, we showed that TB suppresses proliferation of
cultured HUVEC by inhibiting DNA synthesis and arresting
cell-cycle activity through up-regulation of p21. In the present
study, we further investigate the molecular mechanisms
under-lying TB-induced up-regulation of p21 in HUVEC. We
dem-onstrate that TB could cause ERK inactivation, which in turn
increases p53 protein expression and results in the
transactiva-tion of p21 promoter and increase in p21 protein, and finally
leads to growth arrest in HUVEC. Demonstration of TB-induced
inhibition of angiogenesis in vivo together with our previous in
vitro studies strongly suggests potential applications of TB in
anti-angiogenic therapy.
Acknowledgment
This work was supported by research grants from the
National Science Council of the Republic of China (NSC
94-2320-B-038-005, NSC 95-2320-B-038-019).
References
Abdel-Rahman, S.M., Nahata, M.C., 1997. Oral terbinafine: a new antifungal agent. Ann. Pharmacother. 31, 445–456.
Arkell, J., Jackson, C.J., 2003. Constitutive secretion of MMP9 by early-passage cultured human endothelial cells. Cell Biochem. Funct. 21, 381–386. Bussolino, F., Mantovani, A., Persico, G., 1997. Molecular mechanisms of
blood vessel formation. Trends Biochem. Sci. 22, 251–256.
Carmeliet, P., 2005. Angiogenesis in life, disease and medicine. Nature 438, 932–936.
Chen, C.H., Wang, W.J., Kuo, J.C., Tsai, H.C., Lin, J.R., Chang, Z.F., Chen, R.H., 2005. Bidirectional signals transduced by DAPK–ERK interaction promote the apoptotic effect of DAPK. EMBO J. 24, 294–304.
Eastman, A., 2004. Cell cycle checkpoints and their impact on anticancer therapeutic strategies. J. Cell. Biochem. 91, 223–231.
Edamatsu, H., Gau, C.L., Nemoto, T., Guo, L., Tamanoi, F., 2000. Cdk inhibitors, roscovitine and olomoucine, synergize with farnesyltransferase inhibitor (FTI) to induce efficient apoptosis of human cancer cell lines. Oncogene 19, 3059–3068.
el-Deiry, W.S., Tokino, T., Velculescu, V.E., Levy, D.B., Parsons, R., Trent, J.M., Lin, D., Mercer, W.E., Kinzler, K.W., Vogelstein, B., 1993. WAF1, a potential mediator of p53 tumor suppression. Cell 75, 817–825.
Feldkamp, M.M., Lau, N., Guha, A., 1999. The farnesyltransferase inhibitor L-744,832 inhibits the growth of astrocytomas through a combination of antiproliferative, antiangiogenic, and proapoptotic activities. Ann. N.Y. Acad. Sci. 886, 257–260.
Gupta, A.K., Shear, N.H., 1997. Terbinafine: an update. J. Am. Acad. Dermatol. 37, 979–988.
Gupta, M.K., Qin, R.Y., 2003. Mechanism and its regulation of tumor-induced angiogenesis. World J. Gastroenterol. 9, 1144–1155.
Gupta, A.K., del Rosso, J.Q., Lynde, C.W., Brown, G.H., Shear, N.H., 1998. Hepatitis associated with terbinafine therapy: three case reports and a review of the literature. Clin. Exp. Dermatol. 23, 64–67.
Ho, P.Y., Liang, Y.C., Ho, Y.S., Chen, C.T., Lee, W.S., 2004. Inhibition of human vascular endothelial cells proliferation by terbinafine. Int. J. Cancer 111, 51–59.
Ho, P.Y., Zhong, W.B., Ho, Y.S., Lee, W.S., 2006. Terbinafine inhibits endothelial cell migration through suppression of the Rho-mediated pathway. Mol. Cancer Ther. 5, 3130–3138.
Inoki, I., Shiomi, T., Hashimoto, G., Enomoto, H., Nakamura, H., Makino, K., Ikeda, E., Takata, S., Kobayashi, K., Okada, Y., 2002. Connective tissue growth factor binds vascular endothelial growth factor (VEGF) and inhibits VEGF-induced angiogenesis. FASEB J 16, 219–221.
Jerry, D.J., Dickinson, E.S., Roberts, A.L., Said, T.K., 2002. Regulation of apoptosis during mammary involution by the p53 tumor suppressor gene. J. Dairy Sci. 85, 1103–1110.
Kim, S.J., Ju, J.W., Oh, C.D., Yoon, Y.M., Song, W.K., Kim, J.H., Yoo, Y.J., Bang, O.S., Kang, S.S., Chun, J.S., 2002. ERK-1/2 and p38 kinase oppositely regulate nitric oxide-induced apoptosis of chondrocytes in association with p53, caspase-3, and differentiation status. J. Biol. Chem. 277, 1332–1339. Kolch, W., 2000. Meaningful relationships: the regulation of the Ras/Raf/MEK/
ERK pathway by protein interactions. Biochem. J. 351 (Pt 2), 289–305. Kwon, Y.W., Kwon, K.S., Moon, H.E., Park, J.A., Choi, K.S., Kim, Y.S., Jang,
H.S., Oh, C.K., Lee, Y.M., Kwon, Y.G., Lee, Y.S., Kim, K.W., 2004. Insulin-like growth factor-II regulates the expression of vascular endothelial growth factor by the human keratinocyte cell line HaCaT. J. Invest. Dermatol. 123, 152–158.
Lane, D.P., Crawford, L.V., 1979. T antigen is bound to a host protein in SV40-transformed cells. Nature 278, 261–263.
Lee, W.S., Chen, R.J., Wang, Y.J., Tseng, H., Jeng, J.H., Lin, S.Y., Liang, Y.C., Chen, C.H., Lin, C.H., Lin, J.K., Ho, P.Y., Chu, J.S., Ho, W.L., Chen, L.C., Ho, Y.S., 2003. In vitro and in vivo studies of the anticancer action of terbinafine in human cancer cell lines: G0/G1 p53-associated cell cycle arrest. Int. J. Cancer 106, 125–137.
Lee, S.K., Kim, J.M., Lee, M.Y., Son, K.H., Yeom, Y.I., Kim, C.H., Shin, Y., Koh, J.S., Han, D.C., Kwon, B.M., 2006. Confirmation of a linkage between H-Ras and MMP-13 expression as well as MMP-9 by chemical genomic approach. Int. J. Cancer 118, 2172–2181.
Liang, Y.C., Huang, Y.T., Tsai, S.H., Lin-Shiau, S.Y., Chen, C.F., Lin, J.K., 1999. Suppression of inducible cyclooxygenase and inducible nitric oxide synthase by apigenin and related flavonoids in mouse macrophages. Carcinogenesis 20, 1945–1952.
Lin, S.Y., Liu, J.D., Chang, H.C., Yeh, S.D., Lin, C.H., Lee, W.S., 2002. Magnolol suppresses proliferation of cultured human colon and liver cancer cells by inhibiting DNA synthesis and activating apoptosis. J. Cell. Biochem. 84, 532–544.
Magee, T., Marshall, C., 1999. New insights into the interaction of Ras with the plasma membrane. Cell 98, 9–12.
Mansour, S.J., Matten, W.T., Hermann, A.S., Candia, J.M., Rong, S., Fukasawa, K., Vande Woude, G.F., Ahn, N.G., 1994. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science 265, 966–970. Marchetti, A., Doglioni, C., Barbareschi, M., Buttitta, F., Pellegrini, S.,
Bertacca, G., Chella, A., Merlo, G., Angeletti, C.A., Dalla Palma, P., Bevilacqua, G., 1996. p21 RNA and protein expression in non-small cell lung carcinomas: evidence of p53-independent expression and association with tumoral differentiation. Oncogene 12, 1319–1324.
Marshall, C.J., 1995. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell 80, 179–185.
Mendoza-Rodriguez, C.A., Cerbon, M.A., 2001. Tumor suppressor gene p53: mechanisms of action in cell proliferation and death. Rev. Invest. Clin. 53, 266–273.
Moraes, J.R., Stastny, P., 1977. A new antigen system expressed in human endothelial cells. J. Clin. Invest. 60, 449–454.
Nakagami, H., Morishita, R., Yamamoto, K., Taniyama, Y., Aoki, M., Kim, S., Matsumoto, K., Nakamura, T., Higaki, J., Ogihara, T., 2000. Anti-apoptotic action of hepatocyte growth factor through mitogen-activated protein kinase on human aortic endothelial cells. J. Hypertens. 18, 1411–1420.
Nakagami, H., Morishita, R., Yamamoto, K., Taniyama, Y., Aoki, M., Matsumoto, K., Nakamura, T., Kaneda, Y., Horiuchi, M., Ogihara, T., 2001. Mitogenic and
antiapoptotic actions of hepatocyte growth factor through ERK, STAT3, and AKT in endothelial cells. Hypertension 37, 581–586.
Okazaki, K., Sagata, N., 1995. MAP kinase activation is essential for oncogenic transformation of NIH3T3 cells by Mos. Oncogene 10, 1149–1157. Passaniti, A., Taylor, R.M., Pili, R., Guo, Y., Long, P.V., Haney, J.A., Pauly,
R.R., Grant, D.S., Martin, G.R., 1992. A simple, quantitative method for assessing angiogenesis and antiangiogenic agents using reconstituted basement membrane, heparin, and fibroblast growth factor. Lab. Invest. 67, 519–528.
Petranyi, G., Ryder, N.S., Stutz, A., 1984. Allylamine derivatives: new class of synthetic antifungal agents inhibiting fungal squalene epoxidase. Science 224, 1239–1241.
Sepp-Lorenzino, L., Rosen, N., 1998. A farnesyl-protein transferase inhibitor induces p21 expression and G1 block in p53 wild type tumor cells. J. Biol. Chem. 273, 20243–20251.
Uchima, Y., Sawada, T., Nishihara, T., Maeda, K., Ohira, M., Hirakawa, K., 2004. Inhibition and mechanism of action of a protease inhibitor in human pancreatic cancer cells. Pancreas 29, 123–131.
Vogelstein, B., Lane, D., Levine, A.J., 2000. Surfing the p53 network. Nature 408, 307–310.
Wong, K.B., DeDecker, B.S., Freund, S.M., Proctor, M.R., Bycroft, M., Fersht, A.R., 1999. Hot-spot mutants of p53 core domain evince characteristic local structural changes. Proc. Natl. Acad. Sci. U. S. A. 96, 8438–8442. Yu, J., Zhang, L., 2005. The transcriptional targets of p53 in apoptosis control.
Biochem. Biophys. Res. Commun. 331, 851–858.
Yuen, P.H., Ryan, E.A., Devroe, E., Wong, P.K., 2001. A single Glu(62)-to-Lys (62) mutation in the Mos residues of the R7Delta447Gag-tMos protein causes the mutant virus to induce brain lesions. Oncogene 20, 692–703.