國 立 交 通 大 學
統計學研究所
碩 士 論 文
在全基因關聯分析中
利用總體的基因表現量作為穩定表現型
Using Global Gene Expression as Endophenotypes
in a Genome-wide Association Study
研 究 生:蔡佩芳
指導教授:黃冠華 博士
在全基因關聯分析中
利用總體的基因表現量作為穩定表現型
Using Global Gene Expression as Endophenotypes
in a Genome-wide Association Study
研 究 生:蔡佩芳 Student: Pei-Fang Tsai
指導教授:黃冠華 Advisor: Dr. Guan-Hua Huang
國 立 交 通 大 學
統計學研究所
碩 士 論 文
A Thesis
Submitted to institute of Statistics College of Science
National Chiao Tung University in Partial Fulfillment of the Requirements
for the Degree of Master
in Statistics July 2008
Hsinchu, Taiwan, Republic of China
中華民國九十七年七月
在全基因關聯分析中
利用總體的基因表現量作為穩定表現型
研究生:蔡佩芳 指導教授:黃冠華 博士
國立交通大學統計學研究所
摘要
在生物學上,穩定表現型(endophenotype)和疾病有著相同的遺傳路徑,但 穩定表現型卻比診斷上的表現型(phenotype)更為接近其相關的基因,這也顯示 穩定表現型在複雜疾病上基因研究的重要性。穩定表現型為主的基因遺傳分析比 表現型為主的基因遺傳分析更容易找到致病基因。由穩定表現型所發展的指標 (PHE),穩定表現型的基因遺傳性所佔比例,用在判別出有可能的穩定表現型。 在這篇報告裡,氣喘是一種基因間作用和環境因素複雜的疾病,我們利用基 因表現量當作穩定表現型找尋可能的氣喘基因。我們利用指標(PHE)判斷哪些探 針組的基因表現量是穩定表現型,接著作指標(PHE)的檢定。針對多重檢定的問 題,我們利用 q-值來控制錯誤發現率和調整。我們也對每一個基因表現量做全 基因關聯分析,比較基因表現量指標(PHE)中有顯著和沒有顯著之間基因遺傳性 的變化。最後,我們論文中有(1) 評估利用基因表現量當作穩定表現型找尋跟疾 病有相關基因的適當性,(2) 檢驗從基因表現量為主的分析,辨別出的基因和文 獻中提過疾病相關的基因重複的多寡,(3) 從這些基因表現量判定為穩定表現型 中,評估它們基因遺傳的特徵。 關鍵字: 順式作用 (cis effect) ; 穩定表現型 ; 表現量位置 ; 全基因關聯分Using Global Gene Expression as Endophenotypes
in a Genome-wide Association Study
Student: Pei-Fang Tsai Advisor: Dr. Guan-Hua Huang
Institute of Statistics
National Chiao Tung University
ABSTRACT
Endophenotype, which involve the same biological pathways as diseases but
presumably are closer to the relevant gene action than diagnostic phenotypes, have
emerged as an important concept in the genetic studies of complex diseases.
Endophenotype-based genetic analysis is more likely to succeed than
phenotype-based one in terms of search for the susceptibility genes. The index,
proportion of heritability explained (PHE), has been proven useful in identifying
potential endophenotypes.
In this report, we use global gene expression as endophenotypes in search for the
susceptibility genes underlying asthma, which is a disease caused by complex
interactions of genetic and environmental factors. We judge which gene expressions
of probe sets are endophenotypes by using the index PHE and do hypothesis test of
PHE. For the problem of multiple testing, we utilize the q-value to control for the
false discovery rate (FDR) for significance judgment. We also perform genome-wide
association tests for each gene expression and compare various genetic properties
between gene expressions with and without significant PHE values. At the end, this
thesis has (1) evaluated the appropriateness of using global gene expressing as
overlap between genes identified by the gene-expression-based analysis and genes
already identified in the literature, and (3) assessed genetic characteristics of gene
expressions that are identified as the endophenotypes.
Key words: Cis effect ; Endophenotype ; Expression quantitative trait loci ;
誌 謝
這兩年來,首先感謝老師 黃冠華的指導,常常因為一點小問題就跑去找老 師,老師都很熱心地幫忙解決問題和疑惑。最後還讓老師非常頭痛地改我的論 文,對老師感到非常不好意思。也感謝老師一年來的訓練,學到很多看書和抓重 點的技巧,也學習到面對一個全新領域一開始應有的應對方式,這一年來獲益良 多。最後對老師在說一聲 謝謝。也感謝其他老師的教導,讓我對統計更有了解。 接下來要感謝我的同學們,像是同老師的同學們 重耕、彥銘和仲竹,彼此間 會互相幫忙指導和提醒對方事情。另外也感謝夙吟、姿蒨、郃嵐和瑜達聽我練習 和給我意見。也非常感謝其他的同學們,因為有你們讓我的研究所生活很精彩。 最後感謝我的家人,常常給我鼓勵和指導一些論文的事項,謝謝你們! 在此,謹以此篇論文,獻給我親愛的家人和同學,還有陪伴我的好朋友們! 蔡佩芳 2008.07.02
Contents
摘要... I
ABSTRACT ...II
1 INTRODUCTION...1
2 LITERATURE REVIEW...4
2.1 STATISTICAL VALIDATION OF SURROGATE ENDPOINTS...4
2.2 STATISTICAL VALIDATION OF ENDOPHENOTYPE...5
2.2.1 Model...6 2.2.2 Variance ofPHE ...10 2.2.3 The covariance of * * ( ) ( ) ˆt and ˆt q q h h ... 11 2.2.4 Hypothesis test ...14
2.3 THE WHOLE-GENOME ASSOCIATION TEST FOR QUANTITATIVE OUTCOMES...14
2.4 THE PREPROCESSING METHOD FOR GENE EXPRESSION LEVELS...16
2.4.1 Background adjustment...16
2.4.2 Normalization ...16
2.4.3 Summarization ...16
2.4.4 RMA ...16
2.5 THE DIFFERENTIAL EXPRESSION METHODS...18
2.5.1 Fold-change ...18
2.5.2 Two sample t-test...18
2.5.3 SAM (Significance Analysis of Microarrays) ...19
2.6 MULTIPLE TESTING PROCEDURES...20
2.6.1 Type I error rates...21
2.6.2 Adjusted p-values ...22
2.6.3 The q-value...23
2.7 DATASETS...24
3 MATERIALS AND METHODS ...26
3.1 DATASETS...27
3.2 STATISTICAL METHOD OF ENDOPHENOTYPE...27
3.3 THE PREPROCESSING AND DIFFERENTIAL METHOD...29
3.4 GENOME-WIDE ASSOCIATION TESTS FOR GENE EXPRESSIONS...30
3.5 ASTHMA GENES AND OVERLAP RATE...30
3.6.2 The scatter plot of heritability of gene expressions versus PHEs...32
3.6.3...The bar-plot of proportions of probe sets with max significant SNPs’ LOD >6 versus PHEs………32
3.6.4 The bar-plot of number of cis eSNPs <100 kb or cis eSNPs >100 kb or trans versus PHEs...………33
3.6.5 The bar-plot of number of cis eSNPs <100 kb or cis eSNPs >100 kb or trans versus heritability of gene expressions and differential expressions. ...34
3.6.6 The density plot of LOD score for cis eSNPs <100 kb, cis eSNPs >100 kb and trans ...36
4 RESULT...37
4.1 TEST OF PHES AND THE DISTRIBUTION OF DIFFERENT CONDITIONS OF PHES...37
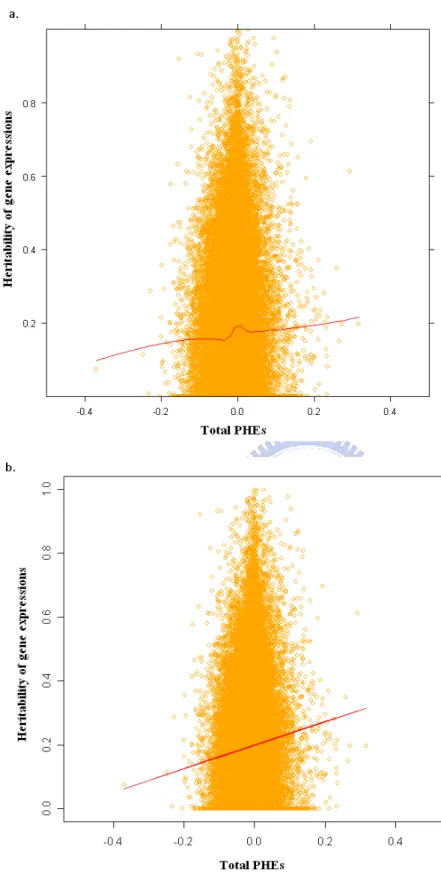
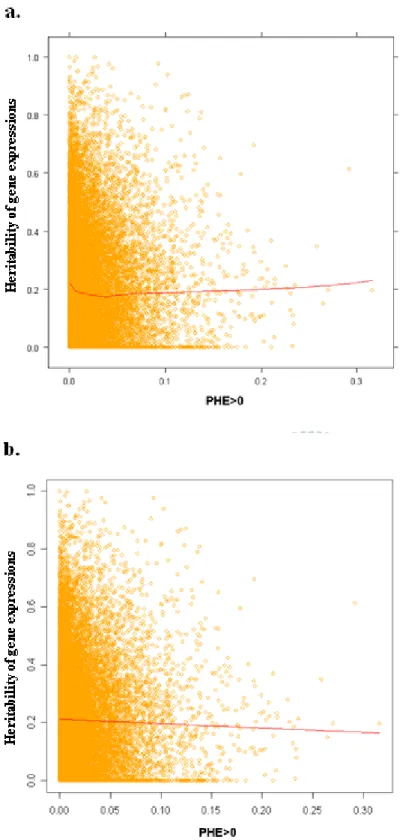
4.2 THE SCATTER PLOT OF HERITABILITY OF GENE EXPRESSION VERSUS PHES...37
4.3 THE OVERLAP RATE...38
4.4 THE RESULT OF ASSOCIATION TEST UNDER FOUR CONDITIONS OF PHES...39
4.4.1 The bar-plot of proportion of probe sets with max significant SNPs’ LOD >6 versus PHEs 39 4.4.2 The bar-plot of the number of cis eSNPs <100 kb or cis eSNPs >100 kb or trans versus PHEs 40 4.4.3 The bar-plot of number of cis eSNPs <100 kb or cis eSNPs >100 kb or trans versus heritability of gene expressions and differential expressions. ...41
4.4.4 The density plot of LOD score for cis eSNPs <100 kb, cis eSNPs >100 kb and trans ...42
5 CONCLUSIONS AND DISCUSSION...43
5.1 CONCLUSIONS...43
5.2 DISCUSSION...44
REFERENCE ...45
APPENDIX I...74
Figures and Tables Content
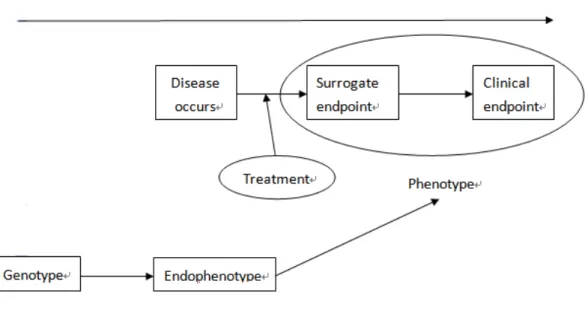
FIGURE 1 A SURROGATE ENDPOINT VERSUS AN ENDOPHENOTYPE IN THE DISEASE PROCESS...48
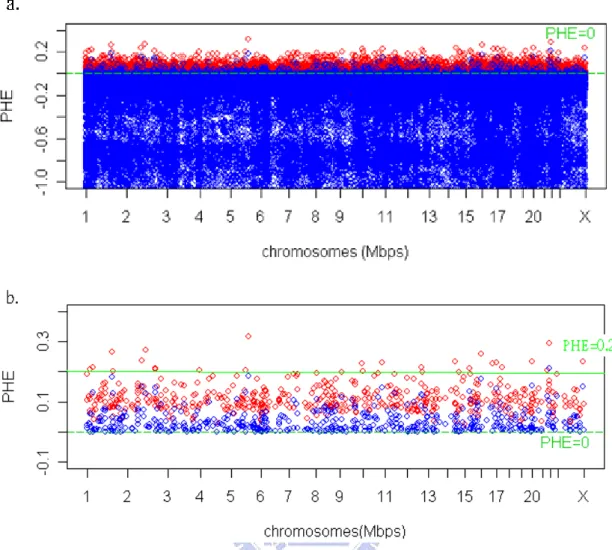
FIGURE 2 THE CONFIDENCE INTERVAL OF PHES ON ALL CHROMOSOMES (RED:PHE, BLUE: THE LOW BOUND OF PHE). ...49
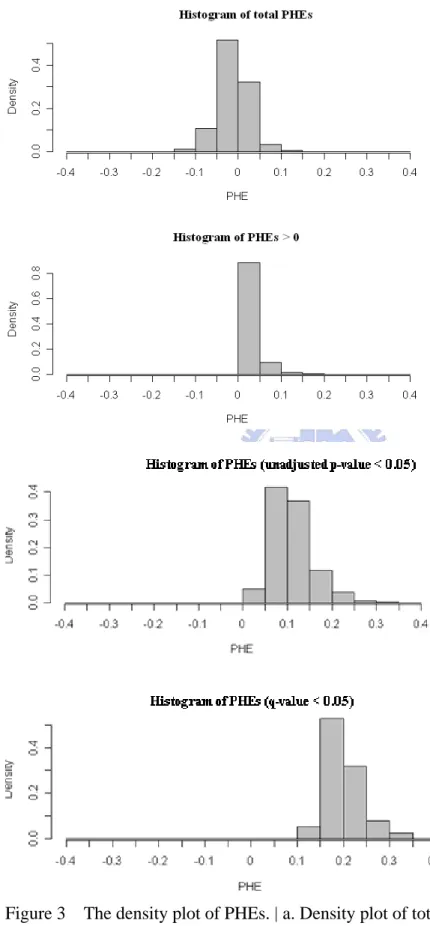
FIGURE 3 THE DENSITY PLOT OF PHES...50
FIGURE 4 THE SCATTER PLOT OF HERITABILITY VERSUS TOTAL PHES. ...51
FIGURE 5 THE SCATTER PLOT OF HERITABILITY VERSUS PHES >0...52
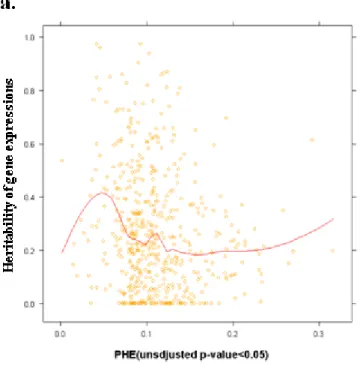
FIGURE 6 THE SCATTER PLOT OF HERITABILITY VERSUS PHES WITH UNADJUSTED P-VALUE <0.05. ...53
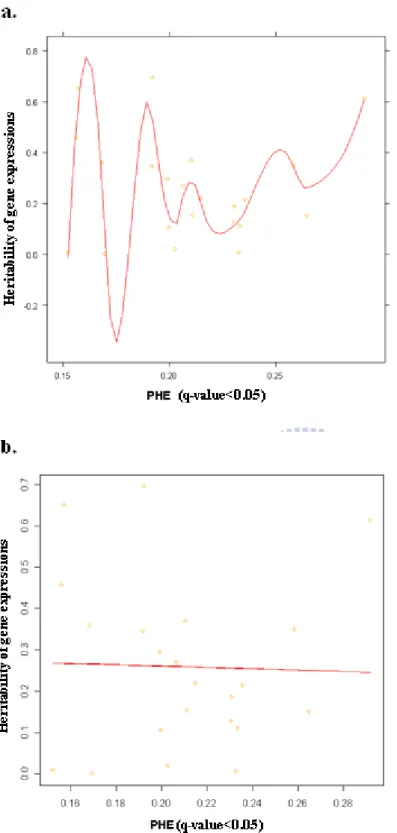
FIGURE 7 THE SCATTER PLOT OF HERITABILITY VERSUS PHES WITH Q-VALUE <0.05. ...54
FIGURE 8 THE BAR-PLOT OF PROPORTION OF PROBE SETS WITH MAX SIGNIFICANT SNPS’LOD>6 VERSUS PHES. ...55
FIGURE 9 THE BAR-PLOT OF THE NUMBER (CIS ESNPS <100KB) VERSUS PHES. ...56
FIGURE 10 THE BAR-PLOT OF THE NUMBER (CIS ESNPS >100KB) VERSUS PHES. ...57
FIGURE 11 THE BAR-PLOT OF THE NUMBER (TRANS) VERSUS PHES. ...58
FIGURE 12 THE BAR-PLOT OF THE NUMBER (CIS ESNPS <100 KB) VERSUS HERITABILITY AND DIFFERENTIAL EXPRESSION. ...59
FIGURE 13 THE BAR-PLOT OF THE NUMBER (CIS ESNPS >100 KB) VERSUS HERITABILITY AND DIFFERENTIAL EXPRESSION. ...60
FIGURE 14 THE BAR-PLOT OF THE NUMBER (TRANS) VERSUS HERITABILITY AND DIFFERENTIAL EXPRESSION. ...61
FIGURE 15 THE DENSITY PLOT OF LOD SCORE FOR CIS ESNPS <100 KB (RED), CIS ESNPS >100 KB (GREEN) AND TRANS (TRANS) BETWEEN X-LIMIT (3,50) AND Y-LIMIT (0,0.8) FOR PROBE SETS WITH TOTAL PHES. ...62
FIGURE 16 THE DENSITY PLOT OF LOD SCORE FOR CIS ESNPS <100 KB (RED), CIS ESNPS >100 KB (GREEN) AND TRANS (BLUE) BETWEEN X-LIMIT (3,25) AND Y-LIMIT (0,0.3) FOR PROBE SETS WITH TOTAL PHES. ...63
FIGURE 17 THE DENSITY PLOT OF LOD SCORE FOR CIS ESNPS <100 KB (RED), CIS ESNPS >100 KB (GREEN) AND TRANS (BLUE) BETWEEN X-LIMIT (3,50) AND Y-LIMIT (0,0.8) FOR PROBE SETS WITH PHES>0. ...64
FIGURE 18 THE DENSITY PLOT OF LOD SCORE FOR CIS ESNPS <100 KB (RED), CIS ESNPS >100 KB (BLUE) AND TRANS (BLUE) BETWEEN X-LIMIT (3,25) AND Y-LIMIT (0,0.5) FOR PROBE SETS WITH PHES>0...65
FIGURE 19 THE DENSITY PLOT OF LOD SCORE FOR CIS ESNPS <100 KB (RED), CIS ESNPS >100 KB (GREEN) AND TRANS (BLUE) BETWEEN X-LIMIT (3,50) AND Y-LIMIT (0,0.8) FOR PROBE SETS WITH PHES WITH UNADJUSTED P-VALUE <0.05. ...66 FIGURE 20 THE DENSITY PLOT OF LOD SCORE FOR CIS ESNPS <100 KB (RED), CIS ESNPS >100 KB
WITH UNADJUSTED P-VALUE <0.05. ...67
FIGURE 21 THE DENSITY PLOT OF LOD SCORE FOR CIS ESNPS <100 KB (RED), CIS ESNPS >100 KB (GREEN) AND TRANS (BLUE) BETWEEN X-LIMIT (3,10) AND Y-LIMIT (0,0.8) FOR PROBE SETS WITH PHES WITH Q-VALUE <0.05. ...68
TABLE 1 GENETIC COMPONENTS OF VARIANCE ASSUMING RANDOM MATING...8
TABLE 2 THE DERIVATIVE OF COVARIANCE COMPONENTS FOR RELATIVE PAIRS...12
TABLE 3 PROBABILITY OF CALLING 1 OR MORE FALSE POSITIVES BY CHANCE. ...21
TABLE 4 TYPE I AND TYPE II ERRORS IN MULTIPLE HYPOTHESIS TESTING. ...21
TABLE 5 THE PHES OF PROBE SETS WITH Q-VALUE <0.05. ...69
TABLE 6 THE PROBE SETS WITH PHES >0.2...71
TABLE 7 GENES OF SIGNIFICANT SNPS (LOD>6) OVERLAP WITH ASTHMA OR ATOPY GENES...72
TABLE 8 THE PROBE SET WITH MAX ESNPS'LOD>6(PHES WITH Q-VALUE <0.05)...73
TABLE 9 CIS ESNPS <100 KB (PHES WITH Q-VALUE <0.05). ...73
1 Introduction
In diseases with classic or Mendelian genetics as their distal causes, genotypes are
usually indicative of phenotypes. However, this degree of genetic certainty does not
exist for complex disease [1]. These “complex” diseases are influenced by multiple
genes, environmental factors and their interactions on phenotypes. As a result, the
direct relationship between phenotype and genotype is disrupted, so that the same
genotype may result in different phenotypes, or different genotypes may result in the
same phenotype. To facilitate the identification of influential genetic markers of
complex disease, endophenotype approach has been advocated.
Endophenotypes are useful for theorizing about clinical phenotypes and mark the
path between genotype and phenotype. The endophenotype is closer to the underlying
gene than the phenotype in the course of disease’s natural history and can increase the
chance of identifying genotype (Figure 1). Huang et al. [2] defined an endophenotype
to be “a trait for which a test of null hypothesis of no genetic heritability implies the
corresponding null hypothesis based on the phenotype of interest” and develop a
formal statistical methodology for accessing the utility of endophenotypes, motivated
by the conditioning strategy used for identifying surrogate endpoints in clinical
research. The methodology is especially useful for the situation where underlying
genotype is unknown and researchers use endophenotypes to increase opportunities of
finding susceptible disease genes. Similar to validating surrogate endpoints, various
indices can be used to validate endophenotypes. One of the indices is the proportion
of heritability explained (PHE) by the endophenotype, similar to PTE introduced
by Freedman et al. [3] in the surrogate endpoint study. The greater the PHE value, the
more likely the intermediate variable is an endophenotype. Hsieh et al. [4] utilized the
There is a problem about signal-SNP association test between SNP genotypes and
case-control status: it cannot discover SNPs that are weakly related to the disease by
itself, but can have great impacts on the disease variability after combining with other
SNPs. A large PHE value represents that endophenotype and phenotype share many
genes. Because endophenotype is closer to genotype, there will be some genes that are
significant in endophenotype, but are not significant in phenotype. We can utilize the
PHE to find SNPs that may be weakly associated to the disease phenotype. Hopefully,
these additional SNPs can increase our chances in searching possible
phenotype-related genes.
Gene expression is a measurement of mRNA and mRNA is transcribed from a
DNA template and carries coding information to the sites of protein synthesis.
Because mRNA is closer to DNA genotype, we use global gene expression as
endophenotypes in search for the susceptibility genes underlying asthma, which is a
disease caused by complex interactions of genetic and environmental factor. In this
thesis, we first get global gene expressions of probe sets in Epstein-Barr virus
Iymphoblastoid cell lines (EBVL) measured with Affymetrix HG-U133 Plus 2.0 chip
[5-6]. Gene expression values are preprocessed by the robust multi-array averaging
(RMA). We judge which gene expressions of probe sets are endophenotypes by using
the index PHE and do hypothesis test of PHE. For the problem of multiple testing, we
utilize the q-value to control for the false discovery rate (FDR) for significance
judgment. We also perform genome-wide association tests for each gene expression
and compare various genetic properties between gene expressions with and without
significant PHE values.
Therefore, this thesis aims at
(1) Evaluating the appropriateness of using global gene expressing as
(2) Examining the overlap between genes identified by the gene-expression-based
analysis and genes already identified in the literature, and
(3) Assessing genetic characteristics of gene expressions that are identified as the
2 Literature Review
2.1 Statistical validation of surrogate endpointsSurrogate endpoints have been frequently utilized in most clinical research, when
the primary endpoint is too difficult or costly or time-consuming to obtain. Clinically
meaningful biomarkers of the disease projected as a surrogate endpoint in a clinical
trial is expected to ultimately demonstrate treatment effect on the primary endpoint if
a treatment effect shown on the markers. Surrogate endpoints have been of clinical
interest for decades, but it was not until Prentice published a seminal paper in 1989
that formal statistical investigation started. Prentice defined a surrogate endpoint to be
“a response variable for which a test of null hypothesis of no relationship to the
treatment groups under comparison is also a valid test of the corresponding null
hypothesis based on the true (clinical) endpoint”.
Prentice’s definition can be written as
( | ) ( ) ( | ) ( )
f S X = f S ⇔ f T X = f T
Where T denotes the status of a primary endpoint, S denotes the status of a
surrogate end-point, X is the treatment variable, f(S) is the distribution of S, and f(S|X)
is the conditional distribution of S given X. Validation of Prentice’s definition
involves the following two criteria:
( | ) ( ) and ( | , ) ( | )
f T S ≠ f T f T S X = f T S
[3, 7-8]. The first criterion states that the surrogate endpoint must be correlated with
the primary clinical endpoint, and the second criterion is that the surrogate endpoint
should fully capture the treatment effect on the treatment effect on the primary
endpoint.
The surrogate endpoint described by Prentice mediates all of the effect of treatment
on the primary endpoint, that is
A more complex, but more likely, situation arises when treatment has a direct effect
on the primary endpoint that is not mediated through the surrogate [9]:
X → →S T
Freedman et al. [3] proposed to focus on the proportion of the treatment effect
mediated through the surrogate. A good surrogate is one that explains large proportion
of that effect. The proposal can be made in the content of generalized linear models
[10]. The net effect of X on T can be assessed through the regression coefficient
T
β in the generalized linear model
[ ( )] T T
g E T =α +β X
Where g(.) is the link function connecting the mean response and covariates, and
the effect of X on T after inclusion of S is the regression coefficient βTSin the following generalized linear model
[ ( )] TS TS TS
g E T =α +β X+γ S
The proportion of the treatment effect (on the primary endpoint) explain (PTE) by
the surrogate is given by
1 TS
T
PTE β
β = −
The 100(1−α)% confidence limits of PTE can be calculated using the delta method.
2.2 Statistical validation of endophenotype Notation:
1,...,
i= I : representing the different family
1,..., i
j= n : representing the jth member of this family
2
A
σ : The variances arising from polygenic additive effects, 2
D
σ : The variance arising from polygenic dominance effects 2
C
σ : The variance arising from the shared environmental effects
2.2.1 Model
Endophenotypes are useful for theorizing about clinical phenotypes and can mark
the path between the genotype and the phenotype. Verification of existence of the
pathway genotype-endophenotype-phenotype is the key of validating endophenotypes.
Analogous to Prentice’s definition [7] that surrogate endpoint to be “a response
variable for which a test of null hypothesis of no relationship to the treatment groups
under comparison is also a valid test of the corresponding null hypothesis based on
the true (clinical) endpoint”, Huang et al. [2] define an endophenotype to be “a trait
for which a test of null hypothesis of no genetic heritability implies the corresponding
null hypothesis based on the phenotype of interest”. More specifically, suppose P is
the phenotype of interest, E is the selected endophenotype, and G represents an underlying genetic structure that fulfills the specified assumptions in calculating
heritability, then the proposed definition is:
( | ) ( ) ( | ) ( )
f E G = f E ⇒ f P G = f P (1) The endophenotype’s definition has two important features [2]. First, “imply” replaces
‘’if and only if’’ statement in Prentice’s definition of surrogate endpoints in avoidance
of a problematic implication arisen in Begg and Leung [11]. This change places
endophenotype in higher upstream of the pathway from genotype to phenotype.
Second, genetic heritability represents the proportion of variability attributable to
genetic factors and can be obtained in a variance component approach [12-13]. This is
genes and allows the likelihood of multiple gene influences.
Suppose the condition
( | , ) ( | )
f P E G = f P E (2)
Huang et al. [2] takes (2) in a variance component model as the operational
criterion for proposed endophenotype definition. It then requires heritability of
phenotype becomes null, conditioning on candidate endophenotype, and implies
genetic heritability of phenotype is captured by endophenotype.
Given an observed phenotype, significance of (2) can be judged through the
following variance component analysis for discrete trait [2, 14]:
2 2 2 2 2 2 2 , , , , (0, ) (0,[ ]) (3) cov( , ) 2 , ij H H ij H ij ij ij ij R ij A D C ij ik ij ik A ij ik D ij ik C P E x G Normal G Normal G G j k α γ τ ε ε σ σ σ σ φ σ σ λ σ = + + + + + + = + Δ + ≠ ∼ ∼
(4) Where αH is a baseline mean, Eij is his/her corresponding specified endophenotype. The termεijis the residual error term representing the effect of non-family factors. The term G is the random effect for the underlying genetic ij
structure. The term φij ik, denotes the kinship coefficient between individual and ij ik : the probability of randomly drawing a single allele in individual ij that is identical by descent (ibd) to a single allele at the same locus
randomly drawn from individualik. The termΔ is the probability that both ij ik, alleles at a locus are shared ibd by individuals ij and .ik the elements,λij ik, , is
simply binary indicator denoting whether two individuals live together (λij ik, =1 ) or apart (λij ik, =0 ).
The (broad sense) heritability ofP , conditional on ij E is ij
2 2 2 2 2 2 A D A D C R h σ σ σ σ σ σ + = + + + (4) The significance of rejecting the hypothesis h=0 indicates the fulfillment of (2).
Table 1 details the term φij ik, andΔ values for selected relative pairs and the total ij ik, genetic variances that these imply [15].
Table 1 Genetic components of variance assuming random mating.
Relationship φ Δ Genetic covariance
same person 1/ 2 1 2 2 A D σ +σ Parent-child 1/ 4 0 2 1/ 2σA Full sibling 1/ 4 1/ 4 2 2 1/ 2σA+1/ 4σD Half sibling 1/ 8 0 2 1/ 4σA Monozygous twins 1/ 2 1 2 2 A D σ +σ Grandparent-grandchild 1/ 8 0 1/ 4σA2 Uncle/aunt-nephew/niece 1/ 8 0 2 1/ 4
σ
A First cousins 1 /16 0 1/ 8σ2ADouble first cousins 1/ 8 1 /16 2 2
1/ 4σA+1/16σD
For a discrete phenotype of ordinal scale, the liability threshold model can be used
in the preceding variance component setting [16-17]. The model postulates the
existence of an unobserved continuous trait (i.e., liability L ), and a set of thresholds ij
1, ,...,2 K 1
t t t − that partition the liability distribution into intervals corresponding to distinct phenotypic states:
1 1 2 1 1, 2, , ij ij ij K ij if L t if t L t P K if t − L < ⎧ ⎪ < < ⎪ = ⎨ ⎪ ⎪ < ⎩
The liabilityL is then assumed to follow the same distribution as theij P in model (3) ij
and heritability can be obtained based on the liability.
Huang et al. [2] have provided the index to evaluate the validation of
endophenotypes that is the proportion of heritability explained ( PHE ) by the
endophenotype defined as 1 NE h PHE h = − (5) WherehNEis the heritability calculated from the variance component analysis (3) without including the endophenotype E with any other covariates. A good ij
endophenotype is one that explains a large proportion of heritability, thus, the
2.2.2 Variance ofPHE
Hsieh et al. [4] redefined
2 ( ) 1 2 2 2 2 2 ( ) 2 2 2 2 2 2 ( ) 3 2 2 2 2 ( ) 2 2 2 2 4 t A A D C R t D A D C R t D A D C R t A D C R h h h h σ σ σ σ σ σ σ σ σ σ σ σ σ σ σ σ σ σ σ = + + + = + + + = + + + = + + +
Where t is representing the different models.
So the broad-sense heritability h≡h1(1)+h2(1). Similarly, hNE ≡h1(2)+h2(2) .
Hsieh et al. [4] use the delta method [4, 18] to evaluate the variance of PHE .
The first-order Taylor approximations give
2 2 4 3 (1) (1) (1) (1) 1 2 1 2 2 2 (2) 1 4
(1
)
(
)
1
( )
(
)
2
( ,
)
1
{
(
)
(
)
2
(
,
)}
{
(
NE NE NE NE NE NE NE h h NE NE h h h h h hh
h
Var
Var
h
h
Var h
Var h
Cov h h
Var h
Var h
Cov h
h
Var h
μ
μ
μ
μ
μ
μ
μ
μ
−
=
≈
+
−
≈
+
+
+
(2) (2) (2) 2 1 2 (1) (2) (1) (2) 1 1 1 2 3 (1) (2) (1) (2) 2 1 2 2)
(
)
2
(
,
)}
2
{
(
,
)
(
,
)
(
,
)
(
,
)}
NE h hVar h
Cov h
h
Cov h
h
Cov h
h
Cov h
h
Cov h
h
μ
μ
+
+
−
+
+
+
Usehˆ1(1) +hˆ2(1)to estimateμh and usehˆ1(2)+hˆ2(2)to estimate
NE h
μ
. The estimators for (1) (1) ( 2) ( 2) 1 2 1 2 ˆ , ˆ ,ˆ , and ˆThe remaining terms, such as (1) (1) (2) (2) (1) (1) (2) (2) 1 2 1 2 1 2 1 2 (1) (2) (1) (2) (1) (2) (1) (2) 1 2 1 2 2 1 2 2 ˆ ˆ ˆ ˆ ˆ ˆ ˆ ˆ ( ), ( ), ( ), ( ), ( , ), ( , ), ˆ ˆ ˆ ˆ ˆ ˆ ˆ ˆ ( , ), ( , ), ( , ), ( , )
Var h Var h Var h Var h Cov h h Cov h h
Cov h h Cov h h Cov h h Cov h h
will be solved in 2.2.3 [4].
2.2.3 The covariance of ˆq( )t and ˆ( )*t*
q
h h
Suppose two models are
'(1) (1) (1) (1) ij ij ij ij P =x β +G +ε and '( 2) ( 2) ( 2) ( 2) ij ij ij ij P =x β +G +ε Where ( ) 2 ( ) ( ) ( ) ( ) ( ) 1 2 3 4 (0, ( ) ) (0, (1 ) ) t t t t t t ij N R N h h h h ε ∼ σ ≡ − − − ( ) 2 2 2 ( ) (0, ( ) ) t t ij A D C G ∼N σ +σ +σ ≡ N(0,h h1( )t 4( )t +h h2( )t 4( )t +h h3( )t 4( )t ), and 2 2 2 , , , ( , ) [ ] (2 )t ij ik ij ik A ij ik D ij ik C Cov G G j≠k = φ σ + Δ σ +λ σ ≡ ( ) ( ) ( ) ( ) ( ) ( ) , 1 4 , 2 4 , 3 4 2 t t t t t t ij ikh h ij ikh h ij ikh h
φ + Δ +λ , andGijus the random effect for the underlying
genetic structure. Assumed I is the total number of family and there aren members in i
the ith family. Leth( )t =(h1( )t ,h2( )t ,h3( )t ,h4( )t ), then we have
(
)
* * ( ) ( ) 1 ' ' ( ) ( ) ( ) ( ) 1( ) 1( ) ( ) ( ) 1( ) ( ) ( ) ( ) ( ) 1 ' ( ) 1( ) ( ) ( ( ) ˆ ˆ ( , ) ˆ ˆ ˆ ˆ t t q q t t t t R t t t t t r r r r r t t t t r q q q q t t t r r r t q Cov h h V W V V W W S V W h h h h V W S V h − − − − = − ⎡ ⎧⎛ ⎞ ⎛ ⎞ ⎛ ⎞ ⎛ ⎞⎫⎤ ∂ ∂ ∂ ∂ ⎪ ⎪ ⎢ ⎜ ⎟ ⎜ ⎟ ⎜ ⎟ ⎜ ⎟ ⎥ ≈⎢ ⎨ − + ⎬⎥ ⎜∂ ⎟ ⎜ ∂ ⎟ ⎜ ∂ ⎟ ⎜ ∂ ⎟ ⎪⎝ ⎠ ⎝ ⎠ ⎝ ⎠ ⎝ ⎠⎪ ⎢ ⎩ ⎭⎥ ⎣ ⎦ ⎛∂ ⎞ ⎜ ⎟ × − ⎜ ∂ ⎟ ⎝ ⎠∑
(
)(
)
* * * ' ( ) ' ) ( ) ( ) 1( ) ( ) 1 ˆ ˆ t R t t t t r r r t r q V S V W h − = ⎡ ⎧ ⎛ ⎞⎫⎤ ⎢ ⎪ ⎜∂ ⎟⎪⎥ − ⎨ ⎬ ⎢ ⎜ ⎟ ⎥ ⎪ ∂ ⎪ ⎢ ⎝ ⎠ ⎥ ⎩ ⎭ ⎣ ⎦∑
(
)
* * * * * * * * * * * * * * * * * 1 ' ' ( ) ( ) ( ) ( ) 1( ) 1( ) ( ) ( ) 1( ) ( ) ( ) ( ) ( ) 1 * * ˆ ˆ 1, 2, 3, 4 1, 2, 3, 4 1, 2 1 t t t t R t t t t t r r r r r t t t t r q q q q V W V V W W S V W h h h h q q t t − − − − = ⎡ ⎧⎛ ⎞ ⎛ ⎞ ⎛ ⎞ ⎛ ⎞⎫⎤ ⎢ ⎪⎪⎜∂ ⎟ ⎜ ∂ ⎟ ⎜∂ ⎟ ⎜∂ ⎟⎪⎪⎥ ⎢ ⎥ × ⎨⎜ ⎟ ⎜ ⎟ − +⎜ ⎟ ⎜ ⎟⎬ ⎢ ⎪⎜ ∂ ⎟ ⎜ ∂ ⎟ ⎜ ∂ ⎟ ⎜ ∂ ⎟⎪⎥ ⎢ ⎪⎩⎝ ⎠ ⎝ ⎠ ⎝ ⎠ ⎝ ⎠⎪⎭⎥ ⎣ ⎦ = = = =∑
, 2 Where ( ) ( ) ( ) ( ) ( ) ( ) ( ) ( ) ( ) 1 1 1 2 1 ( ) ( ) ( ) ( ) ( ) ( ) ( ) ( , ,..., ,..., ) ', ,( ; , ) as given by Covariance after transformation in table 1,
r r r t t t t t t t t t r r r r r r rn rn rn t t t rj rj rj t t t t r r S r r r r r r r r r P x V E S h β β = = − = ( )2 ( ) ( ) ( ) ( ) ( ) ( ) ( ) 2 , , , 4 , and t ij t r r t t t t jm im il jl ij t ij t jm il jm il
for the i jth pairs W
for the i jth and l mth pairs for the i jth pairs
W h h h h σ σ σ σ σ σ σ σ σ σ σ × ⎧ ⎪ = ⎨ + ⎪⎩ ∂ ∂ ∂ = ∂ ∂ ∂ + ∂ ∂ , , jl im
jl im for the i jth and l mth pairs
h h σ σ σ σ ⎧ ⎪ ⎪ ⎨ ∂ ∂ ⎪ + + ⎪ ∂ ∂ ⎩
The table 2 shows the interested derivative of covariance components,
relatedhˆ1and hˆ2 for relative pairs.
Table 2 The derivative of covariance components for relative pairs.
Relationship 1 V h ∂ ∂ 2 V h ∂ ∂ 1 V h ∂ ∂ 2 V h ∂ ∂ Same person 0 0 0 0 Parent-child 1/ 2h4 0 1/ 2 0 Full sibling 1/ 2h4 1/ 4h4 1/ 2 1/ 4 Half sibling 1/ 4h4 0 1/ 4 0 Monozygous twins h 4 h 4 1 1 Grandparent-grandchild 1/ 4h4 0 1/ 4 0 Uncle/aunt-nephew/niece 1/ 4h4 0 1/ 4 0
Based on Table 2, Heish et al. [4] express the result of the above-mentioned as follow:
(
)
* * ( ) ( ) ' ( ) ( ) ( ) 1( ) 1( ) ( ) ( ) 4 ( ) ( ) 1 1 ' ( ) ( ) ( ) 1( ) ( ) 4 ( ) ( ) 4 ( ) ( ) 4 ( ˆ ˆ ( , ) ˆ ˆ ˆ ˆ ˆ ˆ t t q q t t R t r t t t t r r t t r q q t t t r t r t t t q q t t r q Cov h h V W h W W S V h h V V h W h h h V h h − − = − − ⎡ ⎧ ⎛ ⎞ ⎛ ⎞ ∂ ∂ ⎪ ⎢ ⎜ ⎟ ⎜ ⎟ ≈⎢ ⎨ − ⎜ ∂ ⎟ ⎜ ∂ ⎟ ⎪ ⎝ ⎠ ⎝ ⎠ ⎢ ⎩ ⎣ ⎤ ⎫ ⎛∂ ⎞ ⎛∂ ⎞ ⎪⎥ ⎜ ⎟ ⎜ ⎟ + ⎬⎥ ⎜∂ ⎟ ⎜∂ ⎟ ⎪ ⎝ ⎠ ⎝ ⎠ ⎭⎦⎥ ∂ × ∂∑
(
)
(
)
(
)
* * * * * * * * * * * * * * * * ( ) ' ( ) 1( ) ( ) ( ) ( ) ( ) 1( ) 4 ) ( ) 1 ' ( ) ( ) ( ) 1( ) 1( ) ( ) ( ) 4 ( ) ( ) ˆ ˆ ˆ ˆ ˆ ˆ ˆ ˆ t R t t t t t t t r r r r r t t r q t t t r t t t t r r t t r q q V W S V S V W h h V W h W W S V h h − − = − − ⎡ ⎧ ⎛ ⎞ ⎛ ⎞ ⎫⎤ ⎢ ⎪ ⎜ ⎟ ⎜∂ ⎟ ⎪⎥ − − ⎢ ⎨ ⎜ ⎟ ⎜ ⎟ ⎬⎥ ⎢ ⎪ ⎝ ⎠ ⎜ ∂ ⎟ ⎪⎥ ⎢ ⎩ ⎝ ⎠ ⎭⎥ ⎣ ⎦ ⎧ ⎛ ⎞ ⎛ ⎞ ⎪ ⎜∂ ⎟ ⎜ ∂ ⎟ × ⎨ − ⎜ ⎟ ⎜ ⎟ ⎪ ⎝∂ ⎠ ⎝ ∂ ⎠ ⎩∑
* * * * * * * 1 1 ' ( ) ( ) ( ) 1( ) ( ) 4 ( ) ( ) 4 * * ˆ ˆ 1, 2, 3, 4 1, 2, 3, 4 1, 2 1, 2 R t t t r t r t t t q q V V h W h h h q q t t = − − ⎡ ⎢ ⎢ ⎢ ⎣ ⎤ ⎫ ⎛∂ ⎞ ⎛∂ ⎞ ⎪⎥ ⎜ ⎟ ⎜ ⎟ + ⎜ ⎟ ⎜ ⎟ ⎬⎥ ⎪ ∂ ∂ ⎥ ⎝ ⎠ ⎝ ⎠ ⎭⎦ = = = =∑
First cousins 1/ 8h4 0 1/ 8 0Double first cousins 1/ 4h4 1/16h4 1/ 4 1 /16
2.2.4 Hypothesis test
For having more statistical meaning of PHE , we utilize the confidence interval to
get more information about PHE . We hope to find a value that it means that there
exist a useful endophenotype when PHE value is larger than the value. That is, do
one-sided confidence interval.
The hypothesis is 0 1 : : H PHE a H PHE a = ⎧ ⎨ > ⎩
Under null hypothesis and significance level ofα , we rejectH if the lower bound 0 of one-sided confidence interval of PHE ,PHE−Z1−α×s e PHE. .( ), is larger than a .
2.3 The whole-genome association test for quantitative outcomes
For each of the genotyped SNP markers, we are interested in testing whether
observed genotypes and quantitative phenotypes are associated. We let Gijmdenote the observed genotype at makermfor individual j in family i .
We label two allele “A” and “a” and define a genotype score, gijm
0 if / 1 if / 2 if / ijm ijm ijm ijm G a a g G A a G A A ⎧ = ⎪ =⎨ = ⎪ = ⎩
First we built the model for each SNP
( ij) g ij x ij
E P = +μ β g +β x
Hereμis the population mean, βgis the additive effect for each SNP, andβxis a
vector of covariate effects [19].
Chen et al. [19] extend the model with
( ij) g ij x ij
whereg is the expected genotype score and define as ij ( | , , ) 2 ( / | , , ) ( / | , , ) ijm ijm i ijm i ijm i g E g G F P G A A G F P G A a G F θ θ θ = = = + =
whereθ is a vector of intermarker recombination fractions and F is a vector of allele frequencies for each marker. To allow for correlation between different observed
phenotypes within each family, we define the variance-covariance matrix Ω for i family i as 2 2 2 2 2 2 , , , 2 A D C ijk ij ik A ij ik D ij ik C if j k if j k σ σ σ φ σ σ λ σ ⎧ + + = ⎪ Ω = ⎨ + Δ + ≠ ⎪⎩
Chen et al. [19] provide an approach is to first fit a simple variance-components
model to the data (with parametersμ β σ σ, x, A2, R2 but without parametersβ σg, D2). This model provides a vector of fitted values for each family, which they denoteE P( )i (base), and an estimate of the variance-covariance matrix for each family, which they
denoteΩi(base). Using these two quantities, we define the score statistic
[
]
[
]
[
]
2 1 ( ) ( ) 1 ( ) ( ) ' ( ) ( ) ' ( ) base base i i i i i i SCORE base i i i i i i g E g y E y T g E g g E g − − ⎧ − ⎡Ω ⎤ ⎡ − ⎤⎫ ⎨ ⎣ ⎦ ⎣ ⎦⎬ ⎩ ⎭ = ⎡ ⎤ − ⎣Ω ⎦ −∑
∑
Whereg is a vector with expected genotype scores for each individual in the i th
i family, calculated conditional on the available marker data, and E g( i)is a vector with identical elements that give the unconditional expectation of each genotype score.
SCORE
T is approximately distributed as χ2with 1 df. As usual, LOD scores were defined as
2
score / 2 ln(10)
2.4 The preprocessing method for gene expression levels
We interpret the three main steps of data preprocessing.
2.4.1 Background adjustment
Because partial measured probe intensities maybe caused by non-specific
hybridization or the noise in the optical detection system, background adjustment is
essential to rid of these intensities not exactly expressed from genes. Observed probe
intensities need to be adjusted to give the accurate expression levels of specific
hybridization [20]. Some methods make use of MM probes to adjust, but some are
not.
2.4.2 Normalization
During the process of carrying out the microarray experiment involving multiple
arrays, there are many obscuring sources of variation involved, such as physical
problems with the arrays, laboratory conditions, hybridization reactions, labeling, and
scanner difference. In order to compare measurements from different arrays, implying
different tissue, some proper normalization is necessary.
2.4.3 Summarization
Due to Affymetrix platform designing multiple probes to represent a gene,
summarization is needed to combine these probe intensities to a single value. For each
gene, the background adjusted and normalized intensities are used to be summarized
into one measurement that estimates the expression level.
2.4.4 RMA ) ( : , .... , 1 : ,...., 1 ) ( : ,...., 1 gene set probe the ng representi G g gene the in pair probe the ng representi J j sample array different the ng representi I i = = =
RMA [21- 22], Robust Multi-array Analysis, is an expression measure consisting of
three particular preprocessing steps: convolution background correction, quantile
the median polish algorithm. These RMA authors proposed a procedure ignoring the
MM intensities and using only the PM intensities.
The RMA convolution background correction method is motivated by looking at
the distribution of probe intensities. The model observed PM as the sum of a
background intensity bgijgcaused by optical and nonspecific binding, and signal intensitysijg.
, 1, , , 1, , , 1, ,
ijg ijg ijg
PM =bg +s i= … I j= … J g = … G
Under the model above, the background corrected probe intensities will be given by
( ijg)
B PM , where B PM( ijg)≡E s( ijg |PMijg). To obtain a computationally feasible
) (⋅
B we consider the closed-form transformation obtained when assuming thatsijgis distributed exponential and bgijgis distributed normal, and the results obtained using
) (⋅
B work well in practice [21-22].
Next, perform the quantile normalization, which is to make the distribution of
probe intensities for each array the same [22, 23]. In order to summarize the probe
intensities, RMA introduced a log scale linear additive model. The model is:
( ij) i j ij
T PM = +e a +ε ,
where PMijg represents the PM intensity of array i=1,...,I and probe pair j=1,...,J, for any given probe set g. T
( )
⋅ represents the transformation that background corrects, normalizes, and logs the PM intensities, e represents the log2 i scale expression value found on arrays i , aj represents the log scale affinity effects for probes j , and ε represents error [22, 24]. To protect against outlier probes, they ijThe estimate of e as the log scale measure of expression refers to as robust i multi-array average (RMA).
2.5 The differential expression methods 2.5.1 Fold-change
Fold-change analysis is used to identify genes with expression ratios or differences
between a treatment and a control that are outside of a given cutoff or threshold.
Intensity values may be compared using ratio,log (2 ratios , or difference. Biologist ) favors fold-change equal to 2 as the threshold of differential expression.
2.5.2 Two sample t-test
The simplest statistic method for comparing means between two groups is two
sample t-test. The variances of the two samples may be assumed to be equal or
unequal. The approach of unequal variance assumption is also called Welch’s t-test.
For any given gene g, suppose that the number of samples in sample 1 and in sample
2 are M and N respectively. Here we describe the two tests briefly.
Two sample t-test for equal variance: 2 1 1 2 1 2 0 1 2 1 1 2 2 2 2 2 1 1 1: ,...., ~ ( , ) 2 : ,..., ~ ( , ) H : : : ~ , 1 1 ( ) ( ) . 2 g gM g gN M N p M N i i i i p sample X X N sample Y Y N versus H X Y test statistic T S M N X X Y Y where S M N μ σ μ σ μ μ μ μ + − = = = ≠ − + − + − = + −
∑
∑
Two sample t-test for unequal variance (Welch’s t-test): 2 1 1 1 2 1 2 2 0 1 2 1 1 2 2 2 2 2 2 2 1 1 2 2 1: ,...., ~ ( , ) 2 : ,..., ~ ( , ) H : : : ~ ( ), ( ) 1 1 ( ) , ( ) 1 1 ( g gM g gN v X Y M N X i Y i i i X Y sample X X N sample Y Y N versus H X Y
test statistic T approximately S S M N where S X X S Y Y and M N S S M N μ σ μ σ μ μ μ μ ν = = = ≠ − + = − = − − − + =
∑
∑
2 4 4 2 2 ) . ( 1) ( 1) X Y S S M M − +N N−After performing the test and the conclusion leads to rejectH , we consider that 0 this gene is a differentially expressed gene.
2.5.3 SAM (Significance Analysis of Microarrays)
It was proposed by Tusher, Tibshirani and Chu (2001) [22, 25]. The method is
based on a modified version of the standard t-statistic to adjust the high variance
probably caused by a low expression level.
The “relative difference”dg, g=1, ,pgenes:
0 g g g r d s s = +
Here rg is a score, sg is a standard deviation, and s is an exchangeability 0 factor in the denominator to ensure that the variance ofd is independent of gene g
2 1 2 2 1 2 1 2 1 2 1 2 ( ) ( ) 1 1 ( ) , 2 g g g gi g gi g i group i group g r x x x x x x s n n n n ∈ ∈ = − − + − = + × + −
∑
∑
where xg1 and xg2 are defined as the average levels of expression for gene g in group 1 and group 2, and xgi is defined as the expression level for gene g and sample i. Group 1 and 2 have n and 1 n genes, respectively. Then rank all genes by 2 the observed relative difference d and denote the new arrangements asg d( g). B sets
of permutations of the samples are taken to obtain the expected relative difference
* ) ( g
d by a similar way (For more details, see [22, 25-26]). A scatter plot of d( g) vs.
* ) ( g
d is used and the genes apart from the * ) ( ) (g dg
d = line by a distance greater than the threshold Δ are regarded as differentially expressed genes.
2.6 Multiple testing procedures
If thousands of hypotheses are tested simultaneously, the probability of false positives
by chance increases. We use an example to understand the question: when a
two-sample t-test is performed on a gene, the probability by which the gene’s
expression level will be considered significantly different between two groups of
samples is expressed by the p-value. The p-value is the probability that a gene’s
expression levels are different between the two groups due to chance. A p-value of
0.05 signifies a 5% probability that the gene’s mean expression value in one condition
is different than the mean in the other condition by chance alone. If 10,000 genes are
Table 3 probability of calling 1 or more false positives by chance.
Number of genes
tested (N)
False positives
incidence
Probability of calling 1 or more false positives
by chance (100(1 0.95 )− N )
1 1/20 5%
2 1/10 10%
20 1 64%
100 5 99.40%
In microarray data analysis, false positives are genes that are found to be
statistically different between conditions, but are not in reality. We need to adjust
p-values derived from multiple statistical tests to correct for occurrence of false
positives.
2.6.1 Type I error rates
The null hypotheses in M tests,H0(1),…,H M0( ), we conduct 2 2× table to interpret the different number of tests in different conditions.
Table 4 Type I and type II errors in multiple hypothesis testing.
Number of not rejectingH0 Number of rejectingH 0 Number of true Non-differential genes (H is true) 0 U (false positive) (type I error) V 0 M Number of true Differential genes (H is not true) 0 (false negative) (type II error) T S 1 M
Type I error rates is defined as a parameters,θ θ= (FV R, ) , of the joint
distributionFV R, of the numbers of Type I errors V and the number of rejecting hypotheses R . Such a general representation covers the following commonly-used
Type I error rates [20].
(1) Generalized family-wise error rate (gFWER), or probability of at least (k+1)
Type I errors.
( ) Pr( ).
gFWER k ≡ V >k
When k=0, the gFWER is the usual family-wise error rate (FWER), or probability
of at least one Type I error. FWER k( )≡Pr(V >0).
(2) Tail probabilities for the pro portion of false positives (TPPFP) among the
rejected hypotheses,
( ) Pr( / ), (0,1).
TPPFP q ≡ V R>q q∈
(3) False discovery rate (FDR), or expected value of the proportion of false
positives among the rejected hypotheses (Benjamini and Hochberg, 1995),
[
/]
.FDR≡E V R
The convention that V /R≡0 if R=0is used.
2.6.2 Adjusted p-values
Given M null hypotheses being tested H0(1),…,H M0( ) , the adjusted
p-valueP0a( )m , for null hypothesisH m , is defined as the smallest Type I error 0( ) levelα at which one would rejectH m , that is, 0( )
[ ]
{
}
0a( ) inf 0,1 : ( ) ( ) , 1, ,
P m ≡ α∈ T m ∈C m m= … M
Where T m is the test statistic and ( ) C m is the rejection region for test( ) m .It need to have null distribution of ( )T m [22].
hypothesis is stronger. The difference between adjusted p-value and unadjusted
p-values that the unadjusted p-value,p m , for null hypothesis0( ) H m is the smallest 0( ) type I error rate of the single hypothesis testing procedure at which one would
rejectH m0( ) .
2.6.3 The q-value
Storey et al. [27] define a new false discovery rate, pFDR
0
(V | 0) Pr( is true | )
pFDR E R H T C
R
= > = ∈
WhereT is the test statistic and C is the rejection region. The term ’positive’ has been
added to reflect the fact that we are conditioning on the event that positive findings
have occurred.
As a natural extension to pFDR , the q-value has the following definition [27].
Definition 1 For an observed statistic T =t, the q-value of t is defined to be
{ : }
( )
inf {
( )}
C t Cq t
pFDR C
∈=
Definition 2 For a set of hypothesis tests conducted with independent p-values, the q-value of the observed p-value p is
( )
inf{
( )}
p
q p
pFDR
γ≥
γ
=
Where the nested set of rejection regions take the form[0, ]γ .
The q-value was discussed, which is the pFDR analogue of the p-value. Whereas it
can be inconvenient to have to fix the rejection region or the error rate beforehand, the
2.7 Datasets
The dataset we used is from the Gene Expression Omnibus (GEO) in NCBI. The
Gene Expression Omnibus (GEO) is a public repository that archives and freely
distributes microarray and other forms of high-throughput data submitted by the
scientific community. In addition to data storage, a collection of web-based interfaces
and applications are available to help users query and download the experiments and
gene expression patterns stored in GEO.
In 2007, Moffatt et al. [5] brought up this paper about asthma and offered
expression file free to download. Moffatt et al. [5] mentioned that the study subjects
were recruited from family (MRC-A) and case-control panels (MAGICS and UK-C).
The family panel included a 207 predominantly (99 %) nuclear families (MRC-A).
these were recruited through a proband with severe (step 3) childhood onset asthma
and contained 295 sib pairs, 11 half-sib pairs and 3 singletons (counting all possible
sibs). The study included siblings regardless of asthma status. Lymphoblastoid cell
lines (LCLs) were derived from peripheral blood Iymphocytes on probands and
siblings. Cells were harvested at log phase from roller cultures in the first growth after
transformation. Global gene expression in Epstein-Barr virus Iymphoblastoid cell
lines (EBVL) was measured with the Affymetrix HG-U133 Plus 2.0 chip in family
panel. The other one, in case-control panel, included 437 non-asthmatic Caucasian
UK controls (UK-C) children, 728 asthmatic children of German in the Multicentre
Asthmatic Genetics in Childhood Study (MAGICS) study with physician-diagnosed
asthma for comparison with 694 reference children recruited in the cross sectional
International Study of Asthma and Allergies in Childhood (ISAAC) study. The study
genotyped all children in the primary association study with the Illumina Sentrix
HumanHap300 BeadChip. Additional, the study typed the parents and children in the
This paper had an own database, “mRNA by SNP Browser”, that provides
graphical overviews of whole-genome association studies of datasets with very rich
phenotypic information, such as global surveys of gene expression. The software
incorporates a generic eQTL database and provides a graphic interface for browsing
association between 54,675 transcript levels and 406,912 SNPs. For each transcript,
the browser can tabulate and plot association test statistics (p-value<0.001), estimates
of effect size and allele information across the genome. The browser automatically
links results to the UCSC genome browser where users can examine each transcript in
its genomic context. In addition to browsing the results by transcript or by position,
results can be searched for information on specific SNPs. LD and tag information are
3 Materials and Methods
DNA transcribes to mRNA, mRNA translates to protein which may affect the
disease. Because mRNA is closer to DNA and gene expression value is measured in
mRNA, we use global gene expression of probe sets as endophenotypes in search for
the susceptibility genes underlying asthma. Gene expression values are preprocessed
by the robust multi-array averaging (RMA) to adjust for background noises,
normalize expression levels and summarize multiple probs. We judge which gene
expressions of probe sets are endophenotype by using the index PHE and do
hypothesis test of PHE=0 : Endophenotype and phenotype share no genes. For the
problem of multiple testing, we utilize the q-value to control for the false discovery
rate (FDR) for significance judgment. We also perform genome-wide association tests
for each gene expression of probe set.
We then derive SNPs under four conditions; they are SNPs (1) significantly
associated with gene expressions of all probes sets, (2) significantly associated with
gene expressions of probe sets with PHEs greater than zero, (3) significantly
associated with gene expressions of probe sets with PHEs significantly different from
zero at unadjusted p-values smaller than 0.05, and (4) significantly associated with
gene expressions of probe sets with PHEs significantly different from zero at q-values
smaller than 0.05.
Through systematic literature reviews (Hoffjan et al. [28], Ober et al. [29] and
Zhang et al. [30]), we identify 144 genes that have been reported to be associated with
asthma or atopy phenotypes. Among them, there are 25 genes that have been
repeatedly reported in six or more populations [29]. We further identify additional 125
genes that are related to these 25 genes. We then identify the overlap between these
analysis.
Finally, we are interests in realizing the genetic properties of gene expressions that
are identified as the endophenotypes. We use various plots to aid us observing some
phenomenon.
3.1 Datasets
In 2007, Moffatt et al. [5] brought up this paper about asthma. The subjects of this
study can be divided into two master parts: the case-control panel and the family
panel. We are interested in the family panel. There are 404 children from the family
panel. The family panel included 207 predominantly (99 %) nuclear families. These
were recruited through a proband with severe childhood onset asthma. The study
included siblings regardless of asthma status. Global gene expression in Epstein-Barr
virus Iymphoblastoid cell lines (EBVL) was measured with the Affymetrix HG-U133
Plus 2.0 chip in the family panel. The study genotyped 830 offspring and parents with
the Illumina Sentrix HumanHap-I BeadChip and 378 offspring with the Illumina
Sentrix HumanHap300 BeadChip in family panel.
We downloaded the files of Gene expression levels in 404 children from the family
panel. This dataset is available at 8http://www.ncbi.nlm.nih.gov/ego/, the GEO
accession: GSE8052 [6].
3.2 Statistical method of endophenotype
We use global gene expressions of probe sets as endophenotypes. Epstein-Barr
virus Iymphoblastoid cell lines (EBVL) were derived from participants and global
gene expression was measured with Affymetrix HG-U133 Plus 2.0 chip. One of the
indices determining the gene expression of probe set an endophenotype is the
proportion of heritability explained (PHE) by the endophenotype [2].
2 2 2 2 2 2 2 , , , , (0, ) (0,[ ]) cov( , ) 2 , ij H H ij H ij ij ij ij R ij A D C ij ik ij ik A ij ik D ij ik C P E Z G Normal G Normal G G j k α γ τ ε ε σ σ σ σ φ σ σ λ σ = + + + + + + = + Δ + ≠ ∼ ∼
WhereP is the observed phenotype in the jth member of the ith family,ij E is his/her ij
corresponding specified endophenotype, Z is his/her other covariates.ij εij is the
residual error term representing the effect of non-family factors.G is the random ij
effect for the underlying genetic structure. The componentsσ σ2A, D2 and σC2represent the variance arising from polygenic additive effects, polygenic dominance effects and
shared environmental effects, respectively.
The (broad sense) heritability ofP , conditional onij E is ij
2 2 2 2 2 2 A D A D C R h σ σ σ σ σ σ + = + + +
Then we calculate PHEs of all probe sets.
1 NE h PHE h = −
Where the term h is the heritability calculated from the variance component analysis
with disease status as response variable and covariates including the endophenotype
and other covariates. The term hNE is the heritability calculated from the variance component analysis with disease status as response variable and covariates without
endophenotype [2]. The greater of the PHE value, the more likely he intermediate
variable an endophenotype. Hsieh et al. [4] utilize the delta method to evaluate the
variance of PHE. For having more statistical meanings of PHE, We do hypothesis test
The hypothesis is 0 1 : 0 : 0 H PHE H PHE = ⎧ ⎨ > ⎩
UnderH0:PHE= , we suppose the phenotype and endophenotype share no genes 0 and significance level ofα =0.05. We rejectH if the lower bound of one-sided 0
confidence interval of PHE ,PHE−Z1−α×s e PHE. .( ), is larger than 0 and plot the confidence of interval of PHEs over all chromosomes.
If multiple hypotheses are tested simultaneously, the probability of false positives
by chance increases. We utilize the q-value which is a natural pFDR analogue of the
p-value to correct for occurrence of false positives. These q-values were calculated by
applying the QVALUE (8http://faculty.washington.edu/~jstorey/qvalue/) package.
WherehandhNEwere obtained from the results by performing variance component analysis using the SOLAR computer package [14], so PHE is estimated. The estimator ofs e PHE. .( )used delta method [4].
Then we classify PHEs into four conditions, there are total PHEs, PHEs higher than
zero, significant PHEs with unadjusted p-value smaller than the significant level of
5%, and significant PHEs with q-value smaller than the significant level of 5%.
3.3 The preprocessing and differential method
All analyses of expression value used quantile normalization, after performing
robust multi-array averaging (RMA), to remove non-biological variation, enforce
normality, deal with outlier and summary the intensity values [21-22, 24].
We want to know if a disease may be caused by large expression of particular genes
significance analysis of Microarrays (SAM) [22, 25-26]. The term q-value in SAM
output smaller than 0.05, it represents these probe sets may be different between
diseased and normal subset.
3.4 Genome-wide association tests for gene expressions
A database, “mRNA by SNP Browser”, that provides the results of Genome-wide
association test for each gene expression of probe set [31]. Association analysis was
applied with Merlin (FASTASSOC option), after probabilistically inferring missing
genotypes. This database is downloaded at
8
http://www.sph.umich.edu/csg/liang/asthma/.
We find significant SNPs from results of associate test between expression value of
each probe set and all SNPs genotypes with LOD score≥ 6 (about equivalent to the false discovery rate (FDR) = 0.056) [6]. We define these significant SNPs as
“eSNPs”.
3.5 Asthma genes and overlap rate
Asthma is a disease of chronic airway inflammation that affects nearly 155 million
individuals worldwide. Like other atopic diseases, asthma is a complex disorder
caused by interactions between multiple genes of small to modest effect and equally
important environmental factors. Asthma has an important genetic component but no
clear pattern of inheritance and heritability estimates of asthma vary between 36-79%.
To this day, there are over 100 genes that have been reported to be associated with
asthma or related phenotypes. We look for three review papers: (1) “Association
studies for asthma and atopic diseases: a comprehensive review of the literature” [28],
there are 64 genes in 251 papers about asthma or atopy from 1982 to 2002, (2)
![Table 1 details the term φ ij ik , and Δ values for selected relative pairs and the total ij ik , genetic variances that these imply [15]](https://thumb-ap.123doks.com/thumbv2/9libinfo/8379535.178111/18.892.130.656.573.1126/table-details-values-selected-relative-pairs-genetic-variances.webp)